Abstract
In contrast to USH2A, variants in ADGRV1 are a minor cause of Usher syndrome type 2, and the associated phenotype is less known. The purpose of the study was to characterize the retinal phenotype of 18 ADGRV1 patients (9 male, 9 female; median age 52 years) and compare it with that of 204 USH2A patients (111 male, 93 female; median age 43 years) in terms of nyctalopia onset, best corrected visual acuity (BCVA), fundus autofluorescence (FAF), and optical coherence tomography (OCT) features. There was no statistical difference in the median age at onset (30 and 18 years; Mann–Whitney U test, p = 0.13); the mean age when 50% of the patients reached legal blindness (≥1.0 log MAR) based on visual acuity (64 years for both groups; log-rank, p = 0.3); the risk of developing advanced retinal degeneration (patch or atrophy) with age (multiple logistic regression, p = 0.8); or the frequency of cystoid macular edema (31% vs. 26%, Fisher’s exact test, p = 0.4). ADGRV1 and USH2A retinopathy were indistinguishable in all major functional and structural characteristics, suggesting that the loss of function of the corresponding proteins produces similar effects in the retina. The results are important for counseling ADGRV1 patients, who represent the minor patient subgroup.
1. Introduction
Usher syndrome (USH) is a recessively inherited disorder characterized by the combination of retinal degeneration in the form of retinitis pigmentosa (RP), sensorineural hearing impairment, and, sometimes, vestibular dysfunction. Usher syndrome type 2 (USH2) is the most frequent subtype and presents with moderate to severe congenital hearing impairment, the onset of RP in the second decade of life, and normal vestibular function [1]. It has been associated with pathogenic variants in three genes: USH2A, ADGRV1 (previously known as VLGR1 or GPR98), and WHRN, identified in approximately 90%, 9%, and 1% of USH2 patients, respectively [2]. The pathogenic variants in USH2A are not only the most frequent cause of USH2 but also of RP without the associated hearing impairment (i.e., non-syndromic RP), accounting for 12–25% of cases [3,4]. The characteristics of retinal disease associated with USH2A have been described in many studies and are well known [5,6,7,8,9,10,11]. In contrast, the clinical presentations associated with ADGRV1 and WHRN are less well known, as large patient cohorts have not yet been described phenotypically. The purpose of this study is to describe the characteristics of retinal disease in the largest cohort of ADGRV1 patients to date and compare them with those of USH2A patients.
2. Results
2.1. Genetic Findings
The identified variants in USH2A and ADGRV1 genes are listed in Supplementary Table S1. More than 1000 predicted pathogenic or likely pathogenic variants in the USH2A gene have been reported. Comparatively, only 168 predicted pathogenic or likely pathogenic variants in the ADGRV1 gene have been listed in the LOVD3.0 database (https://databases.lovd.nl/shared/genes/GPR98; accessed on 15 August 2021). In this cohort, we identified a total of 27 different ADGRV1 variants in 16 independent patients and 2 additional siblings. Twenty-four of these twenty-seven variants were loss of function variants (LOF) (i.e., twelve frameshift, nine nonsense, four splicing variants, and one large deletion), and three were missense variants. These variants were distributed along the ADGRV1 gene. Of note, 78% of the patients carried biallelic LOF variants that are predicted to undergo nonsense-mediated decay (NMD) [12].
2.2. Disease Onset
There was no significant difference between the age at disease onset between ADGRV1 (median 30 years, range 5–46 years) and USH2A patients (18 years, range 0–55 years) (Mann–Whitney U test, p = 0.13) (Figure 1).
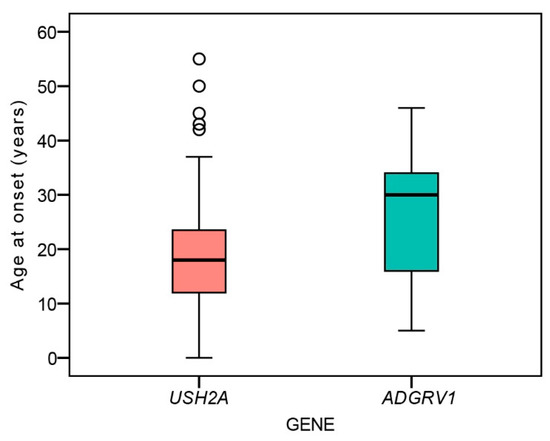
Figure 1.
Boxplot chart showing the ages at onset of nyctalopia in the two patient groups. Horizontal lines represent the median values, boxes represent half of the data for each group/mutation, and whiskers represent the remaining data except in the case of the outliers (circles). Red color represents USH2A patients and green color represents ADGRV1 patients.
2.3. Visual Acuity
Table 1 shows the frequency of legal blindness in the better eye for each genotype. The multiple regression analysis showed a significant association between visual acuity in the better eye and age (p < 0.001) but not genotype (p = 0.3). The Kaplan–Meier survival analysis predicted that the mean age when 50% of the patients reached legal blindness based on visual acuity of 0.1 or less (1.0 logMAR or more) was 64 years for USH2A (95% CI 61–68 years) and 64 years for ADGRV1 (95% CI 60–68 years) (Figure 2). The difference was not significant (log-rank, p = 0.3). The median value was 65 years for USH2A but could not be determined for ADGRV1 due to the fact that all of the patients were censored prior (i.e., all had BCVA > 0.1 at the last follow-up). The median age when 50% of the patients were predicted to reach visual acuity <0.3 (>0.52) on the better eye was 57 and 54 years for USH2A and ADGRV1, respectively. The difference was not significant (log-ank, p = 0.9).

Table 1.
Visual acuity.
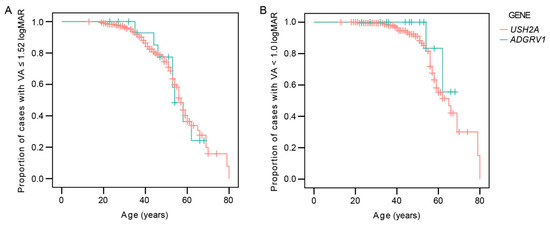
Figure 2.
Kaplan–Meier survival analysis showing the ratio of patients reaching low vision (>1.52 logMAR or <6/20) (A) or legal blindness (≥1.0 log MAR or ≤6/60) (B). Red color represents USH2A patients and green color represents ADGRV1 patients.
2.4. Fundus Autofluorescence Patterns
There was a good interocular symmetry of the FAF patterns; the same FAF pattern was present in 94% (136/145) of USH2A and 88% (14/16) of ADGRV1 patients. The patients with asymmetry had either a combination of a hyperautofluorescent ring and patch (n = 7) or patch and atrophy (n = 4). Table 2 shows the distribution of the different FAF patterns between the two groups on the measured eye. The FAF patterns were qualitatively similar between the two groups (examples in Figure 3). A hyperautofluorescent ring was present in 70% of USH2A and 63% of ADGRV1 patients. A multiple regression was performed to determine whether there was any difference in the autofluorescence patterns between different genetic groups with consideration of age-related disease progression. The multiple logistic regression showed an increased likelihood of advanced structural disease (patch or atrophy) with age (Exp(B) = 1.1, p < 0.001) but no correlation with gene (Exp(B) = 0.9, p = 0.8). Similarly, the multiple linear regression showed a significant association between ring diameter and age (p < 0.001) but not gene (p = 0.3). The survival analysis predicted that 50% of patients progress to advanced degeneration (patch or atrophy) at the median age of 58 years in both groups (95% CI 56–60 years for both) (Figure 4C).
Table 2.
Distribution of different FAF patterns in USH2A and ADGRV1 patient groups.
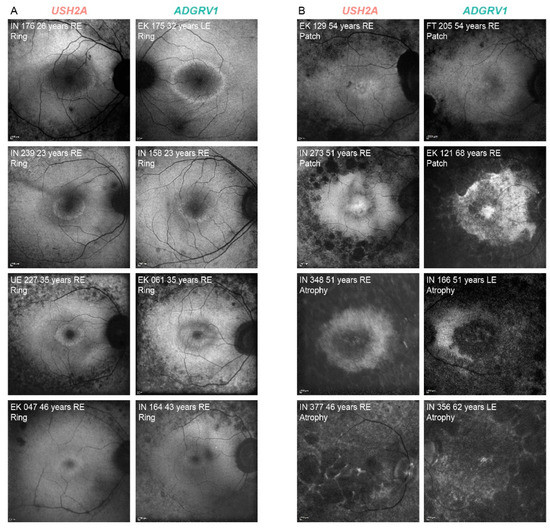
Figure 3.
Representative FAF images of USH2A and ADGRV1 patients. (A). hyperautofluorescent rings with preserved central retina. (B). advanced disease—hyperautofluorescent patch and atrophy. The patient’s ID, age and FAF pattern is stated in the top left corner of each image. The affected gene is stated above each column. Note that almost identical patterns were found in both groups. Red color represents USH2A patients and green color represents ADGRV1 patients.
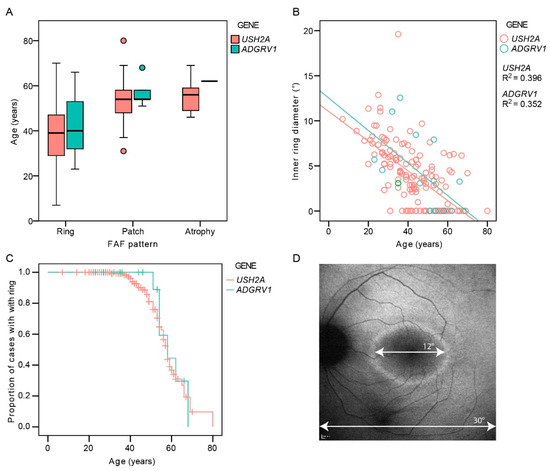
Figure 4.
(A). Boxplot showing ages of patients with different autofluorescence patterns. (B). The diameter of the hyperautofluorescent ring in relation to age and genotype. (C). Kaplan–Meier survival analysis showing the proportion of cases with FAF pattern of hyperautofluorescent ring (preserved central retina) with increasing age. (D). Demonstration of the measurement of the inner ring diameter. Note the similarity between the survival curves representing the structural (C) and functional decline (Figure 2). Red color represents USH2A patients and green color represents ADGRV1 patients.
2.5. Optical Coherence Tomography
CME was present in 26% (35/135) of USH2A and 31% (5/16) of ADGRV1 patients with no statistical difference in frequency between the two groups (Fisher’s Exact Test, p = 0.4). Table 3 shows the number of patients with different degrees of CME. Considering all of the patients, CME was significantly more frequent in patients with a hyperautofluorescent ring pattern (34%, 30/87) than those with patch or atrophy (12%, 5/42) (Fisher’s Exact Test, p < 0.01). An analysis of foveal thickness (FT) with respect to age and genotype was performed on the largest group of patients imaged with the same OCT machine (Spectralis Heidelberg) to avoid measurement differences [13]. The patients with CME were excluded, leaving 83 USH2A and 10 ADGRV1 patients. Multiple regression analysis showed a significant correlation between FT and age (p < 0.001), with FT decreasing with age, but no correlation with gene (p = 0.6) (Figure 5).
Table 3.
Distribution of different degrees of CME in USH2A and ADGRV1 patient groups.
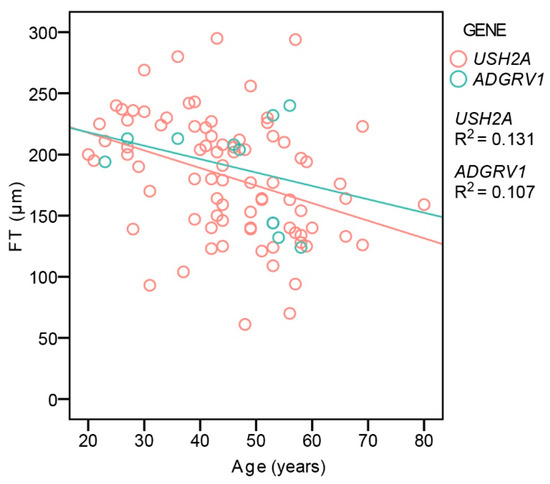
Figure 5.
Central foveal thickness in relation to age and genotype. Red color represents USH2A patients and green color represents ADGRV1 patients.
2.6. Correlation between Gender and Phenotype
The analysis of the same parameters was repeated within each genotypic group to determine whether there was any gender-based difference. There was no significant difference in disease onset, visual acuity, hyperautofluorescent ring diameter, or CME occurrence between male and female patients within each patient group.
2.7. Correlation between of Ethnicity and Phenotype
The study included patient cohorts from Germany, Italy, Spain, and Slovenia. Patient nationality was used as a proxy for ethnicity, and the Kruskal–Wallis test was used to determine whether it had any effect on the measured parameters. There was no significant difference in the median age at onset (17, 17, 19, and 17 years, respectively) and median ring size (4°, 4°, 3°, and 2°, respectively), while there was a significant difference in the median visual acuity (0.6, 0.2, 0.4, and 0.5 logMAR, respectively) and CME occurrence (27%, 10%, 43%, and 31%, respectively) (p < 0.05 for both parameters). There was a significant difference in the median age of the patients from different centers (44, 38, 42, and 53 years, respectively; p < 0.01).
3. Discussion
The present study includes the largest cohort of ADGRV1 patients published to date (n = 18) and provides a detailed overview of the functional and structural parameters of the associated retinal disease. Importantly, data on visual acuity decline are useful in counseling patients. Furthermore, a comparison was made with a large USH2A cohort of patients with USH2, which revealed a striking similarity between the two phenotypes.
3.1. Genotype-Phenotype Correlations
ADGRV1 variants are a rare cause of Usher syndrome, which is reflected by the scarcity of related phenotypic reports. The first paper on ADGRV1 USH2 patients included 13 females from five families, with minimal phenotypic data [14]. The authors posed the question of possible sex predilection/bias towards females, but this was disputed by reports of patients of both sexes identified in four families [15,16,17], while one proposed that males may be more affected [16]. In the present study, the gender distribution was equal (nine male and nine female patients), and there was no significant difference in disease severity between the two genders. Therefore, the previous observations were likely due to chance and the small number of patients. There was, however, a considerable variability in the phenotype regardless of gender, which can be observed, for example, in the range of disease onset (Figure 1) and the diameter of the hyperautofluorescent ring (Figure 4B), suggesting that factors other than gender influence the disease expression.
The majority of the studies involving Usher syndrome patients with ADGRV1 variants included ≤ 5 cases with no or minimal phenotypic data [14,15,16,17,18,19,20,21,22,23,24]. An exception is a report by Schwartz et al. [20], which investigated kinetic and chromatic perimetry, electrophysiology, and OCT parameters and provided a comparison with USH2A. However, the study was also hindered by the small number of ADGRV1 patients (n = 3).
The present study thus provides the first comprehensive overview of ADGRV1 retinopathy. The studied cohort of 18 patients exhibited retinitis pigmentosa, with the median onset at 30 years and a 50% chance of reaching legal blindness (VA ≤ 0.1) at 64 years of age. Similarly, the median age when 50% of the patients progressed to advanced macular degeneration (patch or atrophy autofluorescent patterns) was 58 years, which underscores the association between structure and function.
A great similarity was observed in the comparison group consisting of 204 USH2A patients for all of the measured parameters. It is important to note, however, that only syndromic USH2A patients were included in this comparison as hearing loss was the inclusion criterion for this multicentric study. It has been shown that non-syndromic USH2A patients exhibit milder retinal disease, with delayed visual acuity loss and better-preserved amplitudes on electroretinography [6]. These differences in phenotype are probably related to the effects of different pathogenic variants, as null alleles have been associated with more severe hearing loss and retinal degeneration and are more often observed in syndromic cases [6,7,25,26]. However, a comparison of the structural measurements (ellipsoid zone line length) on retinal imaging between syndromic and non-syndromic USH2A patients showed no significant differences [8], and a large variability may be found even between patients with two null alleles [11].
The correlation between ethnicity and phenotype was examined in a limited fashion by using patient nationality as a proxy for ethnicity. There was no statistical difference in the median age at onset or median ring diameter, the two parameters that probably best describe the RP phenotype. There was a difference in CME occurrence; however, this could be affected by treatment (e.g., systemic acetazolamide, local acetazolamide, or observation). There was also a difference in the median visual acuity; however, this could be affected by opacities in the optic media (cataract or posterior capsular opacification), CME, or different methods of visual acuity measurement (Snellen or ETDRS). Therefore, the current study was not able to show a definitive correlation between ethnicity and phenotype.
3.2. Function of ADGRV1 and USH2A Proteins
Adhesion G protein-coupled receptor V1 (ADGRV1) and usherin (USH2A) are large proteins located at the periciliary region between the inner and outer segments of the photoreceptors and are thought to be important in stabilizing the connecting cilium. The proteins are anchored by a transmembrane domain and have long ectodomains that extend into the gap between the membranes of the connecting cilium and the apical inner segment, and they belong to the same protein complex [27,28,29,30,31,32].
The autosomal recessive inheritance of USH2A and ADGRV1 suggests a loss of function mechanism. No gain of function variant has been reported in Usher syndrome genes, and no case of dominance has been observed, i.e., the parents of affected children are healthy. In support of this, most of the patients in this study harbored LOF variants. These included the frame-shifting variants as they were all predicted to undergo NMD (all were located >50 nucleotides from the last exon-exon junction), and thus expected to result in the absence of protein instead of an aberrant protein [12].
The indistinguishable clinical presentations of the two patient groups suggests that the loss of function of either of the proteins results in a similar functional defect in the retina. Considering the proteins’ shared location, there is a possibility that the proteins physically interact with each other and that the loss of function of either protein could result in a dysfunction of both. The possible interaction between USH2A and ADGRV1 has been schematically proposed by Maerker et al. [29] but has not yet been proven. It is also possible that the loss of either protein disrupts the interactions between other USH proteins in a similar manner. The existence of a quaternary USH2 complex has been proposed, where the third USH2 protein, whirlin (WHRN), and PDZD7, an USH2 modifier, are required for a complex formation with USH2A and ADGRV1. In this model, WHRN prefers to bind to USH2A, whereas PDZD7 prefers to bind to ADGRV1, while interaction between WHRN and PDZD7 is the bridge between USH2A and ADGRV1 [33].
Although both USH2A and ADGRV1 are expressed in rods and cones, it is still not clear why their dysfunction primarily affects rods [29]. Considering that cones are shorter than rods, it is possible that the stabilization of the connecting cilium is not as important for their structural integrity.
3.3. Study Limitations
A limitation of the study is that only USH2A patients with hearing loss were included. Therefore, the findings relate only to those patients. This means that it is possible that there would be differences between ADGRV1 and USH2A patients with non-syndromic RP. This does not exclude the differences between the RP of USH2 patients caused by USH2A or ADGRVL1 defects and non-syndromic RP caused by USH2A defects. Another limitation is that only the retinal phenotype was studied. It would be interesting to expand the study to include hearing and vestibular impairment; however, this was beyond the scope of this study.
4. Materials and Methods
4.1. Patients
The study included 222 USH2 patients who were involved in multicentric study Treatrush [34]. The patients were from Germany (n = 72), France, (n = 69), Slovenia (n = 43), and Italy (n = 38). There were 204 USH2A patients (111 male; median age 43 years, range 1–80 years) and 18 ADGRV1 patients (9 male; median age 52 years, range 23–68 years) with no statistical difference in the median age (Mann–Whitney U test, p = 0.11). Phenotypes of two patients (TR_EK-047 and TR_EK-048) have been described previously [16].
4.2. Genetic Analysis
Genetic analysis was performed by sequential use of three different techniques to analyze selected genomic regions: targeted exome sequencing, comparative genome hybridization, and quantitative exon amplification. The methods and detected variants were described in detail previously by Bonnet et al., Supplement 2 [34].
4.3. Clinical Assessment
Disease onset was defined as the age when patients or patients’ parents first reported night vision problems, and was determined from the hospital records (available for 148 patients). Visual function was measured with either Snellen (n = 116) or ETDRS (n = 83) and converted into logMAR equivalent. Counting finger and hand motion were quantified as 2.0 and 2.3 logMAR [35] and light perception as 2.8 logMAR [36]. Kaplan–Meier survival analysis was performed using best corrected visual acuity (BCVA) on the better eye, equal to or below 0.1 (≤6/60 or ≥1.0 logMAR; legal blindness) or below 0.3 (>6/20 or >0.52 logMAR; low vision), as a threshold. The better eye was used for the survival analysis as it was thought to best reflect the effect of the central visual loss on the patient’s daily life.
Fundus autofluorescence imaging (FAF) was performed in 161 patients using Spectralis imaging system (Heidelberg Engineering, Heidelberg, Germany). FAF pattern was classified as hyperautofluorescent ring, hyperautofluorescent patch, or atrophy, as described previously [9]. The inner horizontal diameter of the hyperautofluorescent ring was measured on FAF using ImageJ (imagej.nih.gov), where the 30° width of the squared FAF image was used as a reference (Figure 4D). The ring diameter was considered to be zero in cases of central patch or atrophy (examples in Figure 3 and Figure S1), as these patterns were previously shown to represent advanced retinal disease following the ring disappearance [9]. Optical coherence tomography (OCT) imaging was performed through the fovea in 151 patients using either Spectralis OCT (Heidelberg Engineering, Heidelberg, Germany; n = 122), Cirrus OCT (Carl Zeiss Meditec, Dublin, CA, USA; n = 23), or Stratus OCT (Carl Zeiss Meditec, Dublin, CA, USA; n = 6). The retinal thickness in the center of the fovea (FT) was measured on the OCT scan using ImageJ, where the 200 μm scale on the OCT image was used for reference. The presence or absence of cystoid macular edema (CME) was determined qualitatively. The right eye was used for analysis, except in seven cases, where poor image quality precluded the measurements.
4.4. Statistical Analysis
Statistical analysis was performed using SPSS software v. 22 (IBM SPSS Statistics; IBM Corporation, Chicago, IL, USA). The median values of the ages and ages at onset were compared using Mann–Whitney U test. Associations of different parameters with gene and age were tested using multiple linear regression (continuous variables) or multiple logistic regression (categorical variables). Fisher’s Exact test was used to compare the frequency of CME between the two patient groups. Kaplan–Meier survival curve was used to determine the mean and/or median age when 50% of patients reached low vision, legal blindness, and advanced retinal degeneration (FAF patterns of hyperautofluorescent patch or atrophy). Log-rank test was used to test for statistical differences between the survival curves of the two patient groups.
5. Conclusions
ADGRV1 and USH2A retinopathy share all the major characteristics of functional and structural impairment, suggesting that the loss of function of the corresponding proteins produces similar effects in the retina. The results are important for counseling ADGRV1 patients, who represent a minor patient subgroup.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms221910352/s1.
Author Contributions
Conceptualization, A.F.; methodology, A.F., C.B., A.K., S.M.-S., D.Z., K.S., F.T., F.S., J.-A.S., I.A., E.Z., M.H. and C.P.; validation, A.F., C.B., A.K., S.M.-S., D.Z., K.S., F.T., F.S., J.-A.S., I.A., E.Z., M.H. and C.P.; formal analysis, A.F., C.B., A.K., S.M.-S., D.Z., K.S., F.T., F.S., J.-A.S., I.A., E.Z., M.H. and C.P.; investigation, A.F., C.B., A.K., S.M.-S., D.Z., K.S., F.T., F.S., J.-A.S., I.A., E.Z., M.H. and C.P.; resources, A.F., C.B., A.K., S.M.-S., D.Z., K.S., F.T., F.S., J.-A.S., I.A., E.Z., M.H. and C.P.; data curation, A.F., C.B., A.K., S.M.-S., D.Z., K.S., F.T., F.S., J.-A.S., I.A., E.Z., M.H. and C.P.; writing—original draft preparation, A.F.; writing—review and editing, A.F., C.B., A.K., S.M.-S., D.Z., K.S., F.T., F.S., J.-A.S., I.A., E.Z., M.H. and C.P.; visualization, A.F.; supervision, M.H. and C.P.; funding acquisition, C.P., A.F., E.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the European Union Seventh Framework Programme under the grant agreement HEALTH-F2-2010-242013 (TREATRUSH), the Slovenian Research Agency (grant ARRS J3-1750), and the Tistou and Charlotte Kerstan Foundation, Tübingen.
Institutional Review Board Statement
The study was carried out following the ethical principles for medical research involving human subjects defined by the WMA Declaration of Helsinki. It was approved by the local ethics committees as stated in [34] (Medical Ethics Committee of the Republic of Slovenia, 55/01/11; 139/01/11).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original data are available upon reasonable request to the corresponding author.
Acknowledgments
We are grateful to the European Reference Network dedicated to Rare Eye Diseases (ERN-EYE) for the support in multicentric research.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Cohen, M.; Bitner-Glindzicz, M.; Luxon, L. The changing face of Usher syndrome: Clinical implications. Int. J. Audiol. 2007, 46, 82–93. [Google Scholar] [CrossRef]
- Jouret, G.; Poirsier, C.; Spodenkiewicz, M.; Jaquin, C.; Gouy, E.; Arndt, C.; Labrousse, M.; Gaillard, D.; Doco-Fenzy, M.; Lebre, A.S. Genetics of Usher Syndrome: New Insights From a Meta-analysis. Otol. Neurotol. 2019, 40, 121–129. [Google Scholar] [CrossRef]
- McGee, T.L.; Seyedahmadi, B.J.; Sweeney, M.O.; Dryja, T.P.; Berson, E.L. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J. Med. Genet. 2010, 47, 499–506. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; McGee, T.L.; Dryja, T.P.; Berson, E.L. Disease course in patients with autosomal recessive retinitis pigmentosa due to the USH2A gene. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5532–5539. [Google Scholar] [CrossRef] [Green Version]
- Pierrache, L.H.; Hartel, B.P.; van Wijk, E.; Meester-Smoor, M.A.; Cremers, F.P.; de Baere, E.; de Zaeytijd, J.; van Schooneveld, M.J.; Cremers, C.W.; Dagnelie, G.; et al. Visual Prognosis in USH2A-Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type IIa Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2016, 123, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Cabral, T.; Schuerch, K.; Duong, J.; Lee, W.; Boudreault, K.; Xu, Y.; Justus, S.; Sparrow, J.R.; Mahajan, V.B.; et al. Electroretinography Reveals Difference in Cone Function between Syndromic and Nonsyndromic USH2A Patients. Sci. Rep. 2017, 7, 11170. [Google Scholar] [CrossRef] [PubMed]
- Fakin, A.; Jarc-Vidmar, M.; Glavač, D.; Bonnet, C.; Petit, C.; Hawlina, M. Fundus autofluorescence and optical coherence tomography in relation to visual function in Usher syndrome type 1 and 2. Vis. Res. 2012, 75, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Fakin, A.; Šuštar, M.; Brecelj, J.; Bonnet, C.; Petit, C.; Zupan, A.; Glavač, D.; Jarc-Vidmar, M.; Battelino, S.; Hawlina, M. Double Hyperautofluorescent Rings in Patients with USH2A-Retinopathy. Genes 2019, 10, 956. [Google Scholar] [CrossRef] [Green Version]
- Zupan, A.; Fakin, A.; Battelino, S.; Jarc-Vidmar, M.; Hawlina, M.; Bonnet, C.; Petit, C.; Glavač, D. Clinical and Haplotypic Variability of Slovenian. Genes 2019, 10, 1015. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Bentaleb-Machkour, Z.; Jouffroy, E.; Rabilloud, M.; Grange, J.D.; Kodjikian, L. Comparison of central macular thickness measured by three OCT models and study of interoperator variability. Sci. World J. 2012, 2012, 842795. [Google Scholar] [CrossRef] [PubMed]
- Weston, M.D.; Luijendijk, M.W.; Humphrey, K.D.; Möller, C.; Kimberling, W.J. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hmani-Aifa, M.; Benzina, Z.; Zulfiqar, F.; Dhouib, H.; Shahzadi, A.; Ghorbel, A.; Rebaï, A.; Söderkvist, P.; Riazuddin, S.; Kimberling, W.J.; et al. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur. J. Hum. Genet. 2009, 17, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebermann, I.; Wiesen, M.H.; Zrenner, E.; Lopez, I.; Pigeon, R.; Kohl, S.; Löwenheim, H.; Koenekoop, R.K.; Bolz, H.J. GPR98 mutations cause Usher syndrome type 2 in males. J. Med. Genet. 2009, 46, 277–280. [Google Scholar] [CrossRef]
- Hilgert, N.; Kahrizi, K.; Dieltjens, N.; Bazazzadegan, N.; Najmabadi, H.; Smith, R.J.; Van Camp, G. A large deletion in GPR98 causes type IIC Usher syndrome in male and female members of an Iranian family. J. Med. Genet. 2009, 46, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yang, L.; Cheng, J.; Imani, S.; Fu, S.; Lv, H.; Li, Y.; Chen, R.; Leung, E.L.; Fu, J. A novel homozygous variant of GPR98 causes usher syndrome type IIC in a consanguineous Chinese family by next generation sequencing. BMC Med. Genet. 2018, 19, 99. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wang, J.; Liu, S.; Liu, M.; Jiang, F. Identification of two novel compound heterozygous mutations of ADGRV1 in a Chinese family with Usher syndrome type IIC. Ophthalmic Genet. 2018, 39, 517–521. [Google Scholar] [CrossRef]
- Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V.; Windsor, E.A.; Sumaroka, A.; Roman, A.J.; Rane, T.; Smilko, E.E.; Bennett, J.; Stone, E.M.; et al. Disease expression in Usher syndrome caused by VLGR1 gene mutation (USH2C) and comparison with USH2A phenotype. Investig. Ophthalmol. Vis. Sci. 2005, 46, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Eandi, C.M.; Dallorto, L.; Spinetta, R.; Micieli, M.P.; Vanzetti, M.; Mariottini, A.; Passerini, I.; Torricelli, F.; Alovisi, C.; Marchese, C. Targeted next generation sequencing in Italian patients with Usher syndrome: Phenotype-genotype correlations. Sci. Rep. 2017, 7, 15681. [Google Scholar] [CrossRef] [PubMed]
- Besnard, T.; Vaché, C.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Odent, S.; Blanchet, P.; Calvas, P.; Hamel, C.; et al. Non-USH2A mutations in USH2 patients. Hum. Mutat. 2012, 33, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Kahrizi, K.; Bazazzadegan, N.; Jamali, L.; Nikzat, N.; Kashef, A.; Najmabadi, H. A novel mutation of the USH2C (GPR98) gene in an Iranian family with Usher syndrome type II. J. Genet. 2014, 93, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Bousfiha, A.; Bakhchane, A.; Charoute, H.; Detsouli, M.; Rouba, H.; Charif, M.; Lenaers, G.; Barakat, A. Novel compound heterozygous mutations in the GPR98 (USH2C) gene identified by whole exome sequencing in a Moroccan deaf family. Mol. Biol. Rep. 2017, 44, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Abadie, C.; Blanchet, C.; Baux, D.; Larrieu, L.; Besnard, T.; Ravel, P.; Biboulet, R.; Hamel, C.; Malcolm, S.; Mondain, M.; et al. Audiological findings in 100 USH2 patients. Clin. Genet. 2012, 82, 433–438. [Google Scholar] [CrossRef]
- Hartel, B.P.; Löfgren, M.; Huygen, P.L.; Guchelaar, I.; Lo-A-Njoe Kort, N.; Sadeghi, A.M.; van Wijk, E.; Tranebjærg, L.; Kremer, H.; Kimberling, W.J.; et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear. Res. 2016, 339, 60–68. [Google Scholar] [CrossRef]
- Adato, A.; Lefèvre, G.; Delprat, B.; Michel, V.; Michalski, N.; Chardenoux, S.; Weil, D.; El-Amraoui, A.; Petit, C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 2005, 14, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maerker, T.; van Wijk, E.; Overlack, N.; Kersten, F.F.; McGee, J.; Goldmann, T.; Sehn, E.; Roepman, R.; Walsh, E.J.; Kremer, H.; et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet. 2008, 17, 71–86. [Google Scholar] [CrossRef]
- Overlack, N.; Kilic, D.; Bauss, K.; Märker, T.; Kremer, H.; van Wijk, E.; Wolfrum, U. Direct interaction of the Usher syndrome 1G protein SANS and myomegalin in the retina. Biochim. Biophys. Acta 2011, 1813, 1883–1892. [Google Scholar] [CrossRef] [Green Version]
- Dona, M.; Slijkerman, R.; Lerner, K.; Broekman, S.; Wegner, J.; Howat, T.; Peters, T.; Hetterschijt, L.; Boon, N.; de Vrieze, E.; et al. Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. Eye Res. 2018, 173, 148–159. [Google Scholar] [CrossRef]
- Michalski, N.; Michel, V.; Bahloul, A.; Lefèvre, G.; Barral, J.; Yagi, H.; Chardenoux, S.; Weil, D.; Martin, P.; Hardelin, J.P.; et al. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci. 2007, 27, 6478–6488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zou, J.; Shen, Z.; Zhang, W.; Yang, J. Whirlin and PDZ domain-containing 7 (PDZD7) proteins are both required to form the quaternary protein complex associated with Usher syndrome type 2. J. Biol. Chem. 2014, 289, 36070–36088. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefèvre, G.M.; Hardelin, J.P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [Green Version]
- Lange, C.; Feltgen, N.; Junker, B.; Schulze-Bonsel, K.; Bach, M. Resolving the clinical acuity categories “hand motion” and “counting fingers” using the Freiburg Visual Acuity Test (FrACT). Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 137–142. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Anderson, R.J.; Tozatti, M.S.; Heckenlively, J.R.; Weleber, R.G.; Edwards, A.O.; Brown, J. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology 1999, 106, 1780–1785. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).