
Optimization of a Lambda-RED Recombination Method for Rapid Gene Deletion in Human Cytomegalovirus

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Recombinant HCMV BAC Construction and Efficiency of the Optimized Method
2.2. Verification of the Deletion of HCMV BAC Constructions
2.3. Recombinant Virus Production and PCR Verification
3. Discussion
4. Materials and Methods
4.1. Bacteria Strains, Virus, Cells and Media
4.2. Plasmid DNA Purification
4.3. Construction of the Parental Strain: Transformation of E. coli DH5α Containing the CMV BADrUL131-Y4 BAC with the Plasmid pKD46
4.4. Preparation of the Linear DNA Kanamycin Resistance Cassettes
4.5. Preparation of the Electrocompetent Cells
4.6. Transformation of the E. Coli DH5α λ pir+BAC+pKD46 Strain with the KanR Cassette
4.7. PCR Amplification and BAC Integrity
4.8. Cell Transfection
4.9. Mutant Virus Propagation and PCR Verification
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Beltran, P.M.J.; Cristea, I.M. The life cycle and pathogenesis of human cytomegalovirus infection: Lessons from proteomics Pierre. Expert Rev. Proteom. 2015, 11, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the gates: Human cytomegalovirus entry and cell tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, 1–6. [Google Scholar] [CrossRef]
- Murphy, E.; Shenk, T.E. Human Cytomegalovirus Genome; Shenk, T.E., Stinski, M.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 325, ISBN 9783540773481. [Google Scholar]
- Boeckh, M.; Geballe, A.P. Science in medicine Cytomegalovirus: Pathogen, paradigm, and puzzle. J. Clin. Investig. 2011, 121, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Seitz, R. Human Cytomegalovirus (HCMV)-Revised. Transfus. Med. Hemotherapy 2010, 37, 365–375. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 1–15. [Google Scholar] [CrossRef]
- Manandhar, T.; Hò, G.G.T.; Pump, W.C.; Blasczyk, R.; Bade-Doeding, C. Battle between host immune cellular responses and HCMV immune evasion. Int. J. Mol. Sci. 2019, 20, 3626. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Ross, S.A.; Fowler, K.B. Congenital Cytomegalovirus Infection: Clinical Outcome. Clin. Infect. Dis. 2013, 57, S178–S181. [Google Scholar] [CrossRef]
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutré, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188. [Google Scholar] [CrossRef]
- Perello, R.; Vergara, A.; Monclus, E.; Jimenez, S.; Montero, M.; Saubi, N.; Moreno, A.; Eto, Y.; Inciarte, A.; Mallolas, J.; et al. Cytomegalovirus infection in HIV-infected patients in the era of combination antiretroviral therapy. BMC Infect. Dis. 2019, 19, 1–6. [Google Scholar] [CrossRef]
- Britt, W.J. Maternal immunity and the natural history of congenital human cytomegalovirus infection. Viruses 2018, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, S.; van Ranst, M.; Maes, P. Genomic and functional characteristics of human cytomegalovirus revealed by next-generation sequencing. Viruses 2014, 6, 1049–1072. [Google Scholar] [CrossRef] [PubMed]
- Martí-Carreras, J.; Maes, P. Human cytomegalovirus genomics and transcriptomics through the lens of next-generation sequencing: Revision and future challenges. Virus Genes 2019, 55, 138–164. [Google Scholar] [CrossRef]
- Suárez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human cytomegalovirus genomes sequenced directly from clinical material: Variation, multiple-strain infection, recombination, and gene loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef]
- Lassalle, F.; Depledge, D.P.; Reeves, M.B.; Brown, A.C.; Christiansen, M.T.; Tutill, H.J.; Williams, R.J.; Einer-Jensen, K.; Holdstock, J.; Atkinson, C.; et al. Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol. 2016, 2, 1–14. [Google Scholar] [CrossRef]
- Van Damme, E.; van Loock, M. Functional annotation of human cytomegalovirus gene products: An update. Front. Microbiol. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.K.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef]
- Patro, A.R.K. Subversion of immune response by human cytomegalovirus. Front. Immunol. 2019, 10, 1–7. [Google Scholar] [CrossRef]
- Balázs, Z.; Tombácz, D.; Szucs, A.; Csabai, Z.; Megyeri, K.; Petrov, A.N.; Snyder, M.; Boldogkoi, Z. Long-Read Sequencing of Human Cytomegalovirus Transcriptome Reveals RNA Isoforms Carrying Distinct Coding Potentials. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Ye, L.; Qian, Y.; Yu, W.; Guo, G.; Wang, H.; Xue, X. Functional Profile of Human Cytomegalovirus Genes and Their Associated Diseases: A Review. Front. Microbiol. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.M.; Yu, D. Human Cytomegalovirus: Bacterial Artificial Chromosome (BAC) Cloning and Genetic Manipulation. Curr. Protoc. Microbiol. 2012, 24, 14E. 4.1–14E. 4.33. [Google Scholar] [CrossRef][Green Version]
- Murphy, K.C.; Campellone, K.G. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 2003, 4, 1–12. [Google Scholar] [CrossRef]
- Wilkinson, G.W.G.; Davison, A.J.; Tomasec, P.; Fielding, C.A.; Aicheler, R.; Murrell, I.; Seirafian, S.; Wang, E.C.Y.; Weekes, M.; Lehner, P.J.; et al. Human cytomegalovirus: Taking the strain. Med. Microbiol. Immunol. 2015, 204, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Dekhtiarenko, I.; Icin-Šain, L.C.; Messerle, M. Use of recombinant approaches to construct human cytomegalovirus mutants. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1119, pp. 59–79. ISBN 9781627037884. [Google Scholar]
- Kalser, J.; Adler, B.; Mach, M.; Kropff, B.; Puchhammer-Stöckl, E.; Görzer, I. Differences in growth properties among two human cytomegalovirus glycoprotein O genotypes. Front. Microbiol. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Borst, E.-M.; Hahn, G.; Koszinowski, U.H.; Messerle, M. Cloning of the Human Cytomegalovirus (HCMV) Genome as an Infectious Bacterial Artificial Chromosome in Escherichia coli: A New Approach for Construction of HCMV Mutants. J. Virol. 1999, 73, 8320–8329. [Google Scholar] [CrossRef] [PubMed]
- Borst, E.M.; Benkartek, C.; Messerle, M. Use of Bacterial Artificial Chromosomes in Generating Targeted Mutations in Human and Mouse Cytomegaloviruses. Curr. Protoc. Immunol. 2007, 77, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Dulal, K.; Silver, B.; Zhu, H. Use of recombination-mediated genetic engineering for construction of rescue human cytomegalovirus bacterial artificial chromosome clones. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Patterson, C.E.; Yang, S.; Zhu, H. Coupling generation of cytomegalovirus deletion mutants and amplification of viral BAC clones. J. Virol. Methods 2004, 121, 137–143. [Google Scholar] [CrossRef]
- Yu, D.; Smith, G.A.; Enquist, L.W.; Shenk, T. Construction of a Self-Excisable Bacterial Artificial Chromosome Containing the Human Cytomegalovirus Genome and Mutagenesis of the Diploid TRL/IRL13 Gene. J. Virol. 2002, 76, 2316–2328. [Google Scholar] [CrossRef]
- Greaves, R.F.; Brown, J.M.; Vieira, J.; Mocarski, E.S. Selectable insertion and deletion mutagenesis of the human cytomegalovirus genome using the Escherichia coli guanosine phosphoribosyl transferase (gpt) gene. J. Gen. Virol. 1995, 76, 2151–2160. [Google Scholar] [CrossRef]
- Dutta, N.; Lashmit, P.; Yuan, J.; Meier, J.; Stinski, M.F. The human cytomegalovirus UL133-138 gene locus attenuates the lytic viral cycle in fibroblasts. PLoS ONE 2015, 10, e0120946. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Enquist, L.W. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 4873–4878. [Google Scholar] [CrossRef]
- King, M.W.; Munger, J. Editing the Human Cytomegalovirus Genome with the CRISPR/ Cas9 System. Virology 2019, 529, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Verdin, E. Viral gene drive in herpesviruses. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Das, S.; Ortiz, D.A.; Gurczynski, S.J.; Khan, F.; Pellett, P.E. Identification of Human Cytomegalovirus Genes Important for Biogenesis of the Cytoplasmic Virion Assembly Complex. J. Virol. 2014, 88, 9086–9099. [Google Scholar] [CrossRef]
- Yu, D.; Ellis, H.M.; Lee, E.C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5978–5983. [Google Scholar] [CrossRef]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005, 33, 1–12. [Google Scholar] [CrossRef]
- Stropes, M.P.M.; Miller, W.E. Functional analysis of human cytomegalovirus pUS28 mutants in infected cells. J. Gen. Microbiol. 2008, 89, 97–105. [Google Scholar] [CrossRef]
- Britt, W.J.; Jarvis, M.; Seo, J.-Y.; Drummond, D.; Nelson, J. Rapid Genetic Engineering of Human Cytomegalovirus by Using a Lambda Phage Linear Recombination System: Demonstration that pp28 (UL99) Is Essential for Production of Infectious Virus. J. Virol. 2004, 78, 539–543. [Google Scholar] [CrossRef]
- Gurczynski, S.J.; Das, S.; Pellett, P.E. Deletion of the Human Cytomegalovirus US17 Gene Increases the Ratio of Genomes per Infectious Unit and Alters Regulation of Immune and Endoplasmic Reticulum Stress Response Genes at Early and Late Times after Infection. J. Virol. 2014, 88, 2168–2182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marchini, A.; Liu, H.; Zhu, H. Human Cytomegalovirus with IE-2 (UL122) Deleted Fails To Express Early Lytic Genes. J. Virol. 2001, 75, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Ligat, G.; Jacquet, C.; Chou, S.; Couvreux, A.; Alain, S.; Hantz, S. Identification of a short sequence in the HCMV terminase pUL56 essential for interaction with pUL89 subunit. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, K.L.; Weyell, A.; Subramanian, N.; Wu, Z.; Sinzger, C. A TB40/E-derived human cytomegalovirus genome with an intact US-gene region and a self-excisable BAC cassette for immunological research. Biotechniques 2017, 63, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef]
- Murphy, K.C. λ Recombination and Recombineering. EcoSal Plus 2016, 7, 1–70. [Google Scholar] [CrossRef]
- Hashimoto-Gotoh, T.; Franklin, F.C.H.; Nordheim, A.; Timmis, K.N. Specific-purpose plasmid cloning vectors I. Low copy number, temperature-sensitive, mobilization-defective pSC101-derived containment vectors. Gene 1981, 16, 227–235. [Google Scholar] [CrossRef]
- Fels, U.; Gevaert, K.; van Damme, P. Bacterial Genetic Engineering by Means of Recombineering for Reverse Genetics. Front. Microbiol. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Noad, R.J.; Stewart, M.; Boyce, M.; Celma, C.C.; Willison, K.R.; Roy, P. Multigene expression of protein complexes by iterative modification of genomic bacmid DNA. BMC Mol. Biol. 2009, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, X.; Mehboob, A.; Hu, X.; Yi, Y.; Li, Y.; Zhang, Z. A construction strategy for a baculovirus-silkworm multigene expression system and its application for coexpression of type I and type II interferons. Microbiologyopen 2020, 9, 1–8. [Google Scholar] [CrossRef]
- Cottingham, M.G. Genetic Manipulation of Poxviruses Using Bacterial Artificial Chromosome Recombineering. In Vaccinia Virus and Poxvirology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 37–57. ISBN 9781617798757. [Google Scholar]
- Yu, Z.; Lu, J.; Yu, H.; Yan, Q.; Zuo, L.; Li, G. A precise excision of the complete Epstein-Barr virus genome in a plasmid based on a bacterial artificial chromosome. J. Virol. Methods 2011, 176, 103–107. [Google Scholar] [CrossRef]
- Muth, D.; Meyer, B.; Niemeyer, D.; Schroeder, S.; Osterrieder, N.; Müller, M.A.; Drosten, C. Transgene expression in the genome of middle east respiratory syndrome coronavirus based on a novel reverse genetics system utilizing red-mediated recombination cloning. J. Gen. Virol. 2017, 98, 2461–2469. [Google Scholar] [CrossRef]
- Hollenback, S.M.; Lyman, S.; Cheng, G. Recombineering-Based Procedure for Creating BAC Transgene Constructs for Animals and Cell Lines. Curr. Protoc. Mol. Biol. 2011, 95, 23.14.1–23.14.28. [Google Scholar] [CrossRef]




{kind=link}
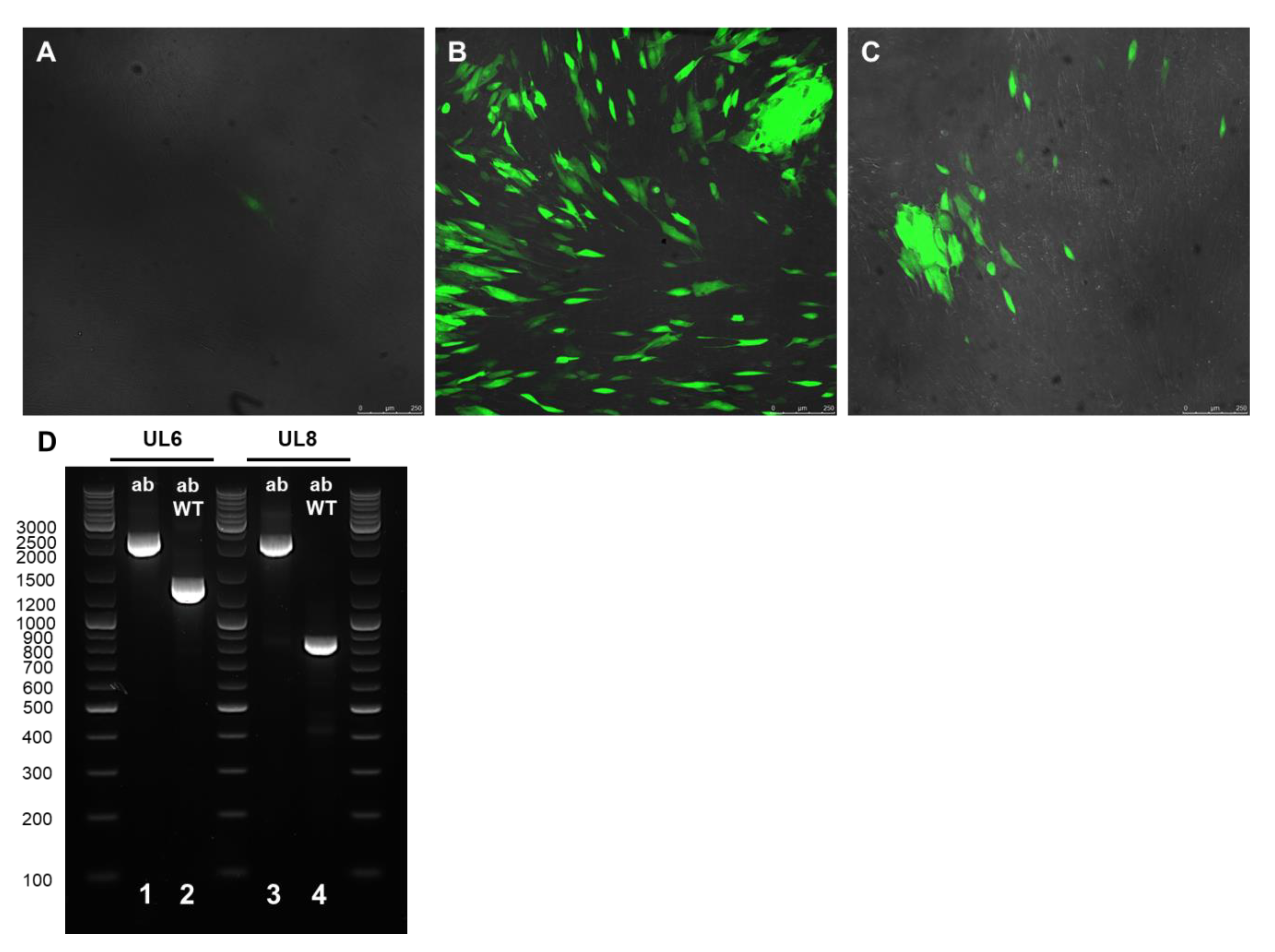{kind=link}
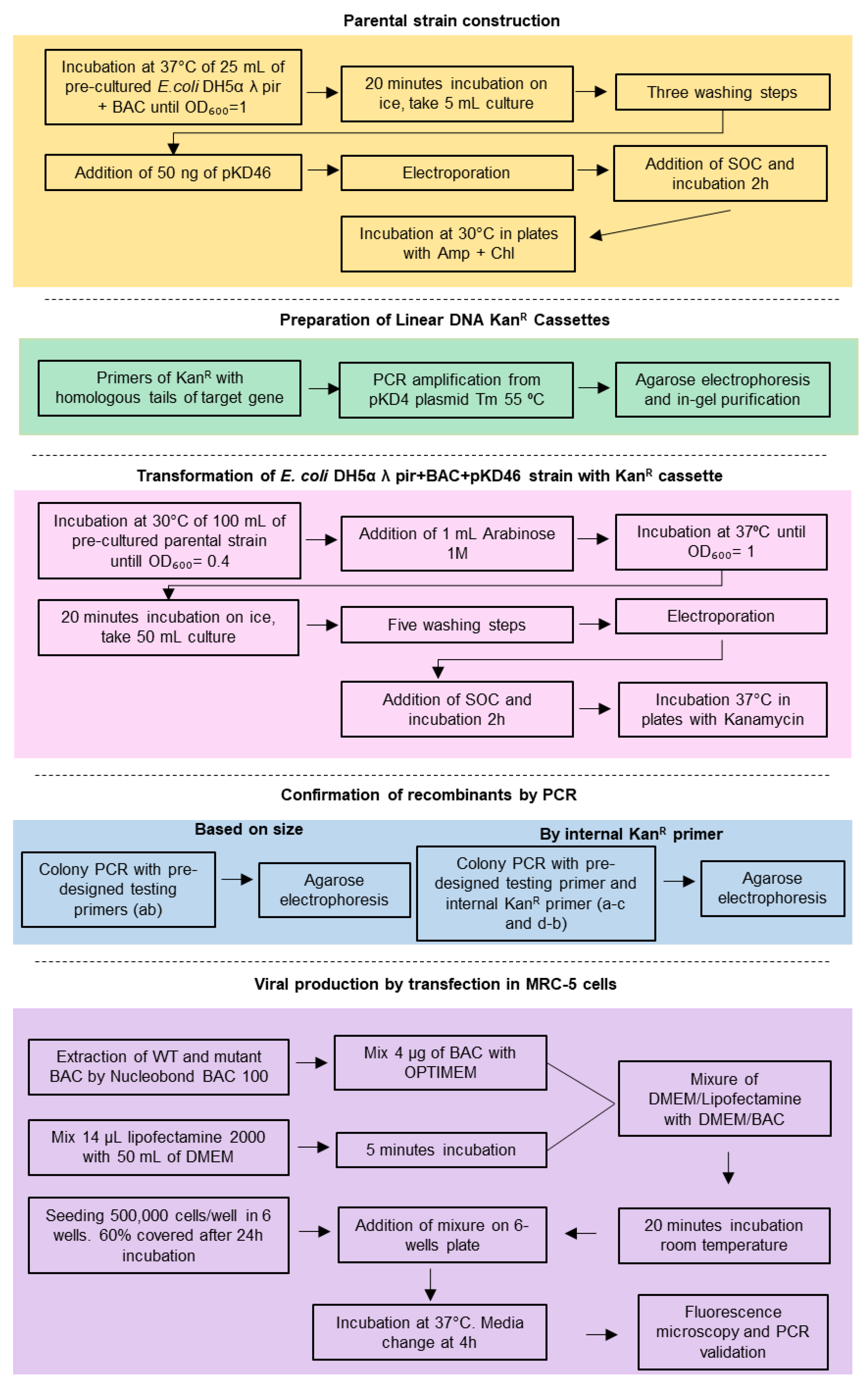{kind=link}
| Gene | Number of Tested Colonies | Positive Colonies (Test 1) | Positive Colonies (Test 2) | Percentage of Positive (%) |
|---|---|---|---|---|
| UL2 | 20 | 4 | 4 | 20 |
| UL5 | 4 | 1 | 1 | 25 |
| UL6 | 7 | 1 | 1 | 14.3 |
| UL7 | 4 | 2 | 2 | 50 |
| UL8 | 16 | 2 | 2 | 12.5 |
| UL9 | 24 | 1 | 1 | 4.2 |
| UL44 | 19 | 2 | 2 | 10.5 |
| UL121 | 55 | 1 | 1 | 1.8 |
| US7 | 10 | 6 | 6 | 60 |
| US9 | 42 | 21 | 21 | 50 |
| US13 | 10 | 1 | 1 | 10 |
| US14 | 4 | 3 | 3 | 75 |
| US16 | 10 | 5 | 5 | 50 |
| US17 | 4 | 3 | 3 | 75 |
| US18 | 22 | 6 | 6 | 27.2 |
| US20 | 15 | 11 | 11 | 73.3 |
| US21 | 15 | 7 | 7 | 46.7 |
| US30 | 15 | 1 | 1 | 6.7 |
| pp65 | 15 | 3 | 3 | 20 |
| IE1 | 15 | 1 | 1 | 6.7 |
| gO | 15 | 7 | 7 | 46.7 |
| gM | 6 | 6 | 6 | 100 |
| Oligonucleotides | Sequence (5′–3′) |
|---|---|
| UL6KanR-F (a) | ATGCATGCTAAGATGAACGGGTGGGCTGGGGTGCGCTTGGTAACTCACTGGCATTACACGTCTTGAG |
| UL6KanR-R (a) | CTACATTAACAAACCACGTTCTTCATCGTCCACGTGGCTTCGCCAGCGTCTAAGGTTTAACGGTTGTG |
| UL8KanR-F (a) | ATGACTAACCCTGGGCTATATGCATCGGAAAATTATAACGGAAATTATGAGCATTACACGTCTTGAG |
| UL8KanR-R (a) | TCACAGCTCCGTGTCCGTCATAAATACTTGTCCGTACTGTTTATTGTCTTTAAGGTTTAACGGTTGTG |
| UL6KanRcp-F (b) | TCCATCTGTCGTTTCTG |
| UL6KanRcp-R (b) | CCTCTCCACGTTTGTAA |
| UL8KanRcp-F (b) | GTTTAGCACCACAACTAC |
| UL8KanRcp-R (b) | CTGGCTTGCTATCTATTT |
| KanRInt-F (c) | GGATCTCCTGTCATCTC |
| KanRInt-R (c) | CATGATATTCGGCAAGC |
| Seq-F (d) | GCATTACACGTCTTGAG |
| Seq-R (d) | TAAGGTTTAACGGTTGTG |
| UL8 | US20 | US21 | gM | gO | IE1 | pp65 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fresh | Frozen | Fresh | Frozen | Fresh | Frozen | Fresh | Frozen | Fresh | Frozen | Fresh | Frozen | Fresh | Frozen | |
| Time (ms) | 4.9 | 4.7 | 4.5 | 3.2 | 4 | 3.3 | 4.3 | 3.8 | 4.3 | 3.7 | 4.2 | 3.9 | 4.2 | 3.6 |
| Number of total colonies | 16 | 190 | 43 | 52 | 27 | 73 | 10 | 21 | 34 | 31 | 26 | 52 | 47 | 23 |
| Number of tested colonies | 16 | 16 | 15 | 15 | 20 | 20 | 6 | 6 | 17 | 17 | 15 | 15 | 16 | 16 |
| Number of positive colonies | 2 | 11 | 11 | 12 | 9 | 17 | 6 | 6 | 8 | 13 | 1 | 5 | 3 | 9 |
| Percentage of positive | 12.5 | 68.75 | 73.33 | 80 | 45 | 85 | 100 | 100 | 47.05 | 76.47 | 6.66 | 33.33 | 18.75 | 56.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Ríos, E.; Gata-de-Benito, J.; López-Siles, M.; McConnell, M.J.; Pérez-Romero, P. Optimization of a Lambda-RED Recombination Method for Rapid Gene Deletion in Human Cytomegalovirus. Int. J. Mol. Sci. 2021, 22, 10558. https://doi.org/10.3390/ijms221910558
García-Ríos E, Gata-de-Benito J, López-Siles M, McConnell MJ, Pérez-Romero P. Optimization of a Lambda-RED Recombination Method for Rapid Gene Deletion in Human Cytomegalovirus. International Journal of Molecular Sciences. 2021; 22(19):10558. https://doi.org/10.3390/ijms221910558
Chicago/Turabian StyleGarcía-Ríos, Estéfani, Julia Gata-de-Benito, Mireia López-Siles, Michael J. McConnell, and Pilar Pérez-Romero. 2021. "Optimization of a Lambda-RED Recombination Method for Rapid Gene Deletion in Human Cytomegalovirus" International Journal of Molecular Sciences 22, no. 19: 10558. https://doi.org/10.3390/ijms221910558
APA StyleGarcía-Ríos, E., Gata-de-Benito, J., López-Siles, M., McConnell, M. J., & Pérez-Romero, P. (2021). Optimization of a Lambda-RED Recombination Method for Rapid Gene Deletion in Human Cytomegalovirus. International Journal of Molecular Sciences, 22(19), 10558. https://doi.org/10.3390/ijms221910558