
A Distinctive NAFLD Signature in Adipose Tissue from Women with Severe Obesity

 , , ,
, , , 
Abstract
1. Introduction
2. Results
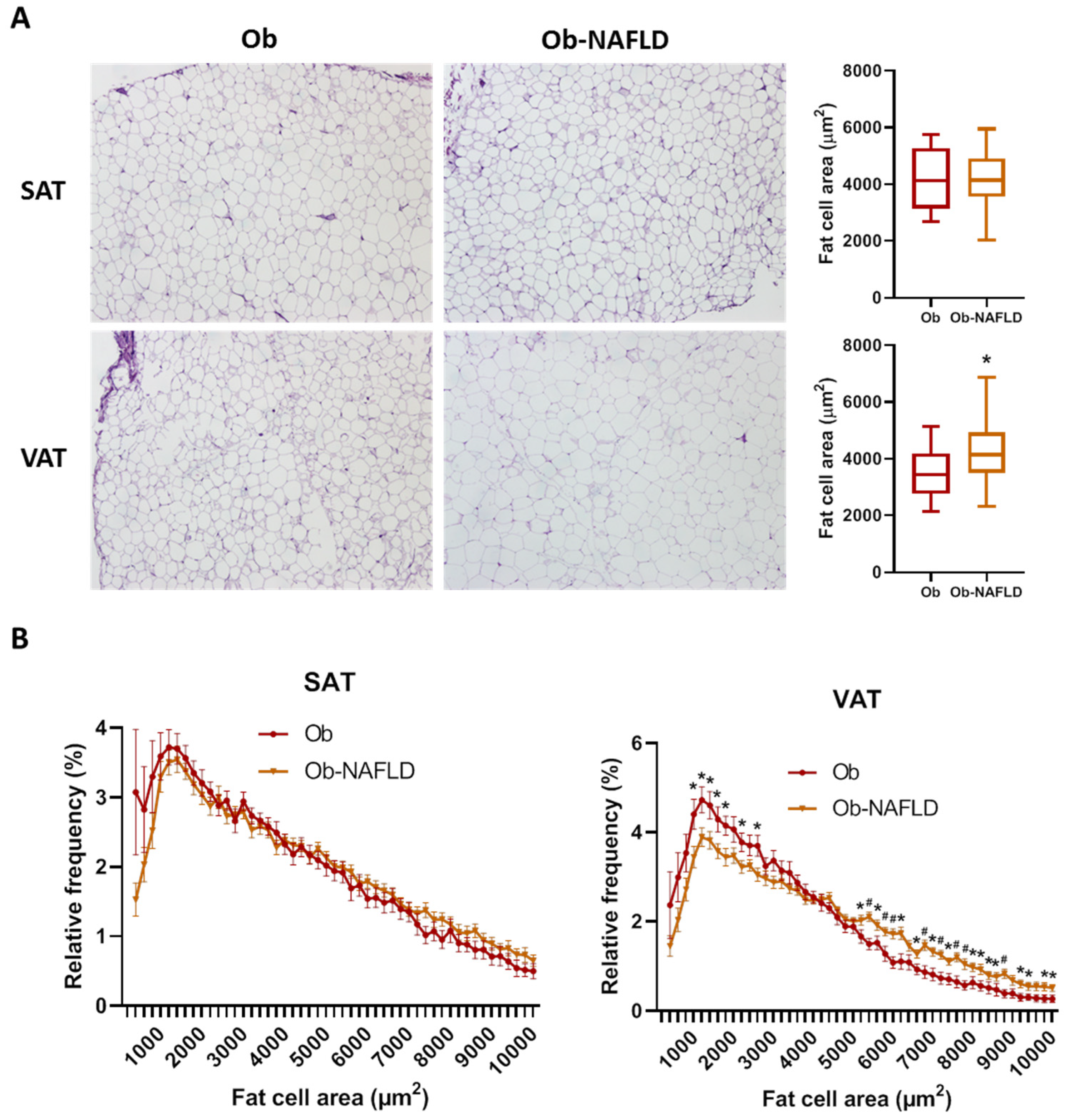
2.1. Adipose Tissue Morphology
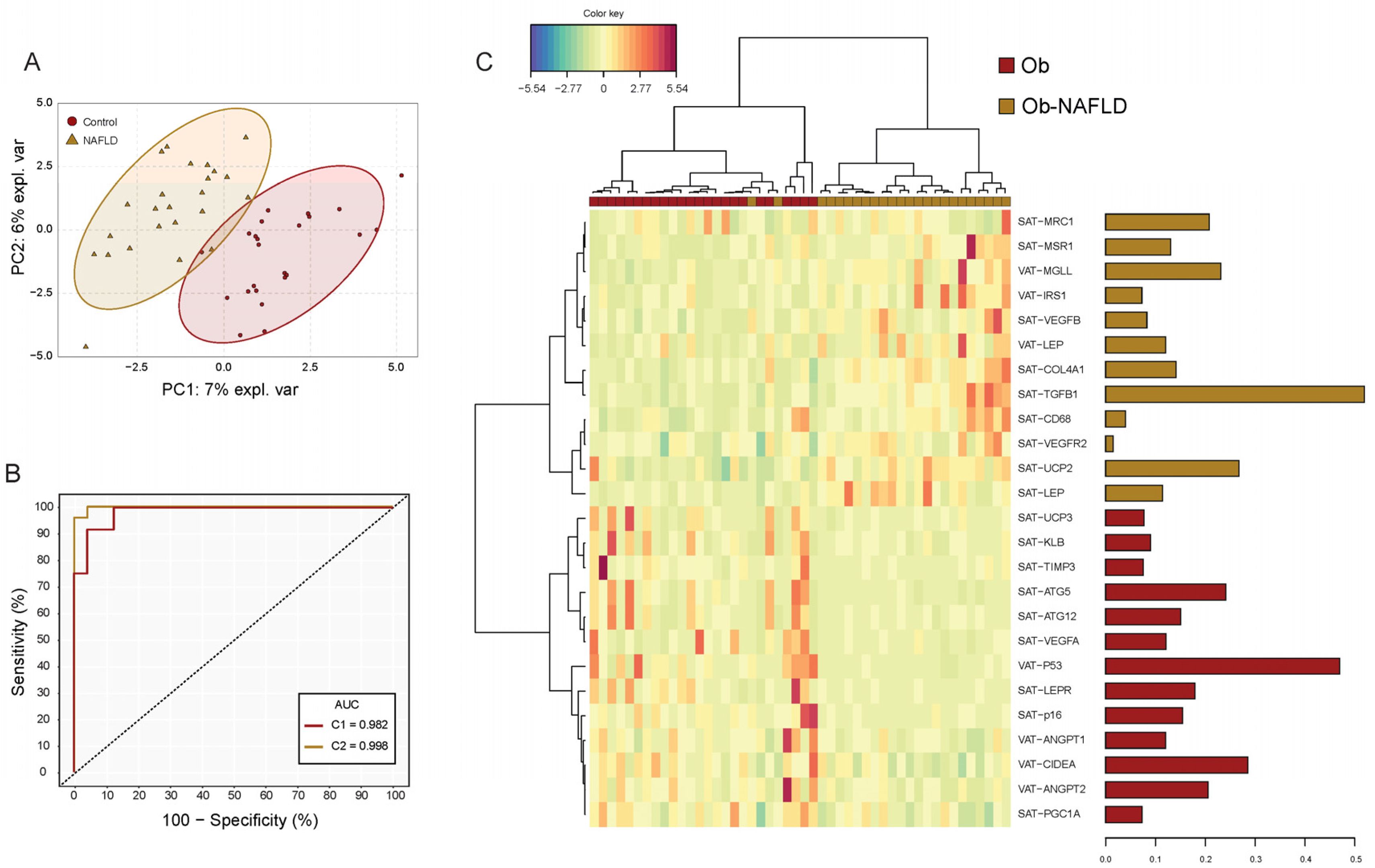
2.2. Differential Gene Expression Analysis
2.3. Principal Component Analysis (PCA) to Identify NAFLD Patients
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Clinical and Anthropometric Data
4.3. Determination of NAFLD
4.4. White Adipose Tissue Biopsies
4.5. Morphometry and Histopathology
4.6. RNA Extraction and Real Time PCR
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-Alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Maury, E.; Brichard, S.M. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell. Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Stanton, M.C.; Chen, S.C.; Jackson, J.V.; Rojas-Triana, A.; Kinsley, D.; Cui, L.; Fine, J.S.; Greenfeder, S.; Bober, L.A.; Jenh, C.H. Inflammatory Signals shift from adipose to liver during high fat feeding and influence the development of steatohepatitis in mice. J. Inflamm. 2011, 8, 1–14. [Google Scholar] [CrossRef]
- Oh, D.Y.; Morinaga, H.; Talukdar, S.; Bae, E.J.; Olefsky, J.M. Increased macrophage migration into adipose tissue in obese mice. Diabetes 2012, 61, 346–354. [Google Scholar] [CrossRef]
- Cho, K.W.; Morris, D.L.; delProposto, J.L.; Geletka, L.; Zamarron, B.; Martinez-Santibanez, G.; Meyer, K.A.; Singer, K.; O’Rourke, R.W.; Lumeng, C.N. An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep. 2014, 9, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Khoo, J.; Hsiang, J.; Taneja, R.; Law, N.M.; Ang, T.L. Comparative effects of liraglutide 3 mg vs structured lifestyle modification on body weight, liver fat and liver function in obese patients with non-alcoholic fatty liver disease: A pilot randomized trial. Diabetes Obes. Metab. 2017, 19, 1814–1817. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Park, B.J.; Kim, Y.J.; Kim, D.H.; Kim, W.; Jung, Y.J.; Yoon, J.H.; Kim, C.Y.; Cho, Y.M.; Kim, S.H.; Lee, K.B.; et al. Visceral adipose tissue area is an independent risk factor for hepatic steatosis. J. Gastroenterol. Hepatol. 2008, 23, 900–907. [Google Scholar] [CrossRef]
- van der Poorten, D.; Milner, K.L.; Hui, J.; Hodge, A.; Trenell, M.I.; Kench, J.G.; London, R.; Peduto, T.; Chisholm, D.J.; George, J. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457. [Google Scholar] [CrossRef]
- Du Plessis, J.; Van Pelt, J.; Korf, H.; Mathieu, C.; Van Der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of Adipose Tissue Inflammation with Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 635.e14–648.e14. [Google Scholar] [CrossRef]
- Munukka, E.; Pekkala, S.; Wiklund, P.; Rasool, O.; Borra, R.; Kong, L.; Ojanen, X.; Cheng, S.M.; Roos, C.; Tuomela, S.; et al. Gut-adipose tissue axis in hepatic fat accumulation in humans. J. Hepatol. 2014, 61, 132–138. [Google Scholar] [CrossRef]
- Tordjman, J.; Divoux, A.; Prifti, E.; Poitou, C.; Pelloux, V.; Hugol, D.; Basdevant, A.; Bouillot, J.L.; Chevallier, J.M.; Bedossa, P.; et al. Structural and inflammatory heterogeneity in subcutaneous adipose tissue: Relation with liver histopathology in morbid obesity. J. Hepatol. 2012, 56, 1152–1158. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, J.; Wang, H.; Guo, D.; Yuan, C.; Liu, B.; Zhong, H.; Li, D.; Li, Y. A novel immune-related genes signature after bariatric surgery is histologically associated with non-alcoholic fatty liver disease. Adipocyte 2021, 10, 424–434. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipose tissue, obesity and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017, 42, 92–108. [Google Scholar] [CrossRef]
- Chrysavgis, L.; Ztriva, E.; Protopapas, A.; Tziomalos, K.; Cholongitas, E. Nonalcoholic fatty liver disease in lean subjects: Prognosis, outcomes and management. World J. Gastroenterol. 2020, 26, 6514–6528. [Google Scholar] [CrossRef] [PubMed]
- Albhaisi, S.; Chowdhury, A.; Sanyal, A.J. Non-alcoholic fatty liver disease in lean individuals. JHEP Reports 2019, 1, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Graham, S.; Wang, X.; Cai, D.; Huang, M.; Pique-Regi, R.; Dong, X.C.; Chen, Y.E.; Willer, C.; et al. Causal relationships between NAFLD, T2D and obesity have implications for disease subphenotyping. J. Hepatol. 2020, 73, 263–276. [Google Scholar] [CrossRef]
- Duwaerts, C.C.; Maher, J.J. Macronutrients and the Adipose-Liver Axis in Obesity and Fatty Liver. CMGH 2019, 7, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H. Multiple organs involved in the pathogenesis of non-alcoholic fatty liver disease. Cell Biosci. 2020, 10, 18. [Google Scholar] [CrossRef]
- Estep, J.M.; Goodman, Z.; Sharma, H.; Younossi, E.; Elarainy, H.; Baranova, A.; Younossi, Z. Adipocytokine expression associated with miRNA regulation and diagnosis of NASH in obese patients with NAFLD. Liver Int. 2015, 35, 1367–1372. [Google Scholar] [CrossRef]
- Lê, K.A.; Mahurkar, S.; Alderete, T.L.; Hasson, R.E.; Adam, T.C.; Kim, J.S.; Beale, E.; Xie, C.; Greenberg, A.S.; Allayee, H.; et al. Subcutaneous adipose tissue macrophage infiltration is associated with hepatic and visceral fat deposition, hyperinsulinemia, and stimulation of NF-κB stress pathway. Diabetes 2011, 60, 2802–2809. [Google Scholar] [CrossRef]
- Cheng, S.; Wiklund, P.; Autio, R.; Borra, R.; Ojanen, X.; Xu, L.; Törmäkangas, T.; Alen, M. Adipose tissue dysfunction and altered systemic amino acid metabolism are associated with non-alcoholic fatty liver disease. PLoS ONE 2015, 10, 89. [Google Scholar] [CrossRef]
- Von Loeffelholz, C.; Pfeiffer, A.F.H.; Lock, J.F.; Lieske, S.; Döcke, S.; Murahovschi, V.; Kriebel, J.; De Las Heras Gala, T.; Grallert, H.; Rudovich, N.; et al. ANGPTL8 (Betatrophin) is Expressed in Visceral Adipose Tissue and Relates to Human Hepatic Steatosis in Two Independent Clinical Collectives. Horm. Metab. Res. 2017, 49, 343–349. [Google Scholar] [CrossRef][Green Version]
- Heebøll, S.; Poulsen, M.K.; Ornstrup, M.J.; Kjær, T.N.; Pedersen, S.B.; Nielsen, S.; Grønbæk, H.; Handberg, A. Circulating sCD36 levels in patients with non-alcoholic fatty liver disease and controls. Int. J. Obes. 2017, 41, 262–267. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Angın, Y.; Arslan, N.; Kuralay, F. Leptin-to-adiponectin ratio in obese adolescents with nonalcoholic fatty liver disease. Turk. J. Pediatr. 2014, 56, 259–266. [Google Scholar]
- Mikami, K.; Endo, T.; Sawada, N.; Igarashi, G.; Kimura, M.; Hasegawa, T.; Iino, C.; Tomita, H.; Sawada, K.; Nakaji, S.; et al. Leptin/adiponectin ratio correlates with hepatic steatosis but not arterial stiffness in nonalcoholic fatty liver disease in Japanese population. Cytokine 2020, 126, 4927. [Google Scholar] [CrossRef]
- Klein, S.; Allison, D.B.; Heymsfield, S.B.; Kelley, D.E.; Leibel, R.L.; Nonas, C.; Kahn, R. Waist circumference and cardiometabolic risk: A consensus statement from Shaping America’s Health: Association for Weight Management and Obesity Prevention; NAASO, the Obesity Society; the American Society for Nutrition; and the American Diabetes Associat. Diabetes Care 2007, 30, 1647–1652. [Google Scholar] [CrossRef]
- Gaddipati, R.; Mitnala, S.; Padaki, N.; Mukherjee, R.M.; Sekaran, A.; Jayaraj-Mansard, M.; Rabella, P.; Rao-Guduru, V.; Reddy-Duwuru, N. Visceral adipose tissue visfatin in nonalcoholic fatty liver disease. Ann. Hepatol. 2010, 9, 266–270. [Google Scholar] [CrossRef]
- Jensen, M.D. Role of body fat distribution and the metabolic complications of obesity. J. Clin. Endocrinol. Metab. 2008, 93, 45. [Google Scholar] [CrossRef] [PubMed]
- Item, F.; Konrad, D. Visceral fat and metabolic inflammation: The portal theory revisited. Obes. Rev. 2012, 13, 30–39. [Google Scholar] [CrossRef]
- Dalmas, E.; Toubal, A.; Alzaid, F.; Blazek, K.; Eames, H.L.; Lebozec, K.; Pini, M.; Hainault, I.; Montastier, E.; Denis, R.G.P.; et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat. Med. 2015, 21, 610–618. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, J.H.; Kim, J.H.; Martinus, R.D.; Park, S.H. Angiopoietin-like protein 2, a chronic inflammatory mediator, is a new target induced by TGF-β1 through a Smad3-dependent mechanism. Biochem. Biophys. Res. Commun. 2013, 430, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Yamamoto, K.; Pandey, M.; Loskutoff, D.J. Elevated expression of transforming growth factor-β in adipose tissue from obese mice. Mol. Med. 1997, 3, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Tichansky, D.S.; Madan, A.K. Transforming Growth Factor β1 release by human adipose tissue is enhanced in obesity. Metabolism 2005, 54, 1546–1551. [Google Scholar] [CrossRef]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Miao, X.; Zhang, B.; Xie, J. p53 as a double-edged sword in the progression of non-alcoholic fatty liver disease. Life Sci. 2018, 215, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Chung, Y.; Lee, J.H.; Chun, J.M.; Park, J.H. The Intricate Role of p53 in Adipocyte Differentiation and Function. Cells 2020, 9, 2621. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. P53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2012, 57, 837–843. [Google Scholar] [CrossRef]
- Panasiuk, A.; Dzieciol, J.; Panasiuk, B.; Prokopowicz, D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J. Gastroenterol. 2006, 12, 6198–6202. [Google Scholar] [CrossRef]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 688. [Google Scholar] [CrossRef]
- Sans, A.; Bonnafous, S.; Rousseau, D.; Patouraux, S.; Canivet, C.M.; Leclere, P.S.; Tran-Van-Nhieu, J.; Luci, C.; Bailly-Maitre, B.; Xu, X.; et al. The Differential Expression of Cide Family Members is Associated with Nafld Progression from Steatosis to Steatohepatitis. Sci. Rep. 2019, 9, 37. [Google Scholar] [CrossRef]
- Zhong, H.; Liu, M.; Ji, Y.; Ma, M.; Chen, K.; Liang, T.; Liu, C. Genipin Reverses HFD-Induced Liver Damage and Inhibits UCP2-Mediated Pyroptosis in Mice. Cell. Physiol. Biochem. 2018, 49, 1885–1897. [Google Scholar] [CrossRef]
- Bagheri, H.; Yaghmaei, P.; Modaresi, M.; Sabbaghian, M.; Ebrahim-Habibi, A. Glycyrrhizin Improves Fatty Liver Symptoms, Increases Adiponectin and Reduces UCP2 Expression in Wistar Rats. Proc. Natl. Acad. Sci. India Sect. B-Biol. Sci. 2020, 90, 191–197. [Google Scholar] [CrossRef]
- Cordero, P.; Gomez-Uriz, A.M.; Campion, J.; Milagro, F.I.; Martinez, J.A. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013, 8, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, 1681. [Google Scholar] [CrossRef]
- Ferhat, M.; Funai, K.; Boudina, S. Autophagy in Adipose Tissue Physiology and Pathophysiology. Antioxid. Redox Signal. 2019, 31, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Wolfs, M.G.M.; Gruben, N.; Rensen, S.S.; Verdam, F.J.; Greve, J.W.; Driessen, A.; Wijmenga, C.; Buurman, W.A.; Franke, L.; Scheja, L.; et al. Determining the association between adipokine expression in multiple tissues and phenotypic features of non-alcoholic fatty liver disease in obesity. Nutr. Diabetes 2015, 5, 73. [Google Scholar] [CrossRef]
- Fuchs, A.; Samovski, D.; Smith, G.I.; Cifarelli, V.; Farabi, S.S.; Yoshino, J.; Pietka, T.; Chang, S.W.; Ghosh, S.; Myckatyn, T.M.; et al. Associations Among Adipose Tissue Immunology, Inflammation, Exosomes and Insulin Sensitivity in People With Obesity and Nonalcoholic Fatty Liver Disease. Gastroenterology 2021, 161, 968–981.e12. [Google Scholar] [CrossRef]
- Hydes, T.; Brown, E.; Hamid, A.; Bateman, A.C.; Cuthbertson, D.J. Current and Emerging Biomarkers and Imaging Modalities for Nonalcoholic Fatty Liver Disease: Clinical and Research Applications. Clin. Ther. 2021, 7, 13. [Google Scholar]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, 5752. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ob (n = 24) | Ob-NAFLD (n = 24) | p-Value | |
|---|---|---|---|
| Age (years) a | 44.88 ± 10.87 | 45.75 ± 8.94 | 0.762 |
| BMI (kg/m2) a | 44.00 ± 5.32 | 44.41 ± 4.75 | 0.778 |
| Waist (cm) | 118.5 (111.3–129.3) | 119.5 (116.3–125.8) | 0.900 |
| Hip (cm) | 137.7 ± 9.49 | 133.2 ± 9.145 | 0.222 |
| Waist-to-hip ratio | 0.88 ± 0.08 | 0.91 ± 0.05 | 0.246 |
| CUN-BAE Index | 54.2 (52.3–55.6) | 54.5 (52.7–56.5) | 0.660 |
| HTN | 5 (20.83%) | 9 (37.50%) | 0.341 b |
| TG (mg/dL) | 189.0 ± 33.02 | 200.0 ± 37.96 | 0.027 |
| Total cholesterol (mg/dL) | 193.82 ± 24.44 | 202.55 ± 36.2 | 0.289 |
| HDL (mg/dL) | 53.75 ± 8.65 | 45.58 ± 7.88 | 0.001 |
| LDL (mg/dL) | 116.3 ± 25.08 | 125.2 ± 29.96 | 0.277 |
| FPG (mg/dL) | 90.5 (88.5–101.0) | 99.0 (90.5–105.8) | 0.264 |
| T2D a | 5 (20.83%) | 5 (20.83%) | >0.999 b |
| HbA1c (%) | 5.5 (5.3–5.7) | 5.6 (5.3–6.1) | 0.493 |
| AST (IU/L) | 20.5 (16.7–23.0) | 21.0 (17.0–27.7) | 0.454 |
| ALT (IU/L) | 21.0 (15.0–25.0) | 22.0 (17.2–40.5) | 0.159 |
| GGT (IU/L) | 19.0 (14.0–31.0) | 24.0 (18.2–38.7) | 0.349 |
| AST:ALT ratio | 1.0 (0.8–1.1) | 0.8 (0.6–1.0) | 0.158 |
| Platelets (×109/L) | 268 (241–306) | 294 (248–382) | 0.125 |
| CRP (mg/dL) | 0.48 (0.34–1.21) | 0.95 (0.38–1.81) | 0.230 |
| FIB-4 Score | 0.71 (0.51–0.86) | 0.69 (0.44–0.91) | 0.778 |
| APRI Score | 0.18 (0.14–0.22) | 0.19 (0.14–0.21) | 0.813 |
| Ob (n = 24) | Ob-NAFLD (n = 24) | p-Value | |
|---|---|---|---|
| SAT fat cell area (µm2) | 4132 ± 1124 | 4260 ± 985 | 0.728 |
| VAT fat cell area (µm2) | 3475 ± 851 | 4174 ± 992 | 0.038 |
| SAT:VAT fat cell area ratio | 1.25 ± 0.33 | 1.03 ± 0.18 | 0.017 |
| SAT Pericellular fibrosis (% area) | 3.27 ± 2.07 | 3.78 ± 2.55 | 0.536 |
| VAT Pericellular fibrosis (% area) | 2.17 ± 1.52 | 2.67 ± 1.67 | 0.418 |
| SAT:VAT fibrosis ratio | 1.15 (0.78–2.04) | 1.09 (0.74–1.76) | 0.808 |
| Ob (n = 24) | Ob-NAFLD (n = 24) | |||||||
|---|---|---|---|---|---|---|---|---|
| Median | Percentile | Median | Percentile | |||||
| Tissue Depot | 25% | 75% | 25% | 75% | FC | p-Value | ||
| SAT | ||||||||
| CD68 | 0.834 | 0.546 | 1.158 | 1.224 | 0.831 | 1.707 | 1.42 | 0.0162 |
| MRC1 | 0.0940 | 0.0615 | 0.147 | 0.158 | 0.0939 | 0.223 | 1.61 | 0.0117 |
| MSR1 | 0.154 | 0.0742 | 0.218 | 0.211 | 0.162 | 0.367 | 1.71 | 0.0171 |
| VEGFB | 0.237 | 0.177 | 0.260 | 0.265 | 0.216 | 0.355 | 1.38 | 0.0321 |
| ANGPT1 | 0.0256 | 0.0165 | 0.0331 | 0.0340 | 0.0254 | 0.0477 | 1.41 | 0.0269 |
| P16 | 0.0307 | 0.0020 | 0.0496 | 0.0060 | 0.0013 | 0.0211 | 0.30 | 0.0264 |
| LEP | 0.707 | 0.570 | 0.915 | 1.240 | 0.624 | 1.764 | 1.75 | 0.0353 |
| LEPR | 0.382 | 0.215 | 1.090 | 0.220 | 0.188 | 0.280 | 0.36 | 0.0105 |
| PGC1A | 0.0067 | 0.0048 | 0.0074 | 0.0048 | 0.0026 | 0.0053 | 0.73 | 0.0167 |
| TGFB1 | 0.135 | 0.103 | 0.160 | 0.184 | 0.137 | 0.265 | 1.70 | 0.0021 |
| UCP2 | 0.283 | 0.194 | 0.443 | 0.460 | 0.344 | 0.570 | 1.44 | 0.0024 |
| COL1A1 | 0.304 | 0.208 | 0.571 | 0.707 | 0.492 | 0.963 | 1.35 | 0.0036 |
| COL4A1 | 1.592 | 1.232 | 1.876 | 2.205 | 1.726 | 3.081 | 1.55 | 0.0070 |
| COL6A1 | 0.0117 | 0.0093 | 0.0166 | 0.0392 | 0.0134 | 0.0847 | 9.44 | 0.0023 |
| TIMP3 | 1.320 | 0.757 | 1.956 | 0.836 | 0.623 | 0.987 | 0.48 | 0.0074 |
| VAT | ||||||||
| HIF1A | 0.348 | 0.259 | 0.434 | 0.244 | 0.178 | 0.374 | 0.81 | 0.0373 |
| ANGPT2 | 0.0273 | 0.0227 | 0.0388 | 0.0209 | 0.0130 | 0.0293 | 0.65 | 0.0246 |
| P53 | 0.0762 | 0.0561 | 0.155 | 0.0507 | 0.0432 | 0.0613 | 0.49 | 0.0019 |
| P16 | 0.0098 | 0.0026 | 0.0170 | 0.0052 | 0.0008 | 0.0096 | 0.35 | 0.0348 |
| LEP | 0.290 | 0.157 | 0.675 | 0.422 | 0.335 | 1.308 | 1.87 | 0.0182 |
| MGLL | 0.389 | 0.242 | 0.467 | 0.592 | 0.391 | 0.688 | 1.67 | 0.0038 |
| ATG12 | 0.0558 | 0.0387 | 0.0707 | 0.0646 | 0.0513 | 0.0953 | 1.32 | 0.0309 |
| CIDEA | 0.0343 | 0.0262 | 0.0405 | 0.0191 | 0.0098 | 0.0348 | 0.63 | 0.0039 |
| Variable | B | S.E. (B) | Exp B (OR) (95% C.I) | Sig. | R2 (%) | Correct Prediction (%) | M. Sig. |
|---|---|---|---|---|---|---|---|
| MODEL 1 | |||||||
| SAT-TGFB1 | 1.397 | 0.642 | 4.04 (1.15–14.23) | 0.030 | 45.2 | 80 | 0.000 |
| VAT-P53 | −1.400 | 0.634 | 0.25 (0.07–0.85) | 0.027 | |||
| Age | −0.016 | 0.040 | 0.98 (0.91–1.06) | 0.683 | |||
| MODEL 2 | |||||||
| SAT-TGFB1 | 110.07 | 8455.23 | 6.33 (0.00–>50.00) | 0.790 | 100 | 100 | 0.000 |
| VAT-P53 | −56.76 | 5653.40 | <0.001 (–) | 0.792 | |||
| VAT-CIDEA | −390.28 | 13264.12 | <0.001 (–) | 0.777 | |||
| SAT-UCP2 | 264.87 | 9103.50 | 10.73 (0.00–>50.00) | 0.777 | |||
| SAT-ATG5 | −54.44 | 2596.91 | <0.001 (–) | 0.783 | |||
| SAT-MRC1 | −97.42 | 7094.69 | <0.001 (–) | 0.789 | |||
| VAT-ANGPT2 | −149.39 | 5819.60 | <0.001 (–) | 0.780 | |||
| Age | 8.92 | 312.56 | 74.90 (0.00–>100.00) | 0.777 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osorio-Conles, Ó.; Vega-Beyhart, A.; Ibarzabal, A.; Balibrea, J.M.; Graupera, I.; Rimola, J.; Vidal, J.; de Hollanda, A. A Distinctive NAFLD Signature in Adipose Tissue from Women with Severe Obesity. Int. J. Mol. Sci. 2021, 22, 10541. https://doi.org/10.3390/ijms221910541
Osorio-Conles Ó, Vega-Beyhart A, Ibarzabal A, Balibrea JM, Graupera I, Rimola J, Vidal J, de Hollanda A. A Distinctive NAFLD Signature in Adipose Tissue from Women with Severe Obesity. International Journal of Molecular Sciences. 2021; 22(19):10541. https://doi.org/10.3390/ijms221910541
Chicago/Turabian StyleOsorio-Conles, Óscar, Arturo Vega-Beyhart, Ainitze Ibarzabal, José María Balibrea, Isabel Graupera, Jordi Rimola, Josep Vidal, and Ana de Hollanda. 2021. "A Distinctive NAFLD Signature in Adipose Tissue from Women with Severe Obesity" International Journal of Molecular Sciences 22, no. 19: 10541. https://doi.org/10.3390/ijms221910541
APA StyleOsorio-Conles, Ó., Vega-Beyhart, A., Ibarzabal, A., Balibrea, J. M., Graupera, I., Rimola, J., Vidal, J., & de Hollanda, A. (2021). A Distinctive NAFLD Signature in Adipose Tissue from Women with Severe Obesity. International Journal of Molecular Sciences, 22(19), 10541. https://doi.org/10.3390/ijms221910541