
Ginsenoside Rh1 Prevents Migration and Invasion through Mitochondrial ROS-Mediated Inhibition of STAT3/NF-κB Signaling in MDA-MB-231 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
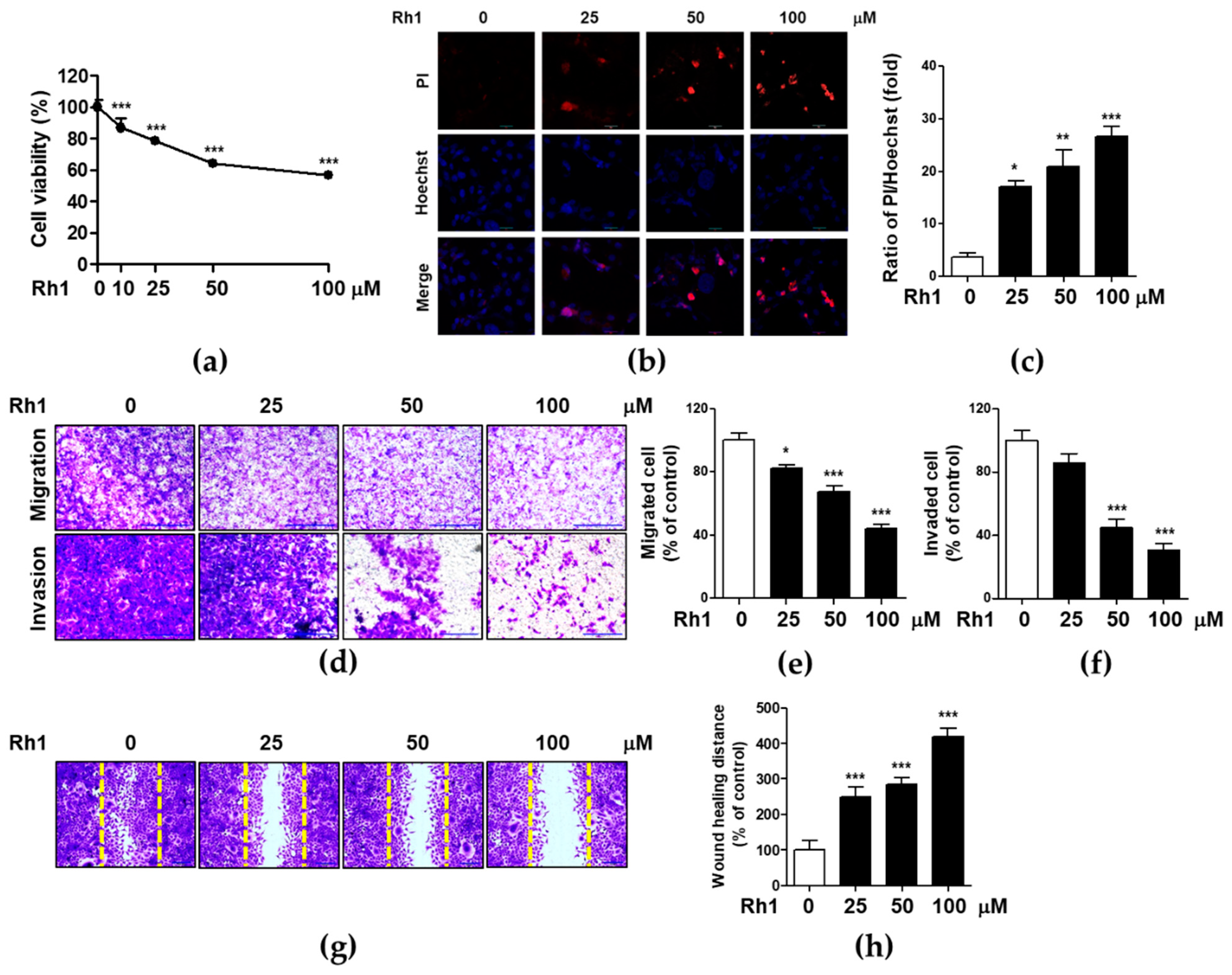
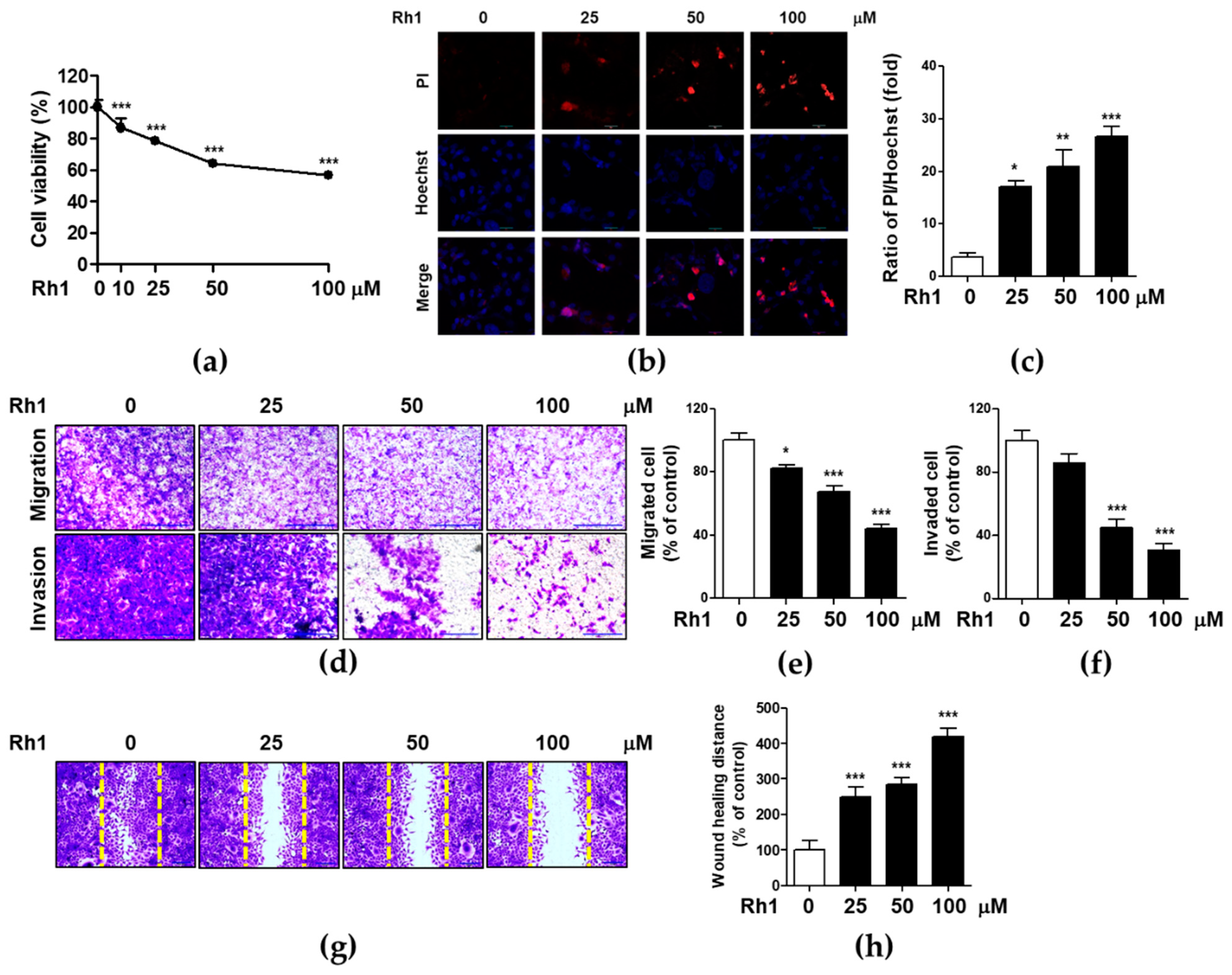
2.1. Rh1 Inhibited Cell Viability, Migration, and Invasion in MDA-MB-231 Cells
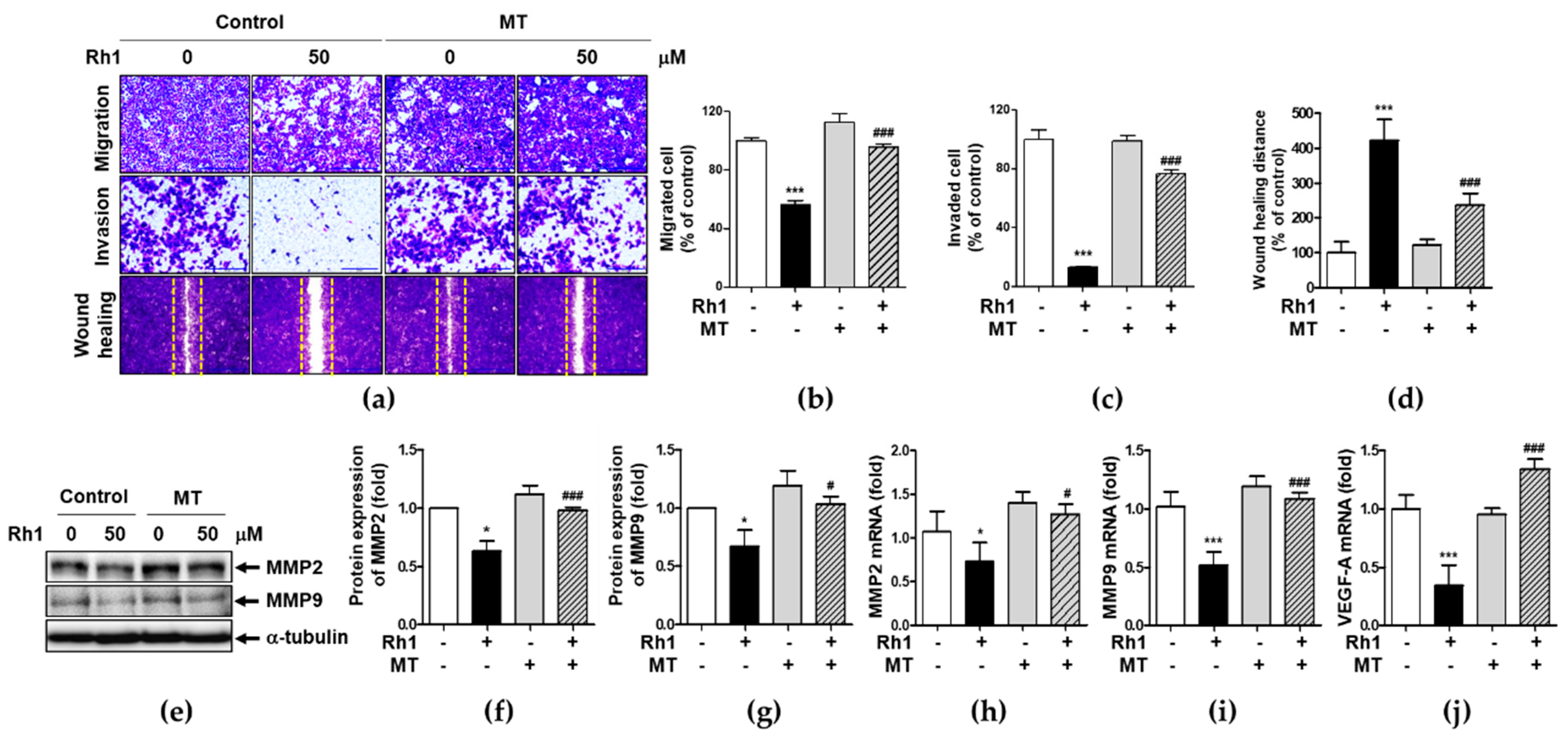
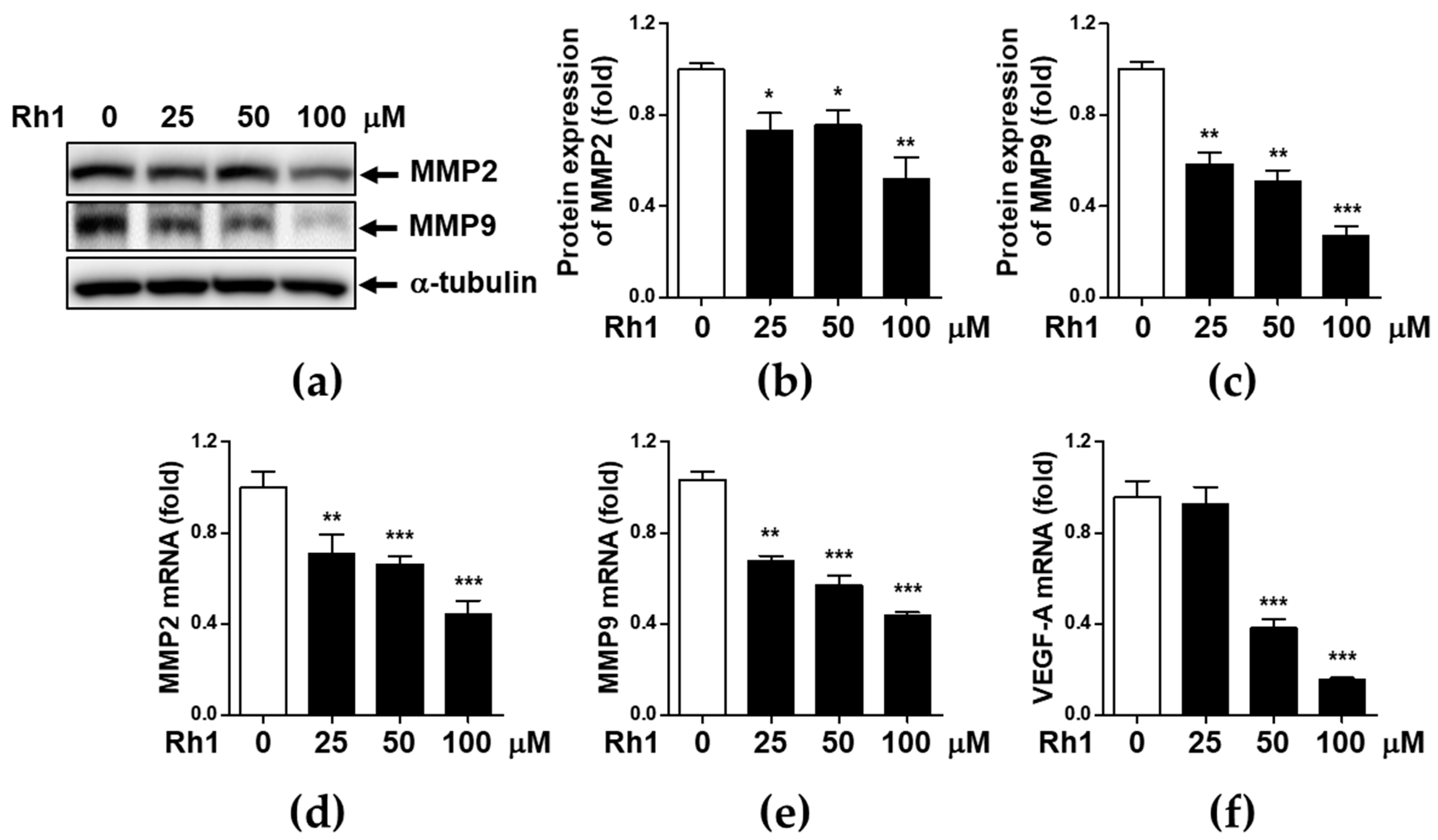
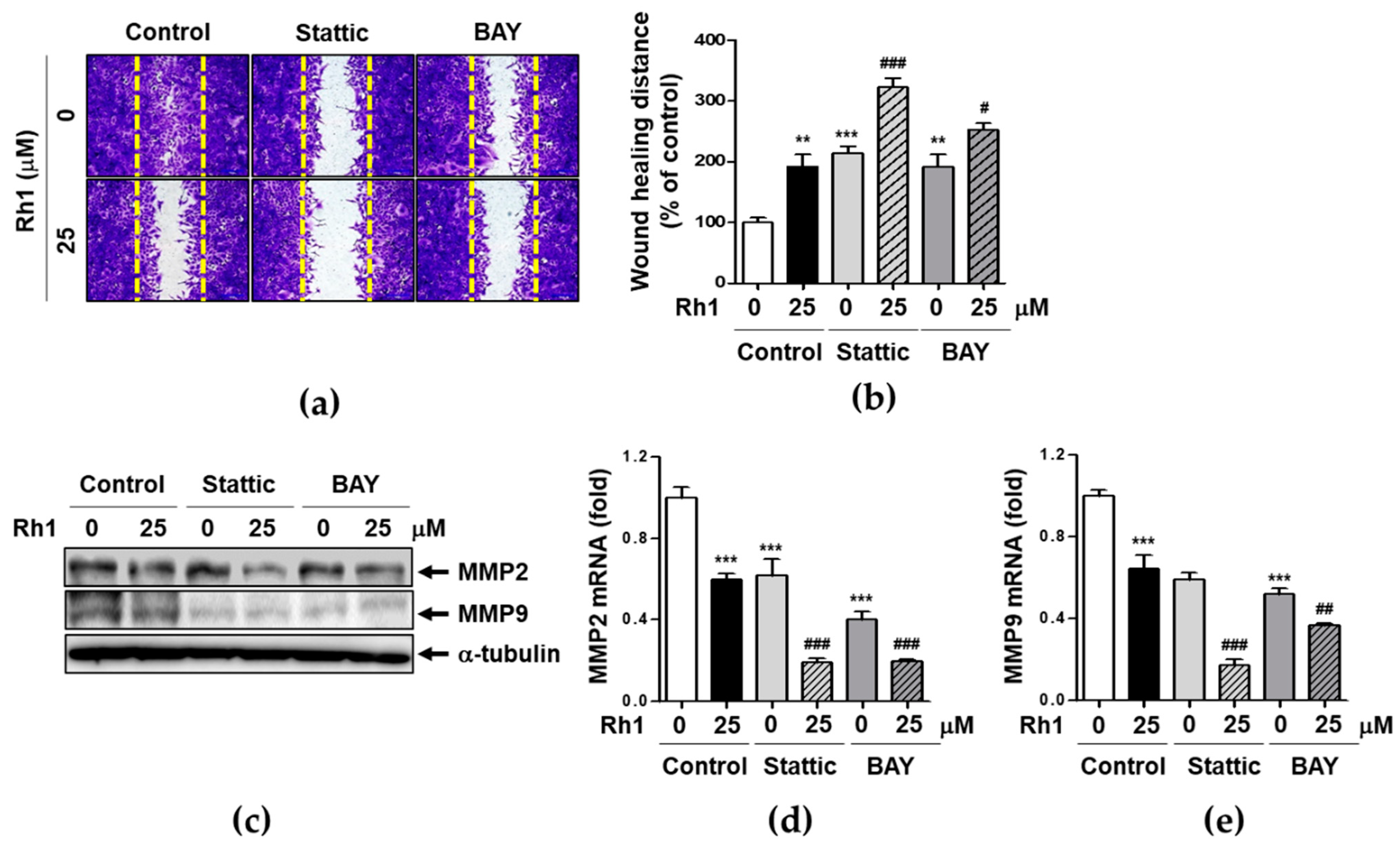
2.2. Rh1 Inhibited Cell Migration via Inhibiting MMP2, MMP9, and VEGF-A
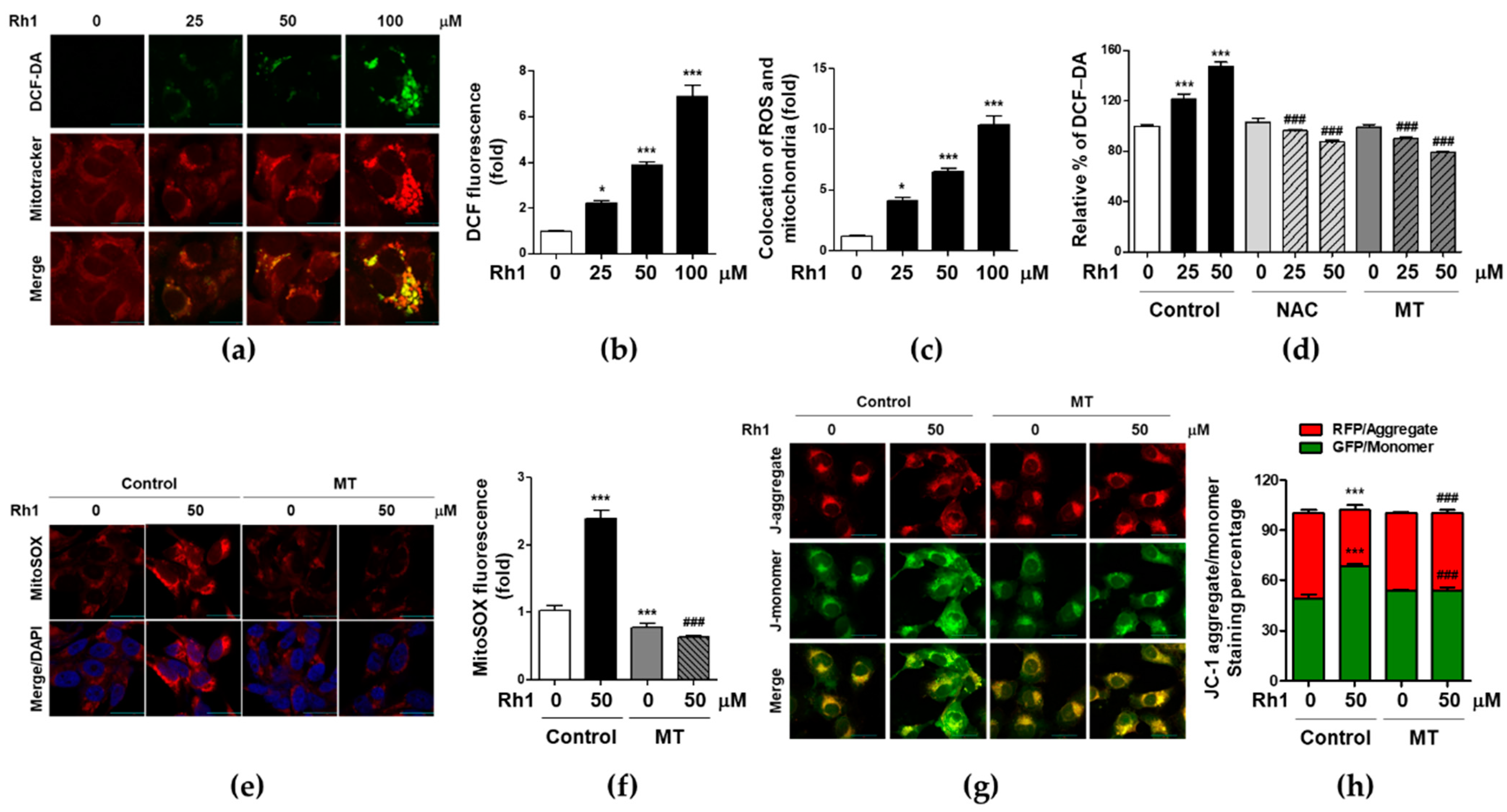
2.3. Rh1 Induced Mitochondrial Dysfunction via Producing Mitochondrial ROS
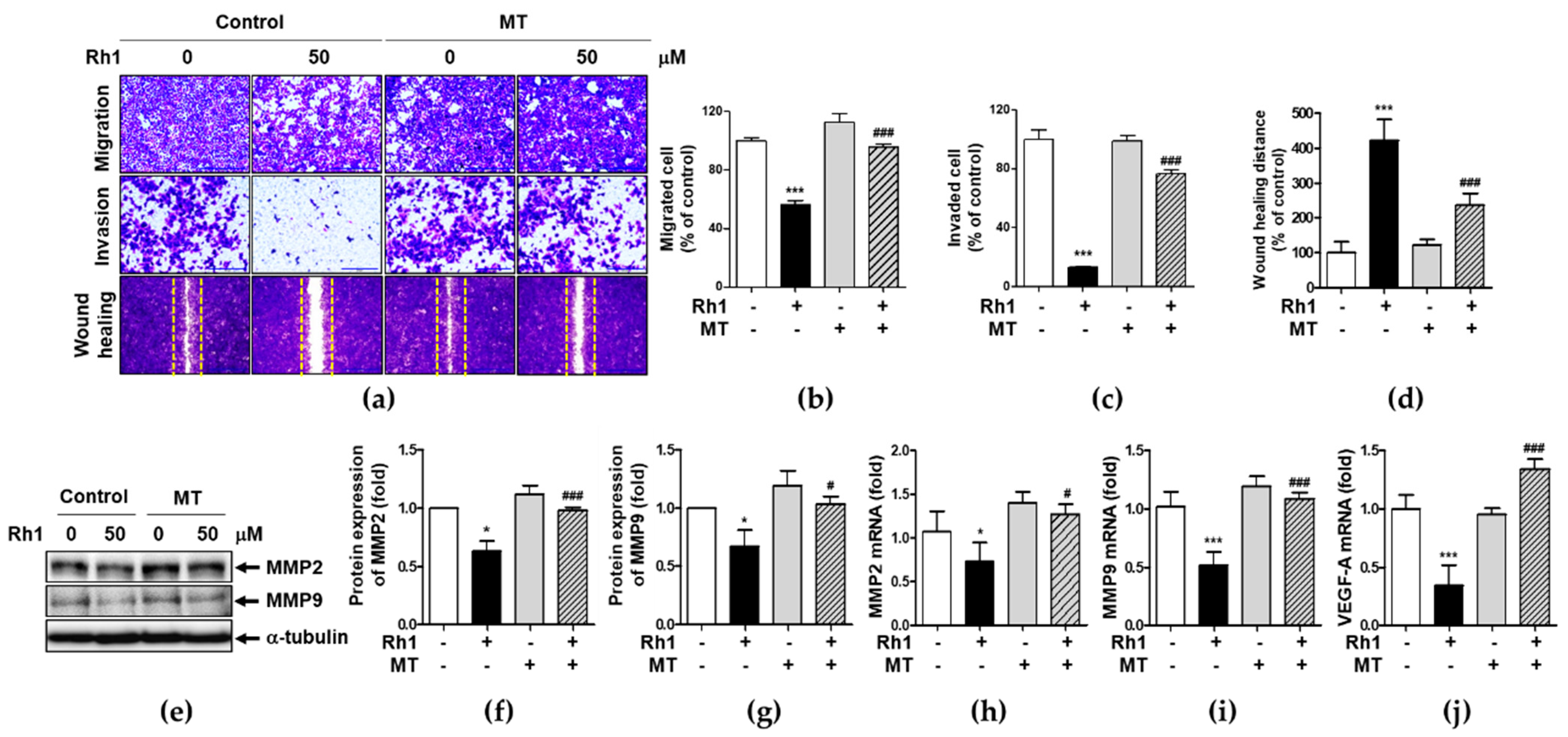
2.4. Mitochondrial ROS Is Associated with Rh1-Inhibited Cell Migration and Invasion
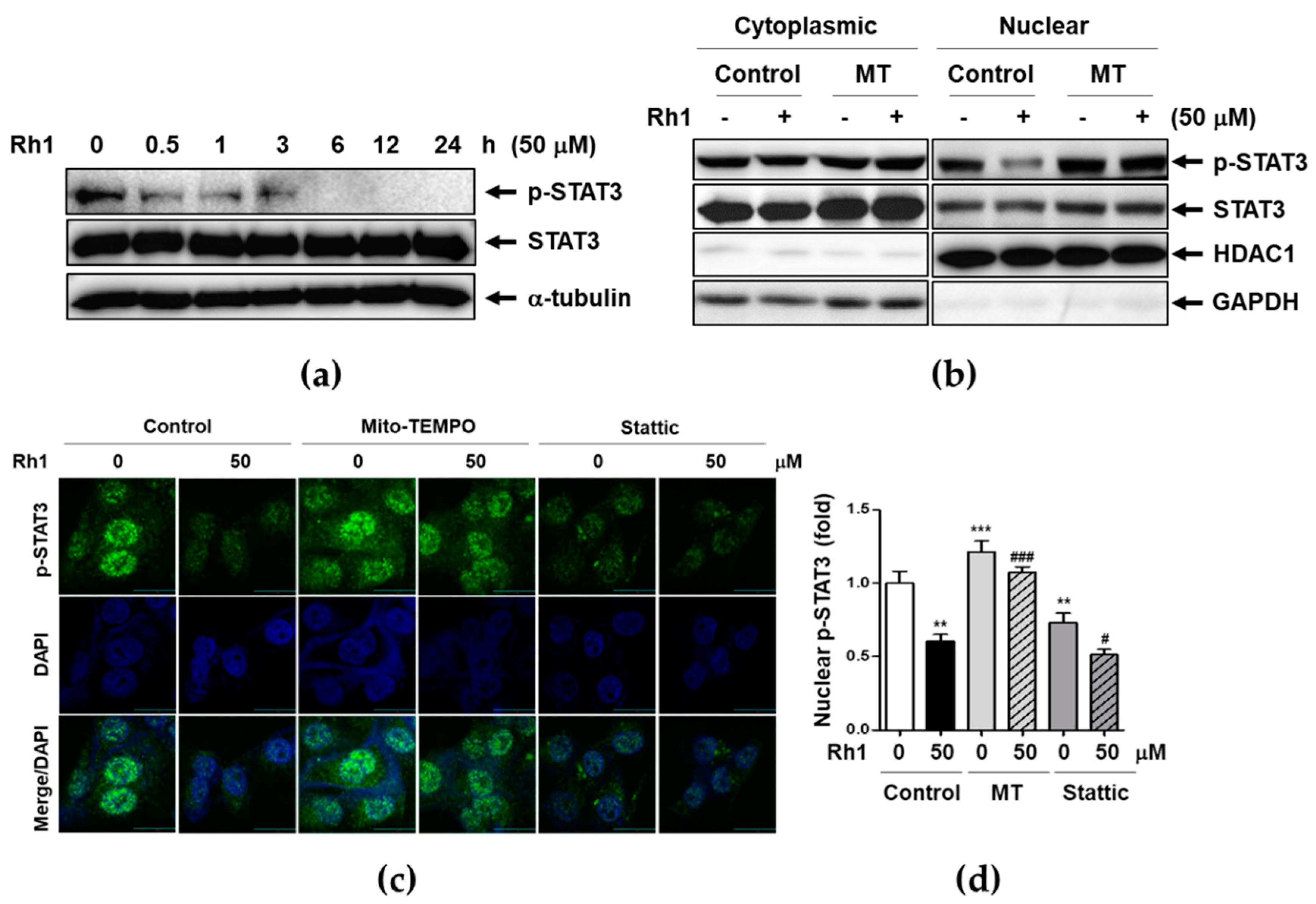
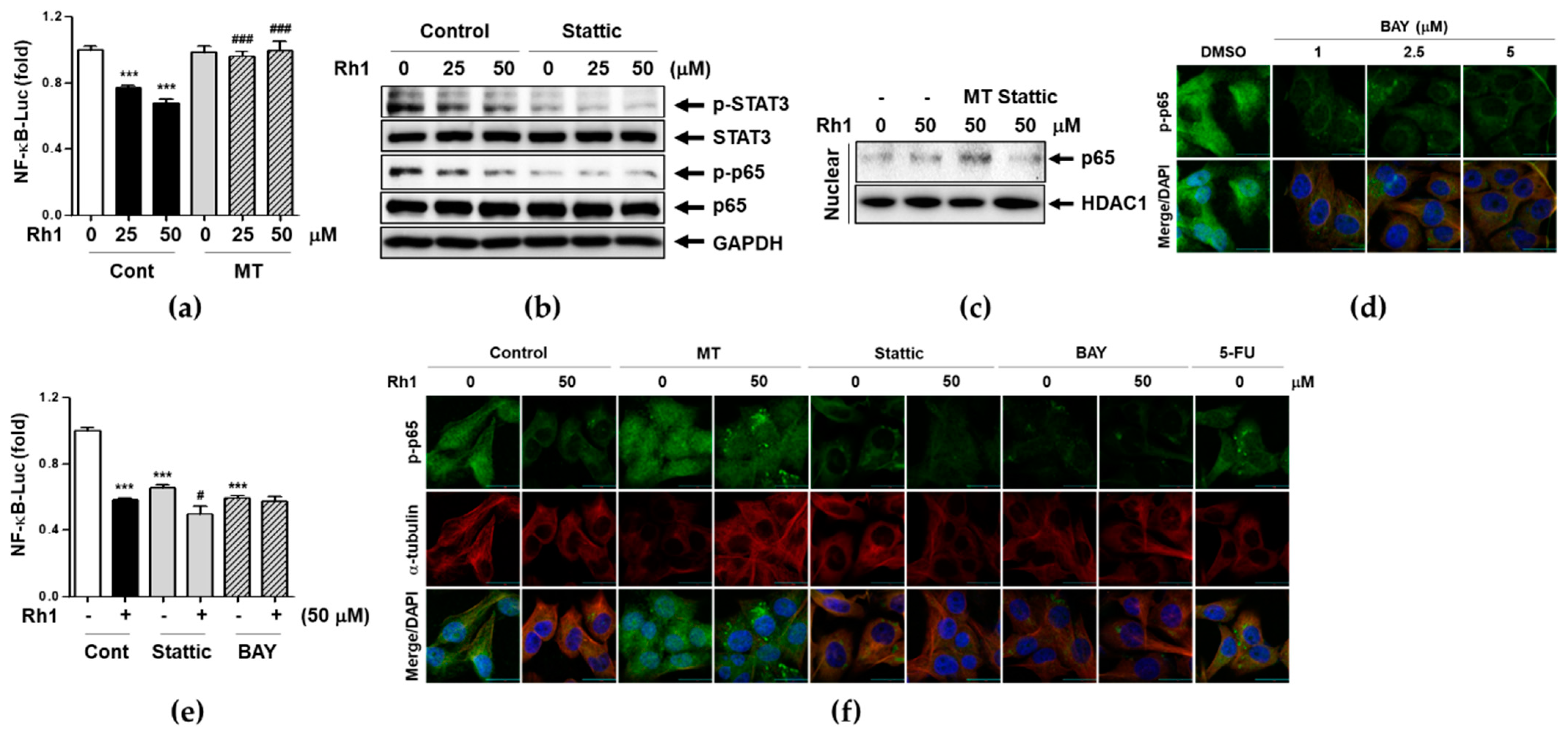
2.5. Rh1 Inhibited STAT3 Activation via Inducing mtROS
2.6. Rh1 Inhibits NF-κB Translocation and Transcriptional Activity through mtROS-induced STAT3 Deactivation
2.7. Rh1 Inhibits Cell Migration via Down-Regulating STAT3/NF-κB Signaling Pathway
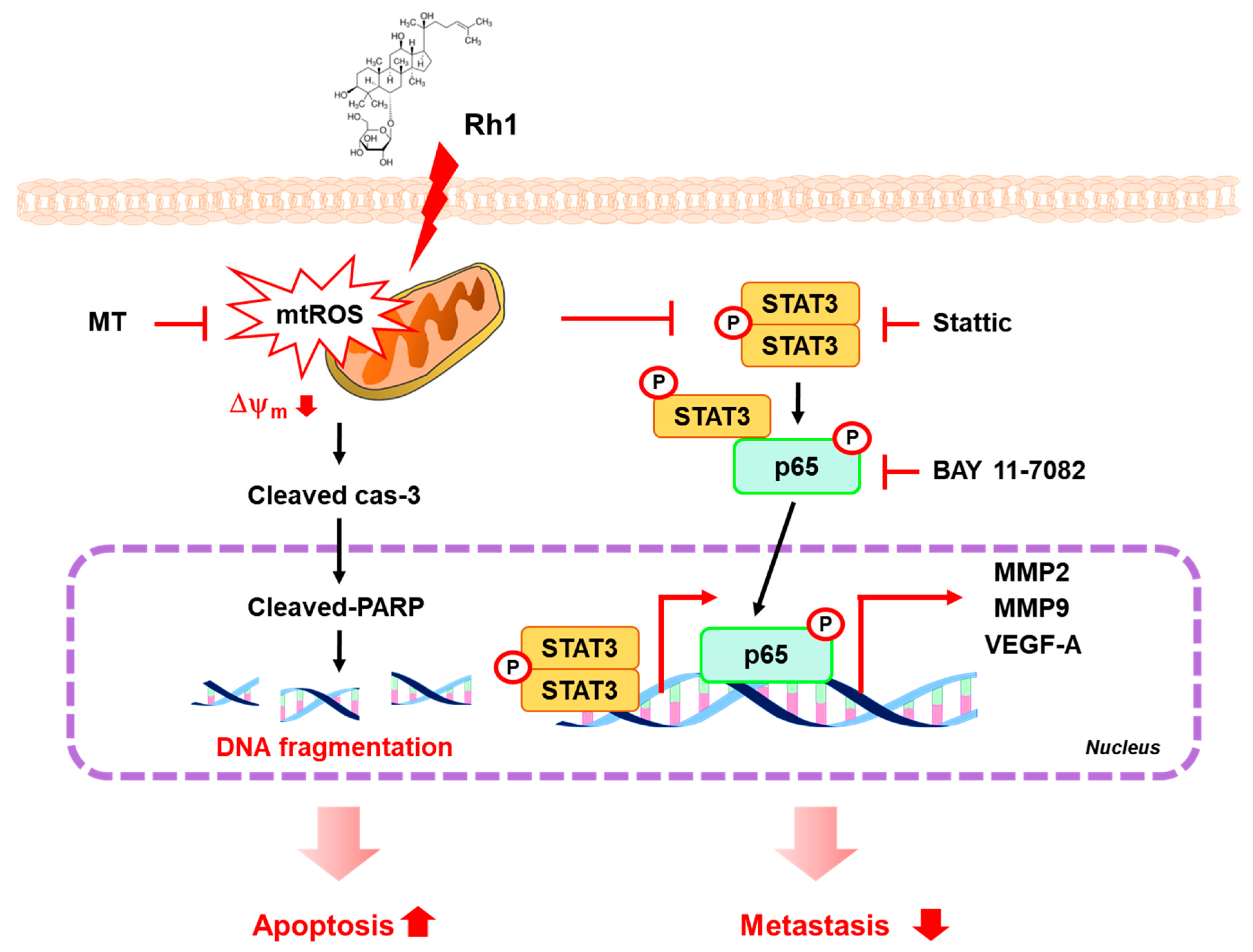
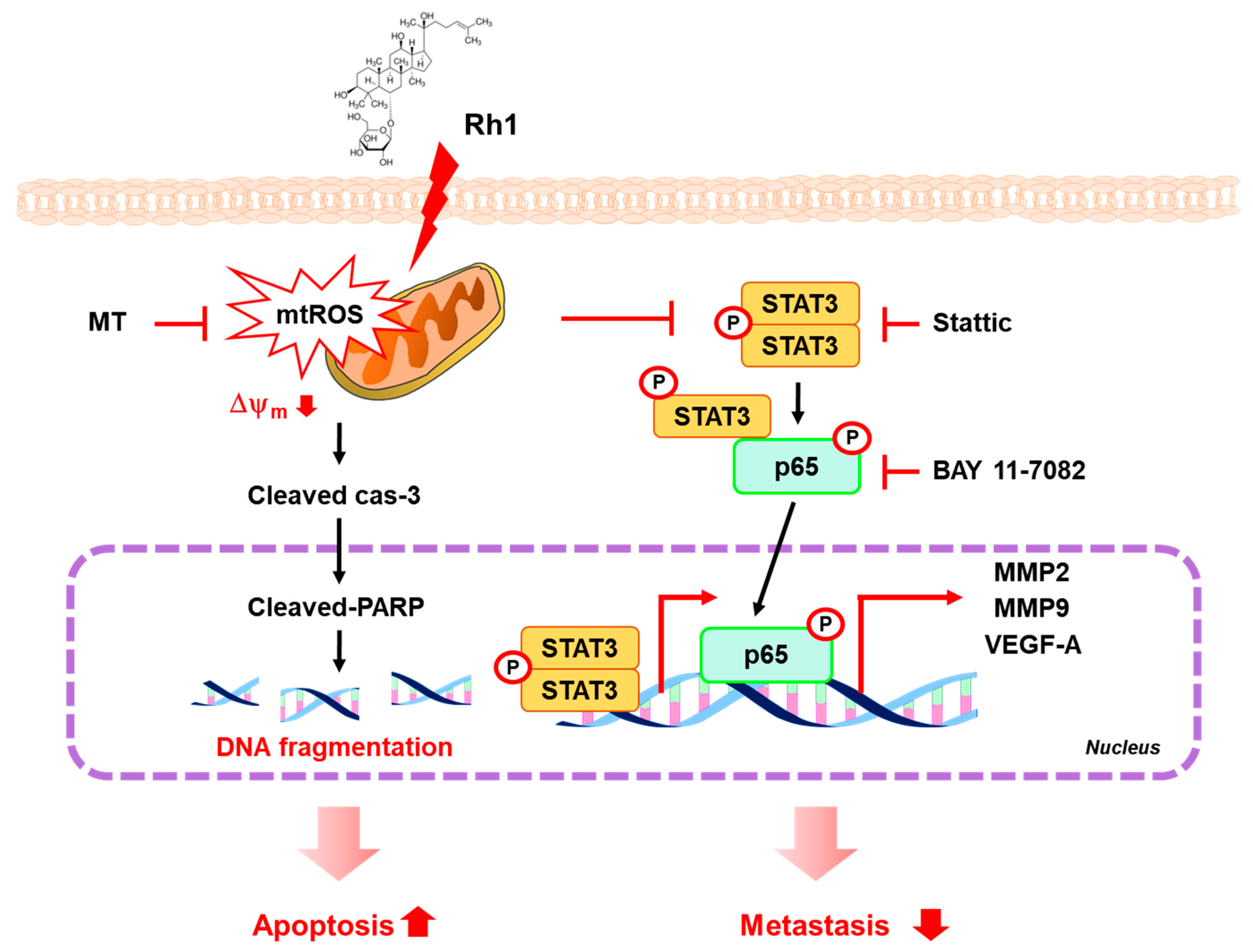
3. Discussion
3.1. Materials
3.2. Cell Culture and Viability Assay
3.3. PI Staining Assay
3.4. Western Blot
3.5. Quantitative Real-Time Polymerase Chain Reaction Assay
3.6. In Vitro Wound-Healing Assay
3.7. Transwell Migration and Invasion Assay
3.8. Measurement of Mitochondria Activity
3.9. Measurement of Mitochondria-Derived ROS
3.10. Measurement of Intracellular ROS Production
3.11. Luciferase Reporter Assay
3.12. Preparation of Cytosolic Extracts and Nuclear Extracts
3.13. Immunofluorescence Assay and Confocal Laser-Scanning Microscopy
3.14. JC-1 Mitochondrial Membrane Potential Assay
3.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BC | Breast cancer |
| BAY | BAY 11-7082 |
| DAPI | 40, 6-diamidino-2-phenylindol |
| DCF-DA | 2′,7′-dichlorodihydrofluoresceindiacetate |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| MT | Mito-TEMPO |
| mtROS | Mitochondria ROS |
| NAC | N-acetyl-cysteine |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PI3K/Akt | Phosphatidylinositol-3-kinase/protein kinase B |
| PTS | 20(S)-protopanaxatriol saponins |
| Rh1 | Ginsenoside Rh1 |
| ROS | Reactive oxygen species |
| STAT | Signal transducer and activator of transcription |
| TNBC | Triple negative breast cancer |
| VEGF | Vascular endothelial growth factor |
References
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast cancer in young women: An overview. Updates Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef]
- Dai, X.; Xiang, L.; Li, T.; Bai, Z. Cancer Hallmarks, Biomarkers and Breast Cancer Molecular Subtypes. J. Cancer 2016, 7, 1281–1294. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.A.; Moon, S.Y.; Park, D.; Lee, D.Y.; Park, J.B.; Lee, C.S. Apoptotic cell clearance in the tumor environment: A potential cancer terapeutic agent. Arch. Pharm. Res. 2019, 42, 658–671. [Google Scholar] [CrossRef]
- Lee, K.L.; Kuo, Y.C.; Ho, Y.S.; Huang, Y.H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers 2019, 11, 1334. [Google Scholar] [CrossRef] [Green Version]
- Boonrao, M.; Yodkeeree, S.; Ampasavate, C.; Anuchapreeda, S.; Limtrakul, P. The inhibitory effect of turmeric curcuminoids on matrix metalloproteinase-3 secretion in human invasive breast carcinoma cells. Arch. Pharm. Res. 2010, 33, 989–998. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Chen, W.; Li, P.; Liu, Y.; Yang, Y.; Ye, X.; Zhang, F.; Huang, H. Isoalantolactone induces apoptosis through ROS-mediated ER stress and inhibition of STAT3 in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 309. [Google Scholar] [CrossRef]
- Alsamri, H.; El Hasasna, H.; Al Dhaheri, Y.; Eid, A.H.; Attoub, S.; Iratni, R. Carnosol, a Natural Polyphenol, Inhibits Migration, Metastasis, and Tumor Growth of Breast Cancer via a ROS-Dependent Proteasome Degradation of STAT3. Front. Oncol 2019, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, C.; Tao, Z.; Zhao, L.; Zhu, Z.; Wu, W.; He, Y.; Chen, H.; Zheng, B.; Huang, X.; et al. Curcumin derivative WZ35 inhibits tumor cell growth via ROS-YAP-JNK signaling pathway in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 460. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Wu, Z.; Hu, X.; Zhang, J.; Gao, Z.; Han, X.; Qin, J.; Li, C.; Wang, Y. The PI3K/Akt/GSK-3beta/ROS/eIF2B pathway promotes breast cancer growth and metastasis via suppression of NK cell cytotoxicity and tumor cell susceptibility. Cancer Biol. Med. 2019, 16, 38–54. [Google Scholar]
- Jin, Y.; Huynh, D.T.N.; Kang, K.W.; Myung, C.S.; Heo, K.S. Inhibition of p90RSK activation sensitizes triple-negative breast cancer cells to cisplatin by inhibiting proliferation, migration and EMT. BMB Rep. 2019, 52, 706–711. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Zu, X.; Liu, K.; Bode, A.M.; Dong, Z.; Liu, Z.; Kim, D.J. Knockdown of Pyruvate Kinase M Inhibits Cell Growth and Migration by Reducing NF-kB Activity in Triple-Negative Breast Cancer Cells. Mol. Cells 2019, 42, 628–636. [Google Scholar]
- Huang, Q.; Li, S.; Zhang, L.; Qiao, X.; Zhang, Y.; Zhao, X.; Xiao, G.; Li, Z. CAPE-pNO2 Inhibited the Growth and Metastasis of Triple-Negative Breast Cancer via the EGFR/STAT3/Akt/E-Cadherin Signaling Pathway. Front. Oncol. 2019, 9, 461. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Lin, Y.; Ukaji, T.; Koide, N.; Umezawa, K. Inhibition of Late and Early Phases of Cancer Metastasis by the NF-kappaB Inhibitor DHMEQ Derived from Microbial Bioactive Metabolite Epoxyquinomicin: A Review. Int. J. Mol. Sci. 2018, 19, 729. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.J.; Zhang, X.J.; Shui, Y.M.; Wan, J.B.; Gao, J.L. Anticancer Activities of Protopanaxadiol- and Protopanaxatriol-Type Ginsenosides and Their Metabolites. Evid Based Complement. Altern. Med. 2016, 2016, 5738694. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Huynh, D.T.N.; Nguyen, T.L.L.; Jeon, H.; Heo, K.S. Therapeutic effects of ginsenosides on breast cancer growth and metastasis. Arch. Pharm. Res. 2020, 43, 773–787. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Sun, Q.; Jiao, W.; Yan, Y.; Zhang, X. Compound K Induces Endoplasmic Reticulum Stress and Apoptosis in Human Liver Cancer Cells by Regulating STAT3. Molecules 2018, 23, 1482. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.O.; Seo, C.H.; Cho, H.H.; Oh, S.; Hong, S.P.; Yoo, H.S.; Hong, J.T.; Oh, K.W.; Lee, Y.M. Ginsenoside compound K inhibits angiogenesis via regulation of sphingosine kinase-1 in human umbilical vein endothelial cells. Arch. Pharm. Res. 2014, 37, 1183–1192. [Google Scholar] [CrossRef]
- Huynh, D.T.N.; Baek, N.; Sim, S.; Myung, C.S.; Heo, K.S. Minor Ginsenoside Rg2 and Rh1 Attenuates LPS-Induced Acute Liver and Kidney Damages via Downregulating Activation of TLR4-STAT1 and Inflammatory Cytokine Production in Macrophages. Int. J. Mol. Sci. 2020, 21, 6656. [Google Scholar] [CrossRef]
- Lyu, X.; Xu, X.; Song, A.; Guo, J.; Zhang, Y.; Zhang, Y. Ginsenoside Rh1 inhibits colorectal cancer cell migration and invasion in vitro and tumor growth in vivo. Oncol. Lett. 2019, 18, 4160–4166. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Choi, Y.J.; Lee, S.G. Ginsenoside Rh1 suppresses matrix metalloproteinase-1 expression through inhibition of activator protein-1 and mitogen-activated protein kinase signaling pathway in human hepatocellular carcinoma cells. Eur. J. Pharm. 2012, 679, 24–33. [Google Scholar] [CrossRef]
- Jung, J.S.; Ahn, J.H.; Le, T.K.; Kim, D.H.; Kim, H.S. Protopanaxatriol ginsenoside Rh1 inhibits the expression of matrix metalloproteinases and the in vitro invasion/migration of human astroglioma cells. Neurochem. Int. 2013, 63, 80–86. [Google Scholar] [CrossRef]
- Yang, Y.F.; Wang, Y.Y.; Hsiao, M.; Lo, S.; Chang, Y.C.; Jan, Y.H.; Lai, T.C.; Lee, Y.C.; Hsieh, Y.C.; Yuan, S.F. IMPAD1 functions as mitochondrial electron transport inhibitor that prevents ROS production and promotes lung cancer metastasis through the AMPK-Notch1-HEY1 pathway. Cancer Lett. 2020, 485, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Esparza-Lopez, J.; Alvarado-Munoz, J.F.; Escobar-Arriaga, E.; Ulloa-Aguirre, A.; de Jesus Ibarra-Sanchez, M. Metformin reverses mesenchymal phenotype of primary breast cancer cells through STAT3/NF-kappaB pathways. BMC Cancer 2019, 19, 728. [Google Scholar] [CrossRef]
- Martincuks, A.; Li, P.C.; Zhao, Q.; Zhang, C.; Li, Y.J.; Yu, H.; Rodriguez-Rodriguez, L. CD44 in Ovarian Cancer Progression and Therapy Resistance—A Critical Role for STAT3. Front. Oncol. 2020, 10, 589601. [Google Scholar] [CrossRef]
- Li, Y.; Gan, C.; Zhang, Y.; Yu, Y.; Fan, C.; Deng, Y.; Zhang, Q.; Yu, X.; Zhang, Y.; Wang, L.; et al. Inhibition of Stat3 Signaling Pathway by Natural Product Pectolinarigenin Attenuates Breast Cancer Metastasis. Front. Pharm. 2019, 10, 1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, D.; Lee, J.H.; Kwak, M.K. NRF2 level is negatively correlated with TGF-beta1-induced lung cancer motility and migration via NOX4-ROS signaling. Arch. Pharm. Res. 2020, 43, 1297–1310. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; He, R.; Xu, Q.; Yang, Y.; Hu, Q.; Hou, H.; Liu, X.; Li, J. Ginsenoside 20(S)-protopanaxadiol inhibits triple-negative breast cancer metastasis in vivo by targeting EGFR-mediated MAPK pathway. Pharm. Res. 2019, 142, 1–13. [Google Scholar] [CrossRef]
- Huynh, D.T.N.; Jin, Y.; Myung, C.S.; Heo, K.S. Ginsenoside Rh1 Induces MCF-7 Cell Apoptosis and Autophagic Cell Death through ROS-Mediated Akt Signaling. Cancers 2021, 13, 1892. [Google Scholar] [CrossRef]
- Jin, S.; Jeon, J.H.; Lee, S.; Kang, W.Y.; Seong, S.J.; Toon, Y.R.; Choi, M.K.; Song, I.S. Detection of 13 Ginsenosides (Rb1, Rb2, Rc, Rd, Re, Rf, Rg1, Rg3, Rh2, F1, Compound K, 20(S)-Protopanaxadiol, and 20(S)-Protopanaxatriol) in Human Plasma and Application of the Analytical Method to Human Pharmacokinetic Studies Following Two Week-Repeated Administration of Red Ginseng Extract. Molecules 2019, 24, 2618. [Google Scholar]
- Lee, S.Y.; Jeong, J.J.; Eun, S.H.; Kim, D.H. Anti-inflammatory effects of ginsenoside Rg1 and its metabolites ginsenoside Rh1 and 20(S)-protopanaxatriol in mice with TNBS-induced colitis. Eur. J. Pharm. 2015, 762, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Menke, K.; Schwermer, M.; Felenda, J.; Beckmann, C.; Stintzing, F.; Schramm, A.; Zuzak, T.J. Taraxacum officinale extract shows antitumor effects on pediatric cancer cells and enhance mistletoe therapy. Complement. Med. 2018, 40, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.Z.; Nisar, M.A.; Alshwmi, M.; Din, S.R.U.; Gamallat, Y.; Khan, M.; Ma, T. Brevilin A Inhibits STAT3 Signaling and Induces ROS-Dependent Apoptosis, Mitochondrial Stress and Endoplasmic Reticulum Stress in MCF-7 Breast Cancer Cells. Onco Targets 2020, 13, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Yeo, I.J.; Park, J.H.; Jang, J.S.; Lee, D.Y.; Park, J.E.; Choi, Y.E.; Joo, J.H.; Joo, K.S.; Jeon, H.O.; Jin, T.H. Inhibitory effect of carnosol on UVB-induced inflammation via inhibition of STAT3. Arch. Pharm. Res. 2019, 42, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.J.; Yan, L.; Zhang, J.; Zhang, W.D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Guo, Q.; Liang, Y.; Zhao, Y.; Tian, X.; Ye, Y.; Tian, J.; Wu, T.; Lu, N. Wogonin induces cellular senescence in breast cancer via suppressing TXNRD2 expression. Arch. Toxicol. 2020, 94, 3433–3447. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Huynh, D.T.N.; Baek, N.; Nguyen, T.L.L.; Heo, K.S. Ginsenoside-Rg2 affects cell growth via regulating ROS-mediated AMPK activation and cell cycle in MCF-7 cells. Phytomedicine 2021, 85, 153549. [Google Scholar] [CrossRef]
- Nguyen, T.L.L.; Huynh, D.T.N.; Jin, Y.; Jeon, H.; Heo, K.S. Protective effects of ginsenoside-Rg2 and -Rh1 on liver function through inhibiting TAK1 and STAT3-mediated inflammatory activity and Nrf2/ARE-mediated antioxidant signaling pathway. Arch. Pharm. Res. 2021, 44, 241–252. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Huynh, D.T.N.; Myung, C.-S.; Heo, K.-S. Ginsenoside Rh1 Prevents Migration and Invasion through Mitochondrial ROS-Mediated Inhibition of STAT3/NF-κB Signaling in MDA-MB-231 Cells. Int. J. Mol. Sci. 2021, 22, 10458. https://doi.org/10.3390/ijms221910458
Jin Y, Huynh DTN, Myung C-S, Heo K-S. Ginsenoside Rh1 Prevents Migration and Invasion through Mitochondrial ROS-Mediated Inhibition of STAT3/NF-κB Signaling in MDA-MB-231 Cells. International Journal of Molecular Sciences. 2021; 22(19):10458. https://doi.org/10.3390/ijms221910458
Chicago/Turabian StyleJin, Yujin, Diem Thi Ngoc Huynh, Chang-Seon Myung, and Kyung-Sun Heo. 2021. "Ginsenoside Rh1 Prevents Migration and Invasion through Mitochondrial ROS-Mediated Inhibition of STAT3/NF-κB Signaling in MDA-MB-231 Cells" International Journal of Molecular Sciences 22, no. 19: 10458. https://doi.org/10.3390/ijms221910458
APA StyleJin, Y., Huynh, D. T. N., Myung, C.-S., & Heo, K.-S. (2021). Ginsenoside Rh1 Prevents Migration and Invasion through Mitochondrial ROS-Mediated Inhibition of STAT3/NF-κB Signaling in MDA-MB-231 Cells. International Journal of Molecular Sciences, 22(19), 10458. https://doi.org/10.3390/ijms221910458