
Tumor Necrosis Factor’s Pathway in Crohn’s Disease: Potential for Intervention


Abstract
:1. Introduction
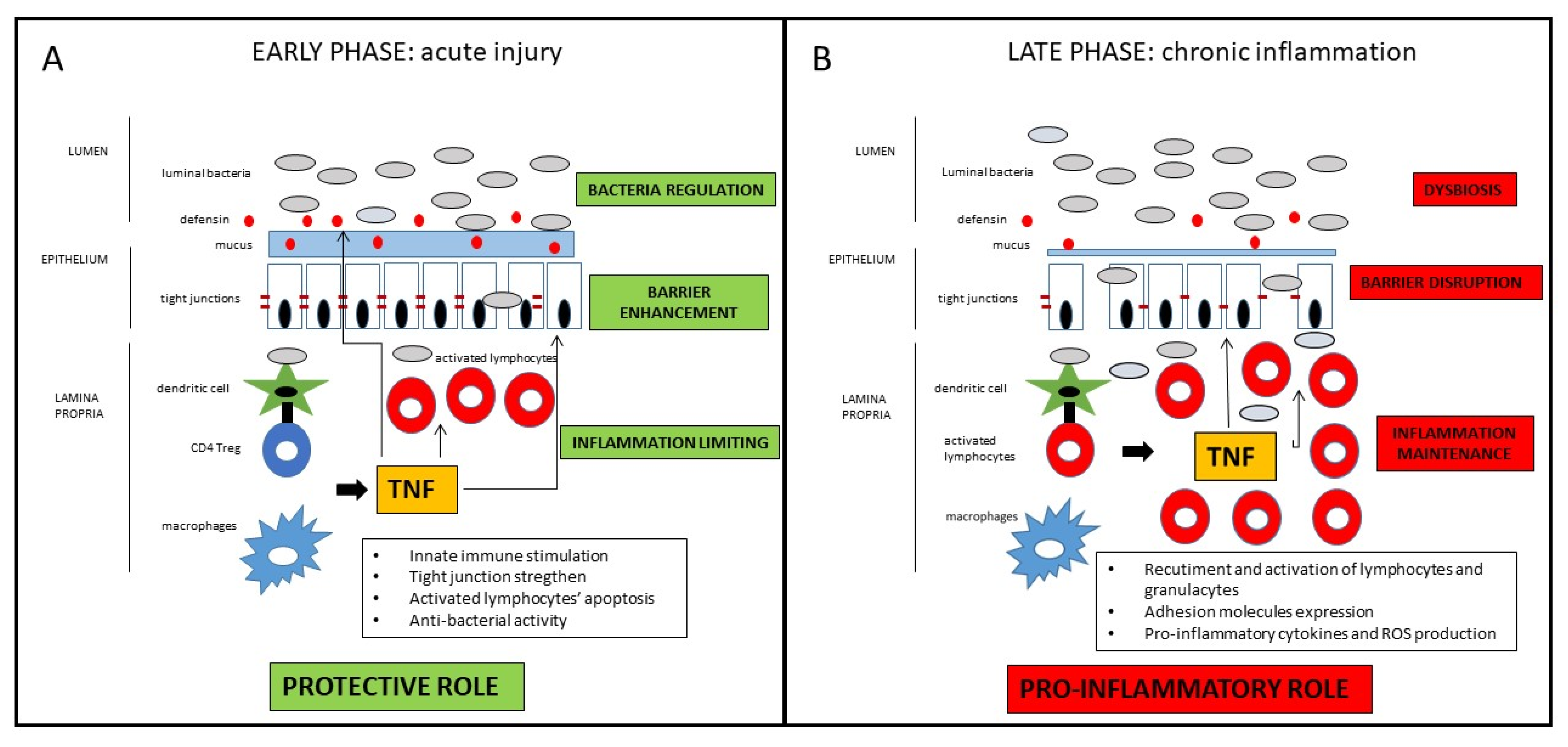
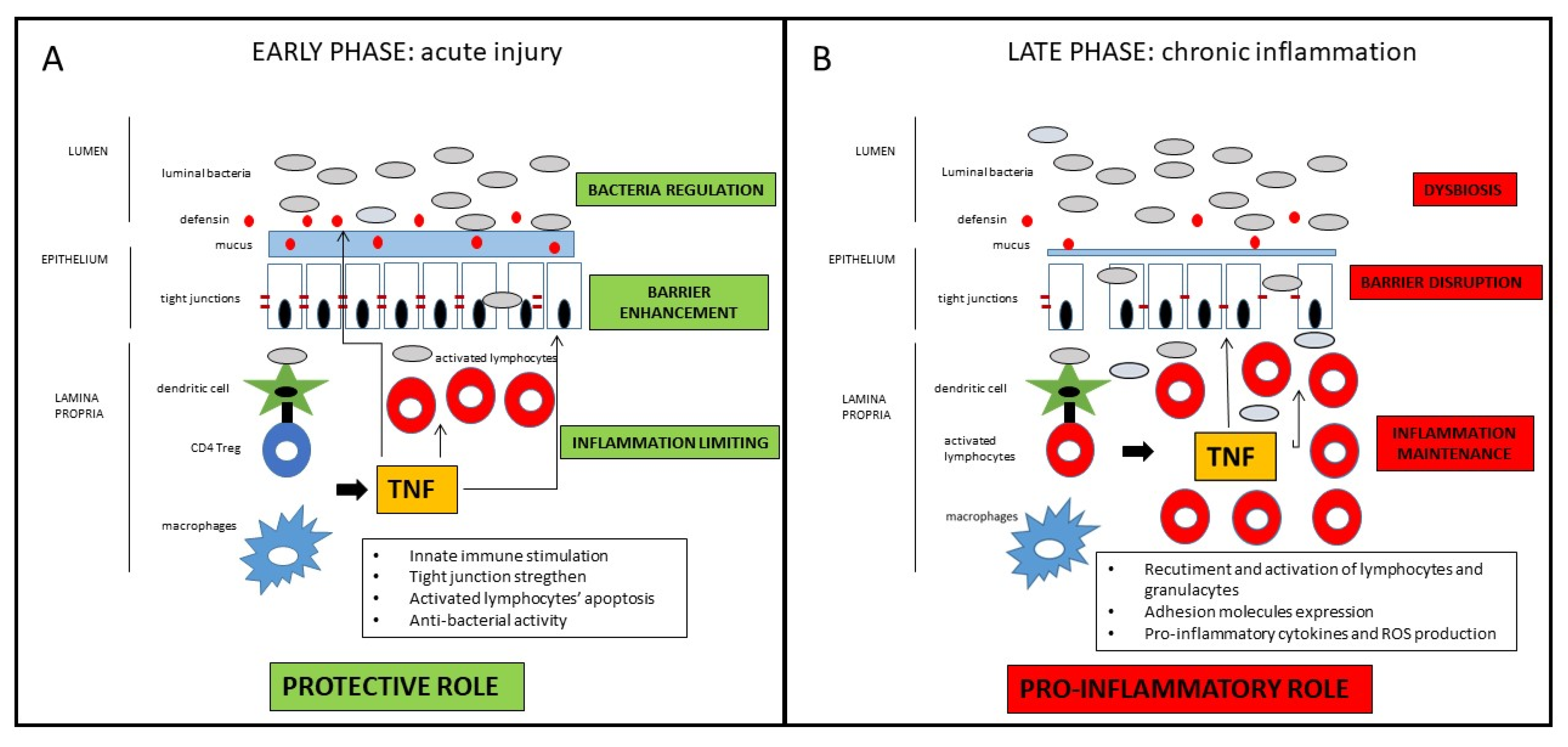
2. TNF in The Pathogenesis of Crohn’s Disease: A Dichotomous Role
2.1. The Classical Pro-Inflammatory Effect of TNF
2.2. Potential Protective and Anti-Inflammatory Effect of TNF
2.3. Potential Clinical Implications
3. The TNF Block as a Therapeutic Target in Crohn’s Disease: Current Drugs Available
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, M.C.; Pagnini, C.; Graziani, M.G. Corticosteroids in Inflammatory Bowel Disease patients: A practical guide for physicians. Curr. Clin. Pharmacol. 2020, 16, 210–218. [Google Scholar] [CrossRef]
- Rezaie, A.; Kuenzig, M.E.; Benchimol, E.I.; Griffiths, A.M.; Otley, A.R.; Steinhart, A.H.; Kaplan, G.G.; Seow, C.H. Budesonide for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2015, 6, CD000296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonovas, S.; Nikolopoulos, G.K.; Lytras, T.; Fiorino, G.; Peyrin-Biroulet, L.; Danese, S. Comparative safety of systemic and low-bioavailability steroids in inflammatory bowel disease: Systematic review and network meta-analysis. Br. J. Clin. Pharmacol. 2018, 84, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Hanauer, S.B.; Sandborn, W.J.; Lichtenstein, G.R. Evolving Considerations for Thiopurine Therapy for Inflammatory Bowel Diseases—A Clinical Practice Update: Commentary. Gastroenterology 2019, 156, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachmann, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohn’s Colitis 2020, 14, 4–22. [Google Scholar] [CrossRef]
- Lichtenstein, L.; Avni-Biron, I.; Ben-Bassat, O. Probiotics and prebiotics in Crohn’s disease therapies. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 81–88. [Google Scholar] [CrossRef]
- Bemelman, W.A.; Warusavitarne, J.; Sampietro, G.M.; Serclova, Z.; Zmora, O.; Luglio, G.; de Buck van Overstraeten, A.; Burke, J.P.; Buskens, C.J.; Colombo, F.; et al. ECCO-ESCP Consensus on Surgery for Crohn’s Disease. J. Crohn’s Colitis 2018, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Travis, S.P. Mucosal healing in inflammatory bowel diseases: A systematic review. Gut 2012, 61, 1619–1635. [Google Scholar] [CrossRef]
- Zallot, C.; Peyrin-Biroulet, L. Deep remission in inflammatory bowel disease: Looking beyond symptoms. Curr. Gastroenterol. Rep. 2013, 15, 315. [Google Scholar] [CrossRef]
- Williet, N.; Sandborn, W.J.; Peyrin-Biroulet, L. Patient-reported outcomes as primary end points in clinical trials of inflammatory bowel disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2014, 12, 1246–1256.e6. [Google Scholar] [CrossRef]
- Pagnini, C.; Pizarro, T.T.; Cominelli, F. Novel Pharmacological Therapy in Inflammatory Bowel Diseases: Beyond Anti-Tumor Necrosis Factor. Front. Pharmacol. 2019, 10, 671. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, M.; Ahmed, S.; Dryden, G. Immunopathophysiology of inflammatory bowel disease: How genetics link barrier dysfunction and innate immunity to inflammation. Innate Immun. 2017, 23, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Stappenbeck, T.S. Genetics and Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Pathol. 2016, 11, 127–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, T.T.; Cominelli, F. Cytokine therapy for Crohn’s disease: Advances in translational research. Annu. Rev. Med. 2007, 58, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Weigmann, B.; Finotto, S.; Glickman, J.; Nieuwenhuis, E.; Iijima, H.; Mizoguchi, A.; Mizoguchi, E.; Mudter, J.; Galle, P.R.; et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J. Exp. Med. 2002, 195, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Kugathasan, S.; Saubermann, L.J.; Smith, L.; Kou, D.; Itoh, J.; Binion, D.G.; Levine, A.D.; Blumberg, R.S.; Fiocchi, C. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut 2007, 56, 1696–1705. [Google Scholar] [CrossRef]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef]
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Ueno, A.; Jeffery, L.; Kobayashi, T.; Hibi, T.; Ghosh, S.; Jijon, H. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 2018, 87, 38–49. [Google Scholar] [CrossRef]
- Muniz, L.R.; Knosp, C.; Yeretssian, G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front. Immunol. 2012, 3, 310. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M.; Madsen, K.; Spiller, R.; Greenwood-Van Meerveld, B.; Verne, G.N. Intestinal barrier function in health and gastrointestinal disease. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2012, 24, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.S.; Reuter, B.K.; Scott, K.G.; Morris, M.A.; Wang, X.M.; Hancock, L.N.; Burcin, T.L.; Cohn, S.M.; Ernst, P.B.; Cominelli, F.; et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J. Exp. Med. 2006, 203, 541–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollander, D.; Vadheim, C.M.; Brettholz, E.; Petersen, G.M.; Delahunty, T.; Rotter, J.I. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann. Intern. Med. 1986, 105, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.H.; Hasnain, S.Z.; Florin, T.H.; McGuckin, M.A. Mucins in inflammatory bowel diseases and colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Salzman, N.H.; Porter, E.; Nuding, S.; Weichenthal, M.; Petras, R.E.; Shen, B.; Schaeffeler, E.; Schwab, M.; Linzmeier, R.; et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 18129–18134. [Google Scholar] [CrossRef] [Green Version]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schaffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Rescigno, M. The microbiota revolution: Excitement and caution. Eur. J. Immunol. 2017, 47, 1406–1413. [Google Scholar] [CrossRef] [Green Version]
- Taurog, J.D.; Richardson, J.A.; Croft, J.T.; Simmons, W.A.; Zhou, M.; Fernandez-Sueiro, J.L.; Balish, E.; Hammer, R.E. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 1994, 180, 2359–2364. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Goboes, K.; Peeters, M.; Hiele, M.; Penninckx, F.; Aerts, R.; Kerremans, R.; Vantrappen, G. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet 1991, 338, 771–774. [Google Scholar] [CrossRef]
- Pagnini, C.; Delle Fave, G.; Bamias, G. Probiotics in inflammatory bowel disease: Pathophysiological background and clinical applications. World J. Immunol. 2013, 3, 31–43. [Google Scholar] [CrossRef]
- Caruso, R.; Lo, B.C.; Nunez, G. Host-microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andoh, A.; Imaeda, H.; Aomatsu, T.; Inatomi, O.; Bamba, S.; Sasaki, M.; Saito, Y.; Tsujikawa, T.; Fujiyama, Y. Comparison of the fecal microbiota profiles between ulcerative colitis and Crohn’s disease using terminal restriction fragment length polymorphism analysis. J. Gastroenterol. 2011, 46, 479–486. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [Green Version]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.M.; MacBeath, G. Cross-talk between receptor tyrosine kinase and tumor necrosis factor-alpha signaling networks regulates apoptosis but not proliferation. Mol. Cell. Proteom. MCP 2012, 11, M111.013292. [Google Scholar] [CrossRef] [Green Version]
- Oceandy, D.; Amanda, B.; Ashari, F.Y.; Faizah, Z.; Azis, M.A.; Stafford, N. The Cross-Talk Between the TNF-alpha and RASSF-Hippo Signalling Pathways. Int. J. Mol. Sci. 2019, 20, 2346. [Google Scholar] [CrossRef] [Green Version]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Kohr, W.J. Human tumor necrosis factor. Methods Enzymol. 1985, 116, 448–456. [Google Scholar] [CrossRef] [PubMed]
- D’Haens, G.R.; van Deventer, S. 25 years of anti-TNF treatment for inflammatory bowel disease: Lessons from the past and a look to the future. Gut 2021, 70, 1396–1405. [Google Scholar] [CrossRef]
- Papadakis, K.A.; Targan, S.R. Tumor necrosis factor: Biology and therapeutic inhibitors. Gastroenterology 2000, 119, 1148–1157. [Google Scholar] [CrossRef]
- Van Assche, G.; Vermeire, S.; Rutgeerts, P. The potential for disease modification in Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 79–85. [Google Scholar] [CrossRef]
- Kojouharoff, G.; Hans, W.; Obermeier, F.; Mannel, D.N.; Andus, T.; Scholmerich, J.; Gross, V.; Falk, W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin. Exp. Immunol. 1997, 107, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Nakagawa, S.; Yamaguchi, T.; Yoshida, N.; Minami, M.; Kita, M.; Imanishi, J.; et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 2003, 18, 560–569. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Hachiya, Y.; Kawada, M.; Nagatani, K.; Ogawa, A.; Sugimoto, K.; Mizoguchi, A.; Podolsky, D.K. TNF receptor type I-dependent activation of innate responses to reduce intestinal damage-associated mortality. Gastroenterology 2008, 134, 470–480. [Google Scholar] [CrossRef]
- Ebach, D.R.; Newberry, R.; Stenson, W.F. Differential role of tumor necrosis factor receptors in TNBS colitis. Inflamm. Bowel Dis. 2005, 11, 533–540. [Google Scholar] [CrossRef]
- Pagnini, C.; Saeed, R.; Bamias, G.; Arseneau, K.O.; Pizarro, T.T.; Cominelli, F. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 454–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corridoni, D.; Pastorelli, L.; Mattioli, B.; Locovei, S.; Ishikawa, D.; Arseneau, K.O.; Chieppa, M.; Cominelli, F.; Pizarro, T.T. Probiotic bacteria regulate intestinal epithelial permeability in experimental ileitis by a TNF-dependent mechanism. PLoS ONE 2012, 7, e42067. [Google Scholar] [CrossRef] [Green Version]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Atreya, R.; Zimmer, M.; Bartsch, B.; Waldner, M.J.; Atreya, I.; Neumann, H.; Hildner, K.; Hoffman, A.; Kiesslich, R.; Rink, A.D.; et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14(+) macrophages. Gastroenterology 2011, 141, 2026–2038. [Google Scholar] [CrossRef] [PubMed]
- Noti, M.; Corazza, N.; Mueller, C.; Berger, B.; Brunner, T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J. Exp. Med. 2010, 207, 1057–1066. [Google Scholar] [CrossRef]
- Urbano, P.C.M.; He, X.; van Heeswijk, B.; Filho, O.P.S.; Tijssen, H.; Smeets, R.L.; Joosten, I.; Koenen, H. TNFalpha-Signaling Modulates the Kinase Activity of Human Effector Treg and Regulates IL-17A Expression. Front. Immunol. 2019, 10, 3047. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, J.; Clanchy, F.I.; Taher, T.E.; Mangat, P.; Jawad, A.S.; Williams, R.O.; Mageed, R.A. TNFalpha in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine 2018, 101, 4–13. [Google Scholar] [CrossRef]
- Tseng, W.-Y.; Huang, Y.-S.; Lin, H.-H.; Luo, S.-F.; McCann, F.; McNamee, K.; Clanchy, F.; Williams, R. TNFR signalling and its clinical implications. Cytokine 2018, 101, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Corridoni, D.; Pizarro, T.T.; Cominelli, F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012, 59, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curciarello, R.; Docena, G.H.; MacDonald, T.T. The Role of Cytokines in the Fibrotic Responses in Crohn’s Disease. Front. Med. 2017, 4, 126. [Google Scholar] [CrossRef] [Green Version]
- Adegbola, S.O.; Sahnan, K.; Warusavitarne, J.; Hart, A.; Tozer, P. Anti-TNF Therapy in Crohn’s Disease. Int. J. Mol. Sci. 2018, 19, 2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targan, S.R.; Hanauer, S.B.; van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N. Engl. J. Med. 1997, 337, 1029–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549. [Google Scholar] [CrossRef]
- Sands, B.E.; Anderson, F.H.; Bernstein, C.N.; Chey, W.Y.; Feagan, B.G.; Fedorak, R.N.; Kamm, M.A.; Korzenik, J.R.; Lashner, B.A.; Onken, J.E.; et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N. Engl. J. Med. 2004, 350, 876–885. [Google Scholar] [CrossRef]
- Colombel, J.F.; Sandborn, W.J.; Reinisch, W.; Mantzaris, G.J.; Kornbluth, A.; Rachmilewitz, D.; Lichtiger, S.; D’Haens, G.; Diamond, R.H.; Broussard, D.L.; et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N. Engl. J. Med. 2010, 362, 1383–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanauer, S.B.; Sandborn, W.J.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.; Panaccione, R.; Wolf, D.; Pollack, P. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: The CLASSIC-I trial. Gastroenterology 2006, 130, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandborn, W.J.; Hanauer, S.B.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.G.; Panaccione, R.; Wolf, D.; Kent, J.D.; Bittle, B.; et al. Adalimumab for maintenance treatment of Crohn’s disease: Results of the CLASSIC II trial. Gut 2007, 56, 1232–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombel, J.F.; Sandborn, W.J.; Rutgeerts, P.; Enns, R.; Hanauer, S.B.; Panaccione, R.; Schreiber, S.; Byczkowski, D.; Li, J.; Kent, J.D.; et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: The CHARM trial. Gastroenterology 2007, 132, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Rutgeerts, P.; Enns, R.; Hanauer, S.B.; Colombel, J.F.; Panaccione, R.; D’Haens, G.; Li, J.; Rosenfeld, M.R.; Kent, J.D.; et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: A randomized trial. Ann. Intern. Med. 2007, 146, 829–838. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Van Assche, G.; Sandborn, W.J.; Wolf, D.C.; Geboes, K.; Colombel, J.F.; Reinisch, W.; Investigators, E.; Kumar, A.; Lazar, A.; et al. Adalimumab induces and maintains mucosal healing in patients with Crohn’s disease: Data from the EXTEND trial. Gastroenterology 2012, 142, 1102–1111.e2. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Stoinov, S.; Honiball, P.J.; Rutgeerts, P.; Mason, D.; Bloomfield, R.; Schreiber, S.; Investigators, P.S. Certolizumab pegol for the treatment of Crohn’s disease. N. Engl. J. Med. 2007, 357, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.; Khaliq-Kareemi, M.; Lawrance, I.C.; Thomsen, O.O.; Hanauer, S.B.; McColm, J.; Bloomfield, R.; Sandborn, W.J.; Investigators, P.S. Maintenance therapy with certolizumab pegol for Crohn’s disease. N. Engl. J. Med. 2007, 357, 239–250. [Google Scholar] [CrossRef]
- Lichtenstein, G.R.; Thomsen, O.O.; Schreiber, S.; Lawrance, I.C.; Hanauer, S.B.; Bloomfield, R.; Sandborn, W.J.; Precise 3 Study, I. Continuous therapy with certolizumab pegol maintains remission of patients with Crohn’s disease for up to 18 months. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2010, 8, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Stidham, R.W.; Lee, T.C.; Higgins, P.D.; Deshpande, A.R.; Sussman, D.A.; Singal, A.G.; Elmunzer, B.J.; Saini, S.D.; Vijan, S.; Waljee, A.K. Systematic review with network meta-analysis: The efficacy of anti-TNF agents for the treatment of Crohn’s disease. Aliment. Pharmacol. Ther. 2014, 39, 1349–1362. [Google Scholar] [CrossRef]
- Bakouny, Z.; Yared, F.; El Rassy, E.; Jabbour, R.; Hallit, R.; Khoury, N.; Honein, K.; Bou Jaoude, J. Comparative Efficacy of Anti-TNF Therapies for The Prevention of Postoperative Recurrence of Crohn’s Disease: A Systematic Review and Network Meta-Analysis of Prospective Trials. J. Clin. Gastroenterol. 2019, 53, 409–417. [Google Scholar] [CrossRef]
- Panes, J.; Rimola, J. Perianal fistulizing Crohn’s disease: Pathogenesis, diagnosis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Van Assche, G.; Gomez-Ulloa, D.; Garcia-Alvarez, L.; Lara, N.; Black, C.M.; Kachroo, S. Systematic Review of Tumor Necrosis Factor Antagonists in Extraintestinal Manifestations in Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2017, 15, 25–36.e27. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Lago, I.; Hoyo, J.D.; Perez-Girbes, A.; Garrido-Marin, A.; Casanova, M.J.; Chaparro, M.; Fernandez-Clotet, A.; Castro-Poceiro, J.; Garcia, M.J.; Sanchez, S.; et al. Early treatment with anti-tumor necrosis factor agents improves long-term effectiveness in symptomatic stricturing Crohn’s disease. United Eur. Gastroenterol. J. 2020, 8, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Hanauer, S.B.; Katz, S.; Safdi, M.; Wolf, D.G.; Baerg, R.D.; Tremaine, W.J.; Johnson, T.; Diehl, N.N.; Zinsmeister, A.R. Etanercept for active Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Gastroenterology 2001, 121, 1088–1094. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Hanauer, S.B.; Present, D.H.; Sutherland, L.R.; Kamm, M.A.; Wolf, D.C.; Baker, J.P.; Hawkey, C.; Archambault, A.; et al. An engineered human antibody to TNF (CDP571) for active Crohn’s disease: A randomized double-blind placebo-controlled trial. Gastroenterology 2001, 120, 1330–1338. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Sandborn, W.J.; Fedorak, R.N.; Rachmilewitz, D.; Tarabar, D.; Gibson, P.; Haagen Nielsen, O.; Wild, G.; Schreiber, S.; Pena Rossi, C.; et al. Onercept for moderate-to-severe Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2006, 4, 888–893. [Google Scholar] [CrossRef]
- Korzenik, J.; Larsen, M.D.; Nielsen, J.; Kjeldsen, J.; Norgard, B.M. Increased risk of developing Crohn’s disease or ulcerative colitis in 17 018 patients while under treatment with anti-TNFalpha agents, particularly etanercept, for autoimmune diseases other than inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 50, 289–294. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, A.; Lucci, M.; Korzenik, J. Inflammatory Bowel Disease Provoked by Etanercept: Report of 443 Possible Cases Combined from an IBD Referral Center and the FDA. Dig. Dis. Sci. 2016, 61, 1772–1774. [Google Scholar] [CrossRef]
- Levin, A.D.; Wildenberg, M.E.; Van den Brink, G.R. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. J. Crohn’s Colitis 2016, 10, 989–997. [Google Scholar] [CrossRef] [Green Version]
- D’Haens, G.; Baert, F.; van Assche, G.; Caenepeel, P.; Vergauwe, P.; Tuynman, H.; De Vos, M.; van Deventer, S.; Stitt, L.; Donner, A.; et al. Early combined immunosuppression or conventional management in patients with newly diagnosed Crohn’s disease: An open randomised trial. Lancet 2008, 371, 660–667. [Google Scholar] [CrossRef]
- Khanna, R.; Bressler, B.; Levesque, B.G.; Zou, G.; Stitt, L.W.; Greenberg, G.R.; Panaccione, R.; Bitton, A.; Pare, P.; Vermeire, S.; et al. Early combined immunosuppression for the management of Crohn’s disease (REACT): A cluster randomised controlled trial. Lancet 2015, 386, 1825–1834. [Google Scholar] [CrossRef]
- Mould, D.R.; Green, B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: Concepts and lessons for drug development. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2010, 24, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Mitrev, N.; Leong, R.W. Therapeutic drug monitoring of anti-tumour necrosis factor-alpha agents in inflammatory bowel disease. Expert Opin. Drug Saf. 2017, 16, 303–317. [Google Scholar] [CrossRef]
- Yanai, H.; Lichtenstein, L.; Assa, A.; Mazor, Y.; Weiss, B.; Levine, A.; Ron, Y.; Kopylov, U.; Bujanover, Y.; Rosenbach, Y.; et al. Levels of drug and antidrug antibodies are associated with outcome of interventions after loss of response to infliximab or adalimumab. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2015, 13, 522–530.e2. [Google Scholar] [CrossRef]
- Steenholdt, C.; Brynskov, J.; Thomsen, O.O.; Munck, L.K.; Fallingborg, J.; Christensen, L.A.; Pedersen, G.; Kjeldsen, J.; Jacobsen, B.A.; Oxholm, A.S.; et al. Individualised therapy is more cost-effective than dose intensification in patients with Crohn’s disease who lose response to anti-TNF treatment: A randomised, controlled trial. Gut 2014, 63, 919–927. [Google Scholar] [CrossRef]
- Velayos, F.S.; Kahn, J.G.; Sandborn, W.J.; Feagan, B.G. A test-based strategy is more cost effective than empiric dose escalation for patients with Crohn’s disease who lose responsiveness to infliximab. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2013, 11, 654–666. [Google Scholar] [CrossRef]
- Stein, B.N.; Pellish, R.S.; Thompson, K.D.; Baptista, V.; Siegel, C.A. Using Therapeutic Drug Monitoring to Identify Variable Infliximab Metabolism in an Individual Patient with Ulcerative Colitis. J. Clin. Gastroenterol. 2016, 50, 66–68. [Google Scholar] [CrossRef]
- Vande Casteele, N.; Ferrante, M.; Van Assche, G.; Ballet, V.; Compernolle, G.; Van Steen, K.; Simoens, S.; Rutgeerts, P.; Gils, A.; Vermeire, S. Trough concentrations of infliximab guide dosing for patients with inflammatory bowel disease. Gastroenterology 2015, 148, 1320–1329.e3. [Google Scholar] [CrossRef]
- D’Haens, G.R.; Vermeire, S.; Lambrecht, G.; Baert, F.J.; Bossuyt, P.; Nachury, M.; Buisson, A.; Bouhnik, Y.; Filippi, J.; Van der Woude, C.J. 692 Drug-level based dosing versus symptom-based dose adaptation in patients with Crohn’s disease: A prospective, randomized multicenter study (TAILORIX). Gastroenterology 2016, 150, S143. [Google Scholar] [CrossRef]
- Pagnini, C.; Arseneau, K.O.; Cominelli, F. Safety considerations when using anti-TNFalpha therapy to treat Crohn’s disease. Expert Opin. Drug Saf. 2015, 14, 31–44. [Google Scholar] [CrossRef]
- Sands, B.E.; Peyrin-Biroulet, L.; Loftus, E.V., Jr.; Danese, S.; Colombel, J.F.; Toruner, M.; Jonaitis, L.; Abhyankar, B.; Chen, J.; Rogers, R.; et al. Vedolizumab versus Adalimumab for Moderate-to-Severe Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Siegel, C.A.; Melmed, G.Y. Predicting response to Anti-TNF Agents for the treatment of crohn’s disease. Ther. Adv. Gastroenterol. 2009, 2, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, N.A.; Heap, G.A.; Green, H.D.; Hamilton, B.; Bewshea, C.; Walker, G.J.; Thomas, A.; Nice, R.; Perry, M.H.; Bouri, S.; et al. Predictors of anti-TNF treatment failure in anti-TNF-naive patients with active luminal Crohn’s disease: A prospective, multicentre, cohort study. Lancet Gastroenterol. Hepatol. 2019, 4, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Arijs, I.; Quintens, R.; Van Lommel, L.; Van Steen, K.; De Hertogh, G.; Lemaire, K.; Schraenen, A.; Perrier, C.; Van Assche, G.; Vermeire, S.; et al. Predictive value of epithelial gene expression profiles for response to infliximab in Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 2090–2098. [Google Scholar] [CrossRef]
- Van den Brande, J.M.; Koehler, T.C.; Zelinkova, Z.; Bennink, R.J.; te Velde, A.A.; Ten Cate, F.J.; van Deventer, S.J.; Peppelenbosch, M.P.; Hommes, D.W. Prediction of antitumour necrosis factor clinical efficacy by real-time visualisation of apoptosis in patients with Crohn’s disease. Gut 2007, 56, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neumann, H.; Neufert, C.; Waldner, M.J.; Billmeier, U.; Zopf, Y.; Willma, M.; App, C.; Munster, T.; Kessler, H.; et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn’s disease. Nat. Med. 2014, 20, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Gortz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef]
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M.; et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFalpha non-response in biopsy and blood of patients with IBD. Gut 2019, 68, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.A.; Bertha, M.; Hofmekler, T.; Chopra, P.; Vatanen, T.; Srivatsa, A.; Prince, J.; Kumar, A.; Sauer, C.; Zwick, M.E.; et al. Dysbiosis, inflammation, and response to treatment: A longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med. 2016, 8, 1–13. [Google Scholar] [CrossRef]
- Gisbert, J.P.; Chaparro, M. Predictors of primary response to biologic treatment (anti-TNF, vedolizumab and ustekinumab) in patients with inflammatory bowel disease: From basic science to clinical practice. J. Crohn’s Colitis 2019, 14, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Hirten, R.; Longman, R.S.; Bosworth, B.P.; Steinlauf, A.; Scherl, E. Vedolizumab and Infliximab Combination Therapy in the Treatment of Crohn’s Disease. Am. J. Gastroenterol. 2015, 110, 1737–1738. [Google Scholar] [CrossRef] [PubMed]
- Yzet, C.; Dupas, J.L.; Fumery, M. Ustekinumab and Anti-TNF Combination Therapy in Patients with Inflammatory Bowel Disease. Am. J. Gastroenterol. 2016, 111, 748–749. [Google Scholar] [CrossRef] [PubMed]
- Bethge, J.; Meffert, S.; Ellrichmann, M.; Conrad, C.; Nikolaus, S.; Schreiber, S. Combination therapy with vedolizumab and etanercept in a patient with pouchitis and spondylarthritis. BMJ Open Gastroenterol. 2017, 4, e000127. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Rath, T.; Geppert, C.I.; Manger, B.; Schett, G.; Neurath, M.F.; Atreya, R. Long-term Combination Therapy with Anti-TNF plus Vedolizumab Induces and Maintains Remission in Therapy-refractory Ulcerative Colitis. Am. J. Gastroenterol. 2017, 112, 1621–1623. [Google Scholar] [CrossRef]
- Sands, B.E.; Kozarek, R.; Spainhour, J.; Barish, C.F.; Becker, S.; Goldberg, L.; Katz, S.; Goldblum, R.; Harrigan, R.; Hilton, D.; et al. Safety and tolerability of concurrent natalizumab treatment for patients with Crohn’s disease not in remission while receiving infliximab. Inflamm. Bowel Dis. 2007, 13, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Drug | Trials (Ref) | Patients | End-Points | Results |
|---|---|---|---|---|
| Infliximab (IFX) | ACCENT I (72) ACCENT II (73) SONIC (74) | 335/573 responders at week 2 282 222 | Remission at week 30 Fistulas’ healing at week 54 Steroid-free remission and mucosal healing at week 26 | IFX 5 mg/Kg: 44/113 (39%) IFX 10 mg/Kg: 50/112 (45%) Placebo: 23/110 (21%) IFX induction + 8 week maintenance: 50/138 (36%) IFX induction + placebo: 27/144 (19%) Remission—IFX + AZA: 96/169 (57%) IFX: 75/169 (44%) AZA: 51/170 (30%) Mucosal healing—IFX + AZA: 47/107 (44%) IFX: 28/93 (30%) AZA 18/109 (17%) |
| Adalimumab (ADA) | CLASSIC I (75) CLASSIC II(76) CHARM (77) GAIN (78) Extend (79) | 299 259 499/854 responders at week 4 325 135 | Remission at week 4 Remission at week 56 Remission at week 26 and 56 Fistulas healing at week 56 Remission rate at week 4 in patients IFX non responders Mucosal healing at week 12 and 52 | ADA 160/80 mg: 27/76 (36%) ADA 80/40 mg: 18/75 (24%) ADA 40/20 mg: 13/74 (18%) Placebo: 9/74 (12%) ADA 40 mg/2 week: 15/19 (79%) ADA 40 mg/week: 15/18 (83%) Placebo: 8/18 (44%) ADA with dose optimization: 93/204 (46%) Week 26—ADA 40 mg/2 week: 68/172 (40%) ADA 40 mg/week: 75/157 (47%) Placebo: 29/170 (17%) Week 56—ADA 40 mg/2 week: 62/172 (36%) ADA 40 mg/week: 65/157 (41%) Placebo: 20/170 (12%) ADA 40 mg/2 week: 10/30 (33%) ADA 40 mg/week: 11/40 (28%) Placebo: 6/47 (13%) ADA: 34/159 (21%) Placebo: 12/166 (7%) Week 12—ADA 40 mg/2 week: 17/62 (27%) Placebo: 8/61 (13%) Week 52—ADA 40 mg/2 week: 15/62 (24%) Placebo: 0/61 (0%) |
| Certolizumab pegol (CZP) | PRECISE1 (80) PRECISE2 (81) PRECISE3 (82) | 655 425 241 from PRECISE2 | Remission at week 6 and 26 Remission at week 26 Remission at week 52 and 80 | Week 6—CZP: 71/329 (22%) Placebo: 57/326 (17%) Week 6 and 26—CZP: 47/327 (14%) Placebo: 32/326 (10%) CZP: 103/215 (48%) Placebo: 61/210 (29%) Week 52—CZP continuous group: 58/141 (41%) Drug-interruption group *: 30/100 (30%) Week 80—CZP continuous group: 51/141 (36%) Drug-interruption group *: 23/100 (23%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagnini, C.; Cominelli, F. Tumor Necrosis Factor’s Pathway in Crohn’s Disease: Potential for Intervention. Int. J. Mol. Sci. 2021, 22, 10273. https://doi.org/10.3390/ijms221910273
Pagnini C, Cominelli F. Tumor Necrosis Factor’s Pathway in Crohn’s Disease: Potential for Intervention. International Journal of Molecular Sciences. 2021; 22(19):10273. https://doi.org/10.3390/ijms221910273
Chicago/Turabian StylePagnini, Cristiano, and Fabio Cominelli. 2021. "Tumor Necrosis Factor’s Pathway in Crohn’s Disease: Potential for Intervention" International Journal of Molecular Sciences 22, no. 19: 10273. https://doi.org/10.3390/ijms221910273
APA StylePagnini, C., & Cominelli, F. (2021). Tumor Necrosis Factor’s Pathway in Crohn’s Disease: Potential for Intervention. International Journal of Molecular Sciences, 22(19), 10273. https://doi.org/10.3390/ijms221910273