
Interleukin-9 Facilitates Osteoclastogenesis in Rheumatoid Arthritis

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
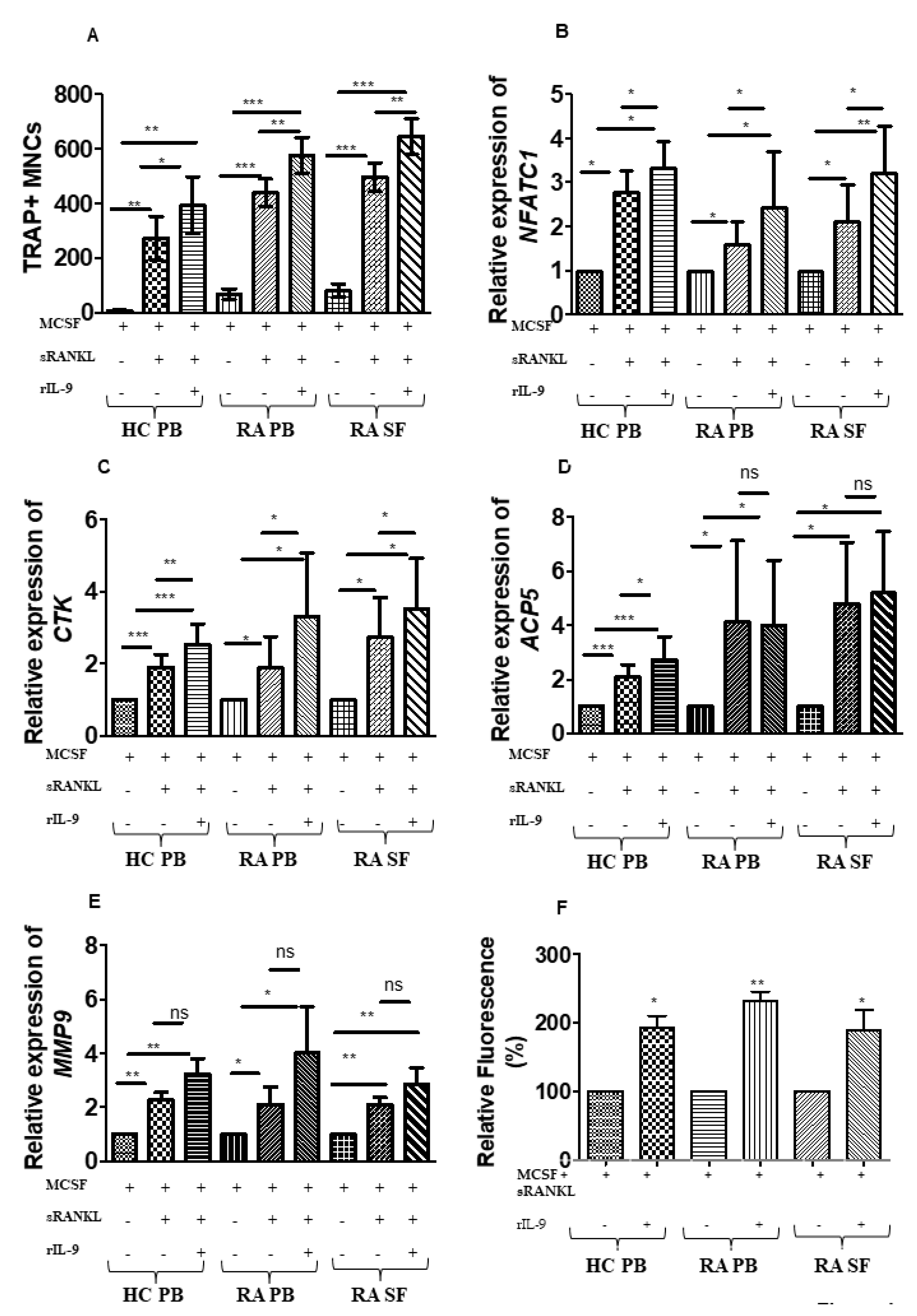
2.2. Influence of IL-9 on M-CSF- and RANKL-Induced Osteoclastogenesis
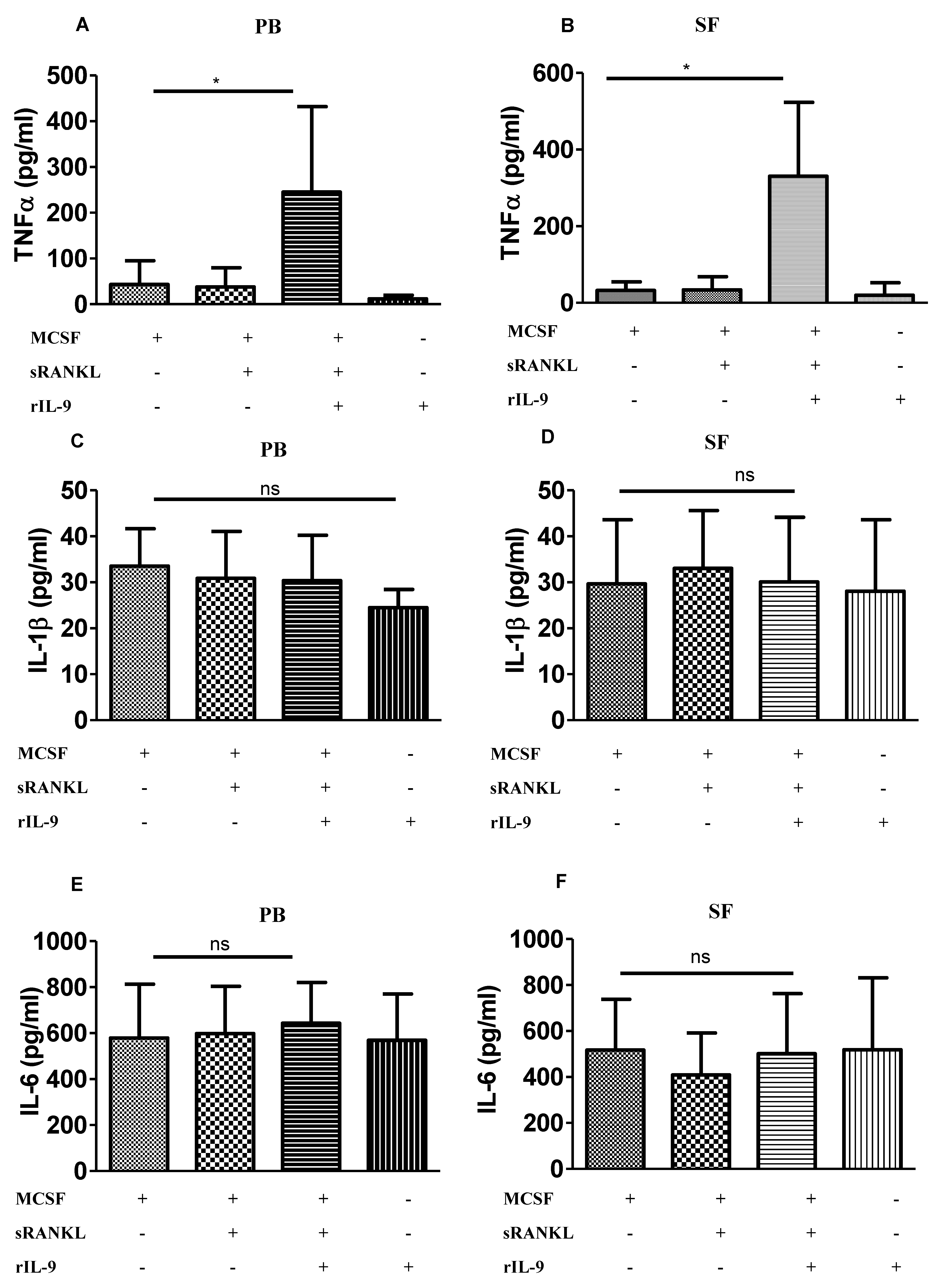
2.3. IL-9 Enhances TNF-α Production during Osteoclastogenesis
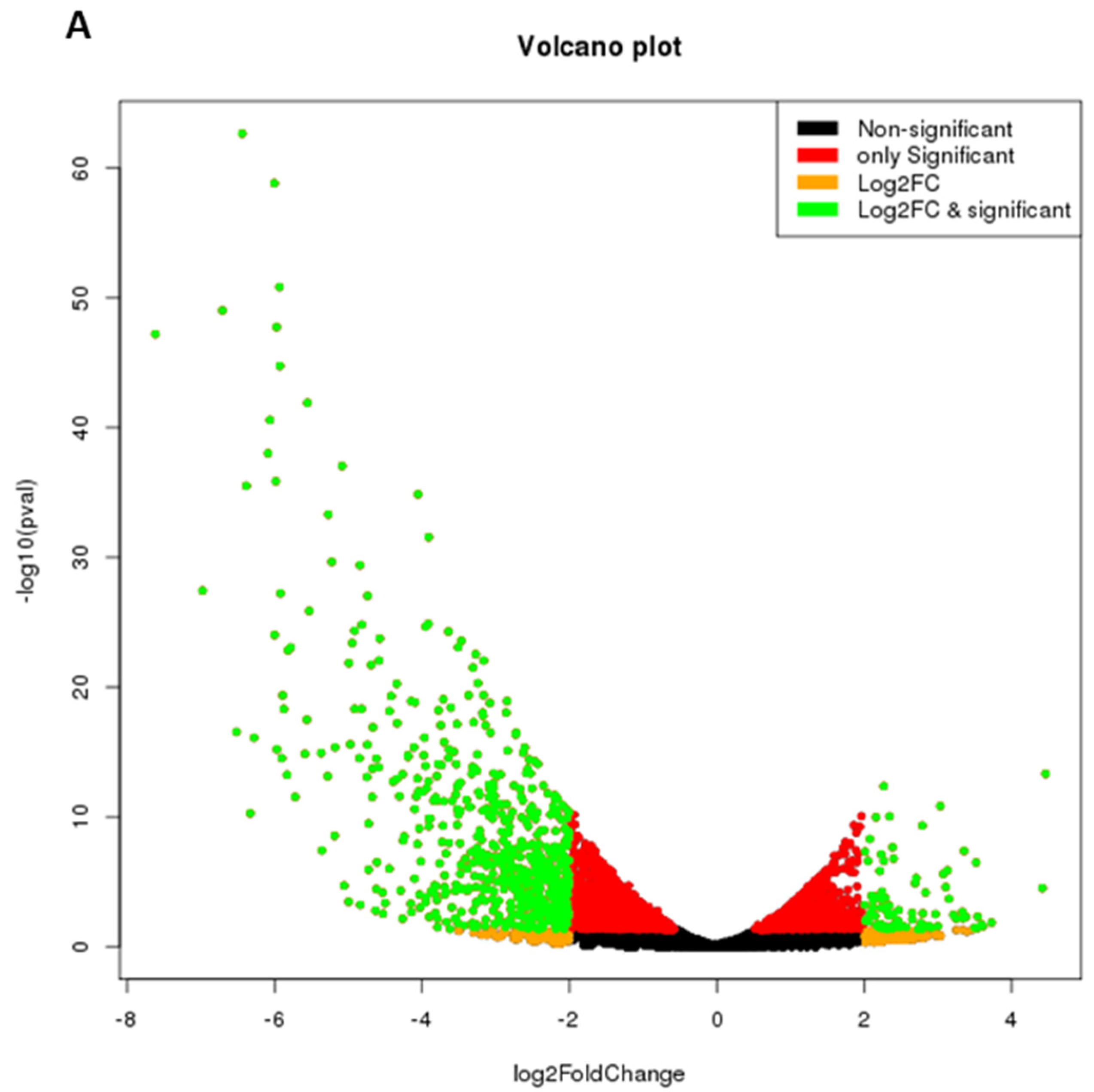
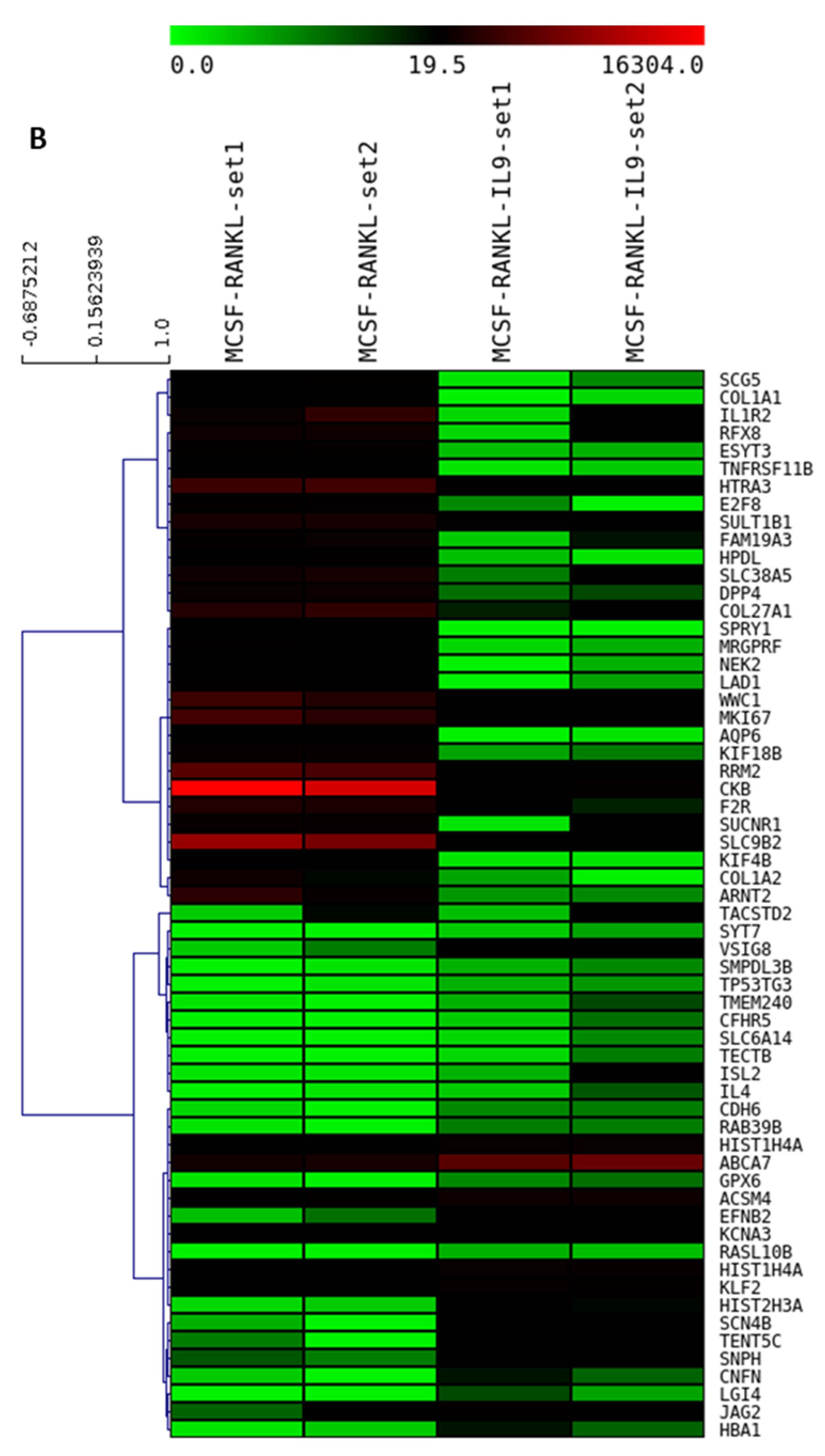
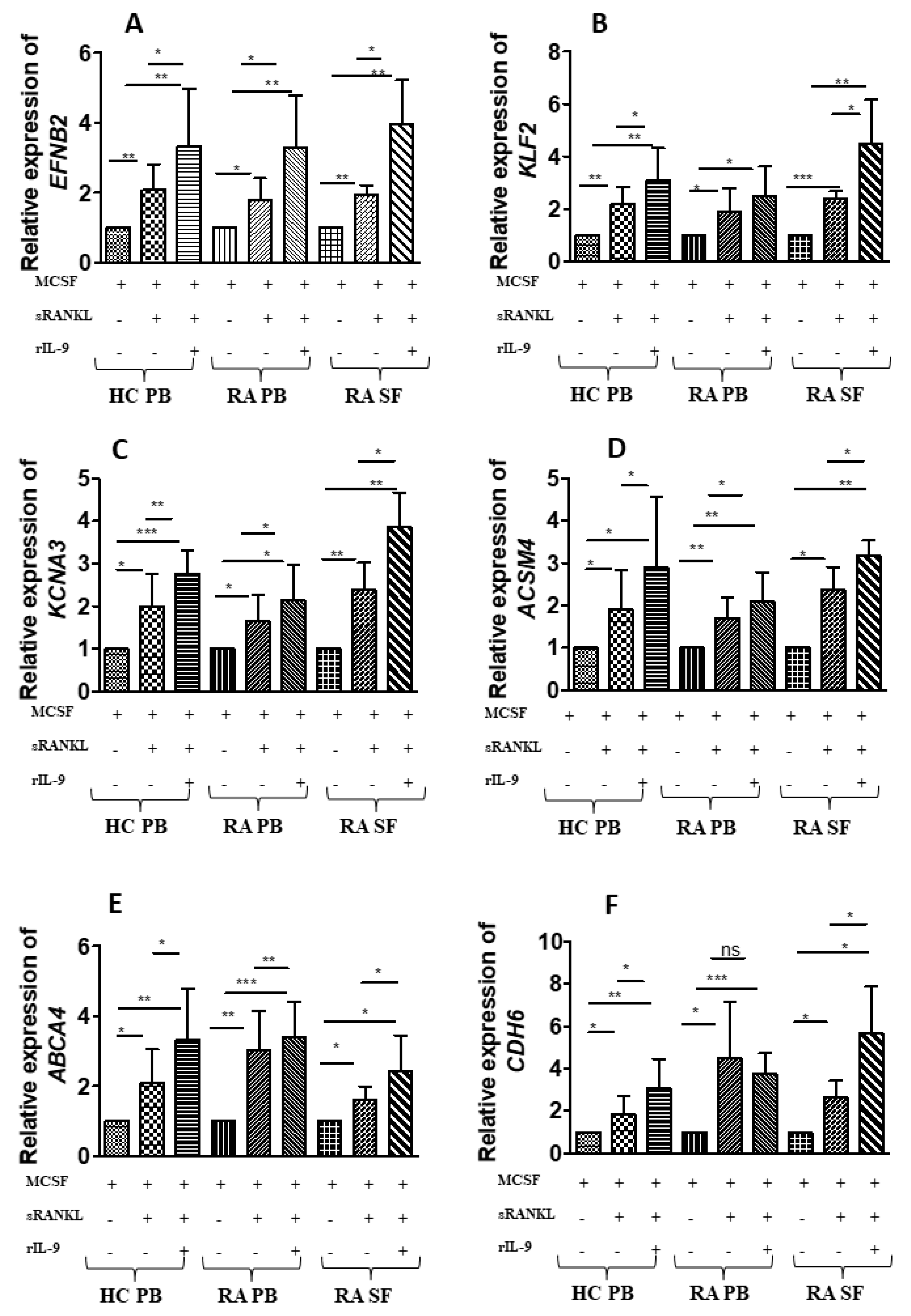
2.4. IL-9 Induces Differential Gene Expression during Osteoclastogenesis in RA
2.5. Validation of the IL-9-Induced Differentially Expressed Genes
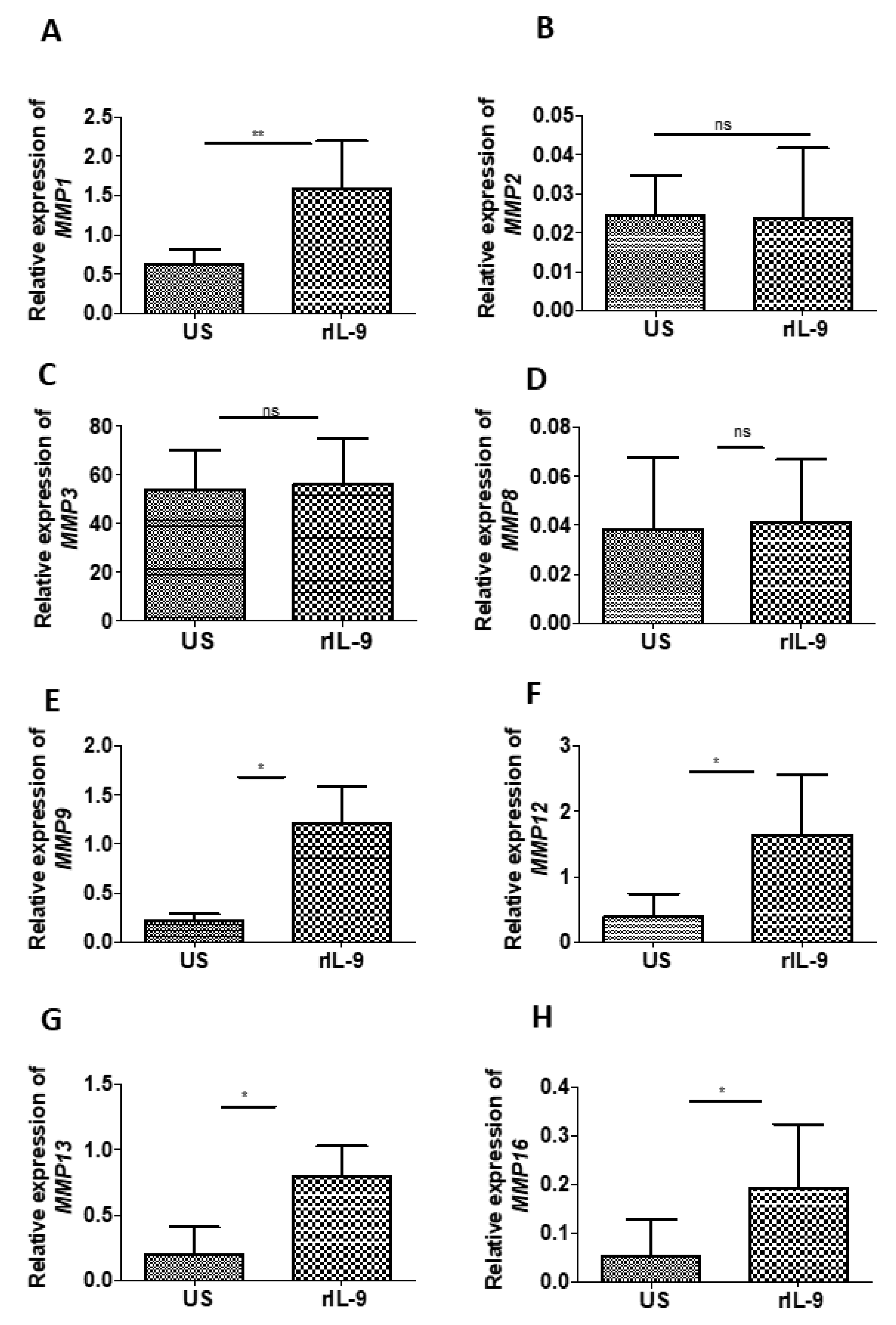
2.6. IL-9 Modulates the Expression of Matrix Metalloproteinases of Bone Cells
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Cell Separation, Culture, and Stimulation
4.3. TRAP Staining
4.4. Bone Explant Culture
4.5. Real Time PCR
4.6. Cytokine ELISA
4.7. Bone Resorption Assay
4.8. RNA Sequencing and Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Rheumatoid arthritis. Cell 1996, 85, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Hoes, J.N.; Bultink, I.E.M.; Lems, W.F. Management of osteoporosis in rheumatoid arthritis patients. Expert Opin. Pharm. 2015, 16, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Cheng, G.; Wang, H.; Feng, Y. Increased risk of vertebral fracture in patients with rheumatoid arthritis: A meta-analysis. Medicine 2016, 95, e5262. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Cells of the synovium in rheumatoid arthritis. Osteoclasts. Arthritis Res. Ther. 2007, 9, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, N.C.; Reinwald, S.; Manning, C.A.; Condon, K.W.; Iwata, K.; Burr, D.B.; Gravallese, E.M. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J. Bone Miner. Res. 2009, 24, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Hattersley, G.; Owens, J.; Flanagan, A.M.; Chambers, T.J. Macrophage colony stimulating factor (M-CSF) is essential for osteoclast formation in vitro. Biochem. Biophys. Res. Commun. 1991, 177, 526–531. [Google Scholar] [CrossRef]
- Tanaka, S.; Takahashi, N.; Udagawa, N.; Tamura, T.; Akatsu, T.; Stanley, E.R.; Kurokawa, T.; Suda, T. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J. Clin. Investig. 1993, 91, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Gravallese, E.M.; Harada, Y.; Wang, J.T.; Gorn, A.H.; Thornhill, T.S.; Goldring, S.R. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am. J. Pathol. 1998, 152, 943–951. [Google Scholar]
- Bromley, M.; Woolley, D.E. Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 1984, 27, 968–975. [Google Scholar] [CrossRef]
- Romas, E.; Bakharevski, O.; Hards, D.K.; Kartsogiannis, V.; Quinn, J.M.; Ryan, P.F.; Martin, T.J.; Gillespie, M.T. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum. 2000, 43, 821–826. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nishikaku, F.; Nakatuka, M.; Koga, Y. Osteoclast-like cells in murine collagen induced arthritis. J. Rheumatol. 1998, 25, 1154–1160. [Google Scholar] [PubMed]
- Pettit, A.R.; Ji, H.; von Stechow, D.; Müller, R.; Goldring, S.R.; Choi, Y.; Benoist, C.; Gravallese, E.M. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am. J. Pathol. 2001, 159, 1689–1699. [Google Scholar] [CrossRef] [Green Version]
- Redlich, K.; Hayer, S.; Ricci, R.; David, J.P.; Tohidast-Akrad, M.; Kollias, G.; Steiner, G.; Smolen, J.S.; Wagner, E.F.; Schett, G. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J. Clin. Investig. 2002, 110, 1419–1427. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Udagawa, N.; Takahashi, N.; Miyaura, C.; Tanaka, S.; Yamada, Y.; Koishihara, Y.; Ohsugi, Y.; Kumaki, K.; Taga, T.; et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc. Natl. Acad. Sci. USA. 1993, 90, 11924–11928. [Google Scholar] [CrossRef] [Green Version]
- Adamopoulos, I.E.; Chao, C.C.; Geissler, R.; Laface, D.; Blumenschein, W.; Iwakura, Y.; McClanahan, T.; Bowman, E.P. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res. Ther. 2010, 12, R29. [Google Scholar] [CrossRef] [Green Version]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Manzo, A.; Vitolo, B.; La Manna, M.P.; Giardina, G.; Sireci, G.; Dieli, F.; Montecucco, C.M.; et al. Potential involvement of IL-9 and Th9 cells in the pathogenesis of rheumatoid arthritis. Rheumatology 2015, 54, 2264–2272. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, K.; Kumar, U.; Das, S.; Chaudhuri, J.; Kumar, P.; Kanjilal, M.; Ghosh, P.; Sircar, G.; Basyal, R.K.; Kanga, U.; et al. Synovial IL-9 facilitates neutrophil survival, function and differentiation of Th17 cells in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 18. [Google Scholar] [CrossRef] [Green Version]
- Dantas, A.T.; Marques, C.D.; da Rocha Junior, L.F.; Cavalcanti, M.B.; Gonçalves, S.M.; Cardoso, P.R.; Mariz Hde, A.; Rego, M.J.; Duarte, A.L.; Pitta Ida, R.; et al. Increased Serum Interleukin-9 Levels in Rheumatoid Arthritis and Systemic Lupus Erythematosus: Pathogenic Role or Just an Epiphenomenon? Dis. Markers 2015, 2015, 519638. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kubatzky, K.F.; Mitra, D.K. An Update on Interleukin-9: From Its Cellular Source and Signal Transduction to Its Role in Immunopathogenesis. Int. J. Mol. Sci. 2019, 20, 2113. [Google Scholar] [CrossRef] [Green Version]
- Leipe, J.; Grunke, M.; Dechant, C.; Reindl, C.; Kerzendorf, U.; Schulze-Koops, H.; Skapenko, A. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010, 62, 2876–2885. [Google Scholar] [CrossRef]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.; Horowitz, M.; Choi, Y. Osteoimmunology: Interactions of the bone and immune system. Endocr. Rev. 2008, 29, 403–440. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Ainola, M.; Valleala, H.; Ma, J.; Ida, H.; Mandelin, J.; Kinne, R.W.; Santavirta, S.; Sorsa, T.; López-Otín, C.; et al. Analysis of 16 different matrix metalloproteinases (MMP-1 to MMP-20) in the synovial membrane: Different profiles in trauma and rheumatoid arthritis. Ann. Rheum. Dis. 1999, 58, 691–697. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu-Raychaudhuri, S.; Abria, C.; Raychaudhuri, S.P. IL-9, a local growth factor for synovial T cells in inflammatory arthritis. Cytokine 2016, 79, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Ferrante, A.; Raimondo, S.; Bignone, R.; Rodolico, V.; Peralta, S.; Van Tok, M.; Cannizzaro, A.; Schinocca, C.; et al. Interleukin-9 Overexpression and Th9 Polarization Characterize the Inflamed Gut, the Synovial Tissue, and the Peripheral Blood of Patients With Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 1922–1931. [Google Scholar] [CrossRef]
- Guggino, G.; Ciccia, F.; Di Liberto, D.; Lo Pizzo, M.; Ruscitti, P.; Cipriani, P.; Ferrante, A.; Sireci, G.; Dieli, F.; Fourniè, J.J.; et al. Interleukin (IL)-9/IL-9R axis drives γδ T cells activation in psoriatic arthritis patients. Clin. Exp. Immunol. 2016, 186, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Rauber, S.; Luber, M.; Weber, S.; Maul, L.; Soare, A.; Wohlfahrt, T.; Lin, N.Y.; Dietel, K.; Bozec, A.; Herrmann, M.; et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 2017, 23, 938–944. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Irie, N.; Takada, Y.; Shimoda, K.; Miyamoto, T.; Nishiwaki, T.; Suda, T.; Matsuo, K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006, 4, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Laha, D.; Deb, M.; Das, H. KLF2 (kruppel-like factor 2 [lung]) regulates osteoclastogenesis by modulating autophagy. Autophagy 2019, 15, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Beier, E.; Sheu, T.; Zhang, H.; Zuscik, M.J.; Puzas, E.J.; Boyce, B.F.; Mooney, R.A.; Xing, L. High-fat diet causes bone loss in young mice by promoting osteoclastogenesis through alteration of the bone marrow environment. Calcif. Tissue Int. 2015, 96, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelton, K.; Krieder, J.; Joiner, D.; Freeman, M.R.; Goldstein, S.A.; Solomon, K.R. Hypercholesterolemia promotes an osteoporotic phenotype. Am. J. Pathol. 2012, 181, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanbe, T.; Tomofuji, T.; Ekuni, D.; Azuma, T.; Tamaki, N.; Yamamoto, T. Oral administration of vitamin C prevents alveolar bone resorption induced by high dietary cholesterol in rats. J. Periodontol. 2007, 78, 2165–2170. [Google Scholar] [CrossRef]
- Prieto-Potin, I.; Roman-Blas, J.A.; Martinez-Calatrava, M.J.; Gomez, R.; Largo, R.; Herrero-Beaumont, G. Hypercholesterolemia boosts joint destruction in chronic arthritis. An experimental model aggravated by foam macrophage infiltration. Arthritis Res. Ther. 2013, 15, R81. [Google Scholar] [CrossRef] [Green Version]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.; Ueda, K.; Yokoyama, S. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsel-Nitschke, P.; Jehle, A.W.; Shan, J.; Cao, G.; Bacic, D.; Lan, D.; Wang, N.; Tall, A.R. Potential role of ABCA7 in cellular lipid efflux to apoA-I. J. Lipid Res. 2005, 46, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Lan, D.; Gerbod-Giannone, M.; Linsel-Nitschke, P.; Jehle, A.W.; Chen, W.; Martinez, L.O.; Tall, A.R. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J. Biol. Chem. 2003, 278, 42906–42912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.O.; Klett, E.L.; Coleman, R.A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Mbalaviele, G.; Nishimura, R.; Myoi, A.; Niewolna, M.; Reddy, S.V.; Chen, D.; Feng, J.; Roodman, D.; Mundy, G.R.; Yoneda, T. Cadherin-6 mediates the heterotypic interactions between the hemopoietic osteoclast cell lineage and stromal cells in a murine model of osteoclast differentiation. J Cell Biol. 1998, 141, 1467–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Kerloc’h, A.; Rotival, M.; Xu, X.; Zhang, Q.; D’Souza, Z.; Kim, M.; Scholz, J.C.; Ko, J.H.; Srivastava, P.K.; et al. Kcnn4 is a regulator of macrophage multinucleation in bone homeostasis and inflammatory disease. Cell Rep. 2014, 8, 1210–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ågren, M.S.; Schnabel, R.; Christensen, L.H.; Mirastschijski, U. Tumor necrosis factor-α-accelerated degradation of type I collagen in human skin is associated with elevated matrix metalloproteinase (MMP)-1 and MMP-3 ex vivo. Eur. J. Cell Biol. 2015, 94, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecil, G.G.; Larsen, P.H.; Corley, S.M.; Herx, L.M.; Besson, A.; Goodyer, C.G.; Yong, V.W. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J. Neurosci. Res. 2000, 61, 212–224. [Google Scholar] [CrossRef]
- Tasaki, K.; Shintani, Y.; Saotome, T.; Andoh, A.; Fujiyama, Y.; Hozawa, S.; Bamba, T. Pro-inflammatory cytokine-induced matrix metalloproteinase-1 (MMP-1) secretion in human pancreatic periacinar myofibroblasts. Pancreatology 2003, 3, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, E.; Jonsson, K.; Saxne, T.; Eberhardt, K. Course of radiographic damage over 10 years in a cohort with early rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 611–616. [Google Scholar] [CrossRef]
- Lipsky, P.E.; van der Heijde, D.M.; St Clair, E.W.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R. For the Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. Infliximab and methotrexate in the treatment of rheumatoid arthritis. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Klareskog, L.; van der Heijde, D.; de Jager, J.P.; Gough, A.; Kalden, J.; Malaise, M. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: Double-blind randomised controlled trial. Lancet 2004, 363, 675–681. [Google Scholar] [CrossRef]
- Breedveld, F.C.; Weisman, M.H.; Kavanaugh, A.F.; Cohen, S.B.; Pavelka, K.; van Vollenhoven, R. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006, 54, 26–37. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age/Sex | Duration of Disease (Years) | DAS28 ESR | ESR (mm/1st Hour) | CRP(mg/lit) | Treatment | Joint Erosions |
|---|---|---|---|---|---|---|---|
| 1 | 43/F | 9.4 | 6.1 | 56 | 43.6 | Mtx | YES |
| 2 | 54/F | 16.7 | 5.9 | 34 | 11.9 | Mtx, Inf | YES |
| 3 | 31/M | 4.2 | 4.5 | 22 | 13.4 | Mtx | NO |
| 4 | 45/F | 8.4 | 4.2 | 59 | 39.1 | Inf | YES |
| 5 | 49/F | 5.9 | 4.6 | 31 | 24.9 | Ada, Mtx | NO |
| 6 | 56/F | 11.5 | 6.2 | 42 | 28.2 | Mtx | YES |
| 7 | 36/M | 6.5 | 6.5 | 47 | 33.9 | Mtx | YES |
| 8 | 41/F | 8.4 | 5.1 | 41 | 27.3 | Lfu | YES |
| 9 | 47/M | 10. | 5.2 | 11 | 1.3 | Eta | NO |
| 10 | 29/M | 8.5 | 4.2 | 21 | 4.3 | Mtx, Inf | YES |
| 11 | 39/M | 4.9 | 5.6 | 29 | 5.6 | Mtx | YES |
| 12 | 46/F | 9.8 | 6.3 | 27 | 3.8 | Mtx, Inf | YES |
| 13 | 54/F | 13.2 | 5.7 | 17 | 0.4 | Mtx, Inf | YES |
| 14 | 48/F | 11.5 | 4.1 | 15 | 1.9 | Mtx, Inf | YES |
| 15 | 58/F | 17.5 | 4.0 | 24 | 2.5 | Mtx, Inf | YES |
| Gene | Primer Sequence |
|---|---|
| EFNB2 | Forward primer: 5′ TCCAACAAGACGTCCAGAAC 3′ Reverse primer: 5′ CGTCTGTGCTAGAACCTGGAT 3′ |
| KCNA3 | Forward primer: 5′ TTGGTCTGCCTATGCCCTTG 3′ Reverse primer: 5′ CAGCCCACTTGCAAAACAGG 3′ |
| CDH6 | Forward primer: 5′ TTTCGTTTTCCTTGGCCCCT 3′ Reverse primer: 5′ CGCCGTGTTGTCTTTGTTGT 3′ |
| ABCA7 | Forward primer: 5′ TGAGGTCAGATACGGAGGCT 3′ Reverse primer: 5′ TTTTCAGGACACGGTCGAGG 3′ |
| KLF2 | Forward primer: 5′ GTGGGCATTTTTGGGCTACC 3′ Reverse primer: 5′ CCCAGTTCCAAGCAACCAGA 3′ |
| ACSM4 | Forward primer: 5′ ACTGCTGCCACGATAAGAGG 3′ Reverse primer: 5′ GCCCAATACGGTACCCAGAG 3′ |
| MMP1 | Forward primer: 5′ AGCCATCACTTACCTTGCACT 3′ Reverse primer: 5′ CTGGGAAGCTGTGAGACACC 3′ |
| MMP2 | Forward primer: 5′ ATCCAGACTTCCTCAGGCGG 3′ Reverse primer: 5′ CCATTAGCGCCTCCATCGTAG 3′ |
| MMP3 | Forward primer: 5′ TGAAATTGGCCACTCCCTGG 3′ Reverse primer: 5′GGAACCGAGTCAGGTCTGTG 3′ |
| MMP8 | Forward primer: 5′CTCCCTGAAGACGCTTCCAT 3′ Reverse primer: 5′TCCAGGTAGTCCTGAACAGT 3′ |
| MMP9 | Forward primer: 5′ GTACTCGACCTGTACCAGCG 3′ Reverse primer: 5′ AGAAGCCCCACTTCTTGTCG 3′ |
| MMP12 | Forward primer: 5′ AACCAACGCTTGCCAAATCC 3′ Reverse primer: 5′ TTTCCCACGGTAGTGACAGC 3′ |
| MMP13 | Forward primer: 5′ TTGTTGCTGCGCATGAGTTC 3′ Reverse primer: 5′AAGTGGCTTTTGCCGGTGTA 3′ |
| MMP16 | Forward primer: 5′CTACTGCTGACCCTGTCTTCG 3′ Reverse primer: 5′ GCTTCAATGGATGGACGAGC 3′ |
| NFATc1 | Forward primer: 5′ CCGTAGGCTTGTTCATAG 3′ Reverse primer: 5′ GTAACCTATTGATGTGCTATTC 3′ |
| CTSK | Forward primer: 5′ CAAATCCATCCTGCTCTTC 3′ Reverse primer: 5′ TATCACCACATCTGCTTCA 3′ |
| ACP5 | Forward primer: 5′ CTTCCACTATGGGACTGA 3′ Reverse primer: 5′ CCTCGATGTAAGTGACAG3′ |
| GAPDH | Forward primer: 5′ AGTCAGCCGCATCTTCTTTT 3′ Reverse primer: 5′ CCCAATACGACCAAATCCGT 3′ |
| Sample | Number of Reads |
|---|---|
| MCSF_RANKL1_set1_R1 | 31,668,998 |
| MCSF_RANKL1_set1_R2 | 31,668,998 |
| MCSF_RANKL2_set2_R1 | 34,530,684 |
| MCSF_RANKL2_set2_R2 | 34,530,684 |
| MCSF_RANKL_IL9_set1_R1 | 30,689,185 |
| MCSF_RANKL_IL9_set1_R2 | 30,689,185 |
| MCSF_RANKL_IL9_set2_R1 | 39,599,229 |
| MCSF_RANKL_IL9_set2_R2 | 39,599,229 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kar, S.; Gupta, R.; Malhotra, R.; Sharma, V.; Farooque, K.; Kumar, V.; Chakraborty, S.; Mitra, D.K. Interleukin-9 Facilitates Osteoclastogenesis in Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 10397. https://doi.org/10.3390/ijms221910397
Kar S, Gupta R, Malhotra R, Sharma V, Farooque K, Kumar V, Chakraborty S, Mitra DK. Interleukin-9 Facilitates Osteoclastogenesis in Rheumatoid Arthritis. International Journal of Molecular Sciences. 2021; 22(19):10397. https://doi.org/10.3390/ijms221910397
Chicago/Turabian StyleKar, Santanu, Ranjan Gupta, Rajesh Malhotra, Vijay Sharma, Kamran Farooque, Vijay Kumar, Sushmita Chakraborty, and Dipendra Kumar Mitra. 2021. "Interleukin-9 Facilitates Osteoclastogenesis in Rheumatoid Arthritis" International Journal of Molecular Sciences 22, no. 19: 10397. https://doi.org/10.3390/ijms221910397
APA StyleKar, S., Gupta, R., Malhotra, R., Sharma, V., Farooque, K., Kumar, V., Chakraborty, S., & Mitra, D. K. (2021). Interleukin-9 Facilitates Osteoclastogenesis in Rheumatoid Arthritis. International Journal of Molecular Sciences, 22(19), 10397. https://doi.org/10.3390/ijms221910397