1. Introduction
The myeloid C-type lectin-like inhibitory receptor 12A (CLEC12A) regulates immune responses in various pathological contexts, including gout, rheumatoid arthritis, and viral infection [
1,
2,
3,
4,
5]. In knock-out mouse models of gout and rheumatoid arthritis, a diminution in CLEC12A expression enhances inflammation and disease severity [
6,
7]. Similarly, low levels of CLEC12A expression by circulating neutrophils and monocytes from early rheumatoid arthritis patients correlate with higher disease activity [
8]. On the other hand, interferon production is significantly downregulated in lymphocytic choriomeningitis virus infected CLEC12A KO mice, resulting in an increased viral load and liver damage [
9]. Thus, CLEC12A differentially regulates myeloid cell responses in a stimulus-dependent manner.
CLEC12A has an extracellular C-type lectin-like domain (CTLD), the defining feature of C-type lectin receptors (CLRs) [
10,
11,
12,
13]. While CTLDs typically bind a diverse array of carbohydrate ligands and some have evolved to bind proteins, endogenous ligands that specifically bind the CLEC12A CTLD remain unidentified [
10,
11,
12,
13,
14]. CLEC12A CTLD is linked to the transmembrane domain by a stalk region that mediates receptor oligomerisation in other CLRs [
5] The short cytoplasmic tail of CLEC12A comprises an immunoreceptor tyrosine-based inhibitory motif (ITIM) through which it regulates intracellular signaling pathways.
The paradigm of inhibitory receptor signaling involves ligand-induced receptor clustering leading to ITIM phosphorylation and the recruitment of phosphatases that inhibit activation signal transduction pathways [
15,
16]. In the absence of a known natural ligand, antibody-induced CLEC12A clustering has identified key events in CLEC12A signaling. Antibody-mediated crosslinking induces CLEC12A translocation to flotillin-rich membrane domains, where ITIM is phosphorylated in a Src-dependent manner [
17]. Phosphatases recruited by CLEC12A include SHP-1 and SHP-2 [
5]. Signaling proteins with reduced phosphorylation after CLEC12A clustering and internalisation include components of the MAP kinase and PI3K-Akt pathways [
17]. CLEC12A regulates the monosodium urate-crystal (MSU)-induced release of IL-8 by neutrophils through the p38/PI3K-Akt signaling pathway [
17,
18]. In addition to regulating cytokine release, reactive oxygen species production is enhanced in CLEC12A KO mice [
9].
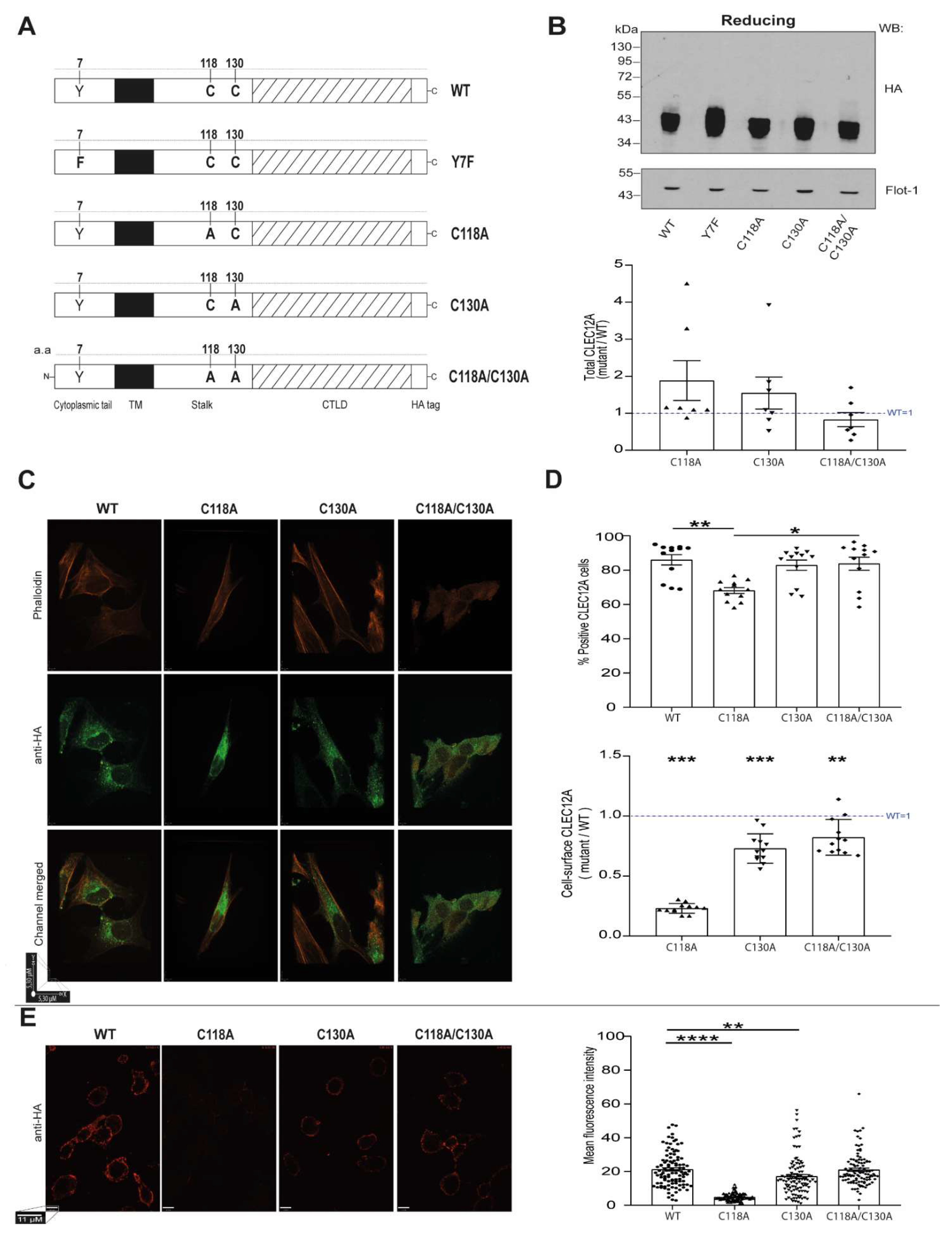
While the role of CLEC12A in several inflammatory diseases is well established and the signaling pathways it modulates are partially defined, little is known about the molecular determinants of CLEC12A function. A potentially essential domain for CLEC12A function is its stalk region as it harbours two cysteine residues. While it is presumed that these cysteine residues have receptor multimerisation properties, this has not been demonstrated experimentally. Herein, we tested the hypothesis that the two cysteine residues in the stalk domain of CLEC12A regulate its expression, internalisation, signaling and/or function through their ability to induce receptor oligomerisation.
3. Discussion
Disulfide bonds are the most common covalent linkages in proteins [
21]. They are particularly frequent in extracellular proteins such as CLRs whose CTLD fold depends on highly conserved cysteines [
14]. In addition to the CTLD, some CLRs such as CLEC12A also habour cysteine residues in their stalk domain that are postulated to be essential for receptor oligomerisation [
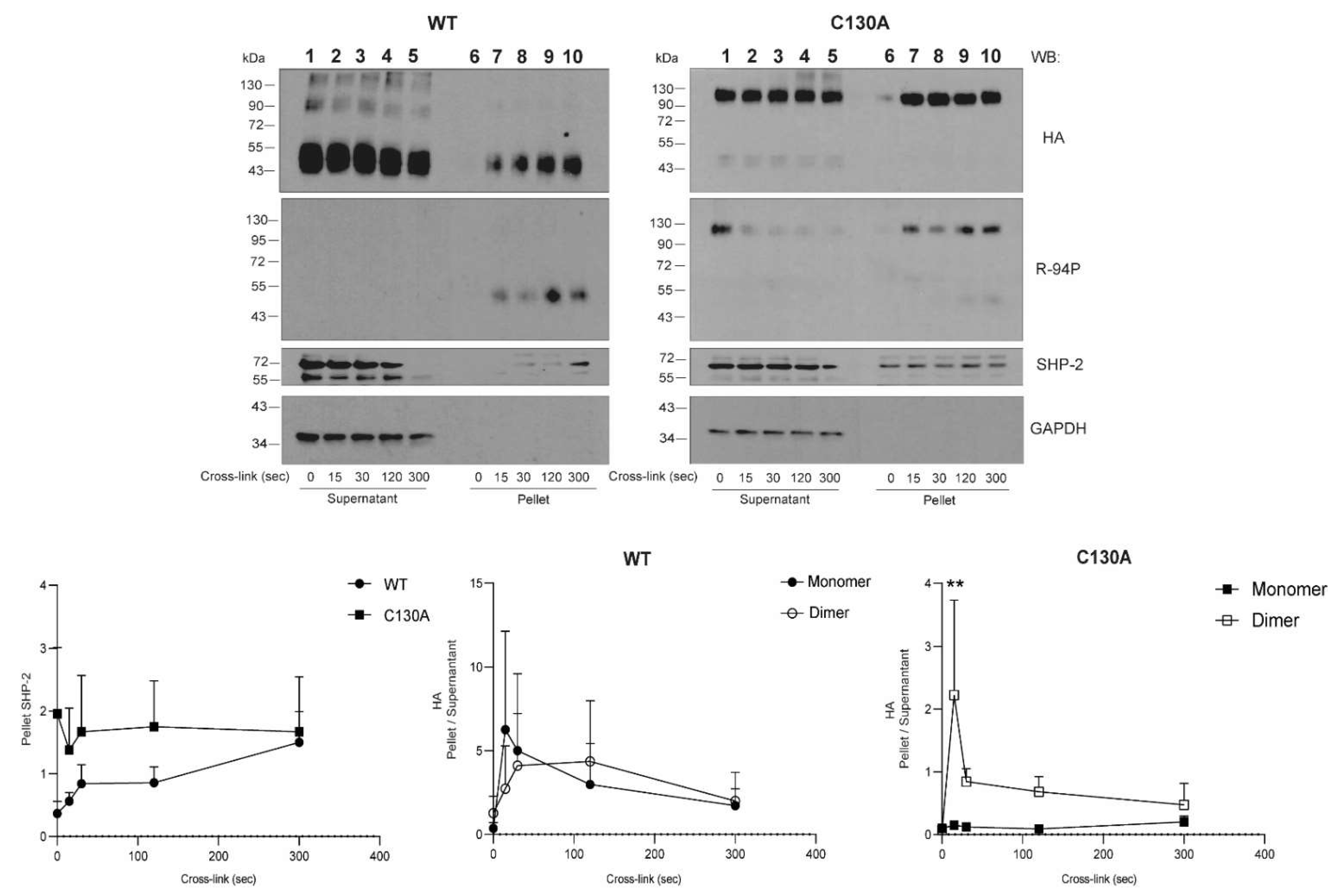
24]. The aim of this study was to elucidate the role of the stalk domain cysteines in CLEC12A expression and early signaling events. Our findings provide the first demonstration of the crucial roles for the two CLEC12A stalk cysteine residues in regulating receptor subcellular localisation, cell-surface expression and signaling. While the loss of C118 disrupts CLEC12A transport through cytoplasmic compartments to the cell-surface expression, mutation of C130 enhances CLEC12A oligomerisation and subsequently its phosphorylation and signaling. These findings are particularly pertinent to the modulation of myeloid cell activation by CLEC12A during inflammation due to the significant changes in the redox environment. In support of this notion, we show that CLEC12A oligomerisation is enhanced in the presence of hydrogen peroxide suggestive that the stalk cysteines have redox regulatory capacity. Moreover, we show that CLEC12A induces flotillin-1 oligomerisation and that flotillin is essential for CLEC12A signaling.
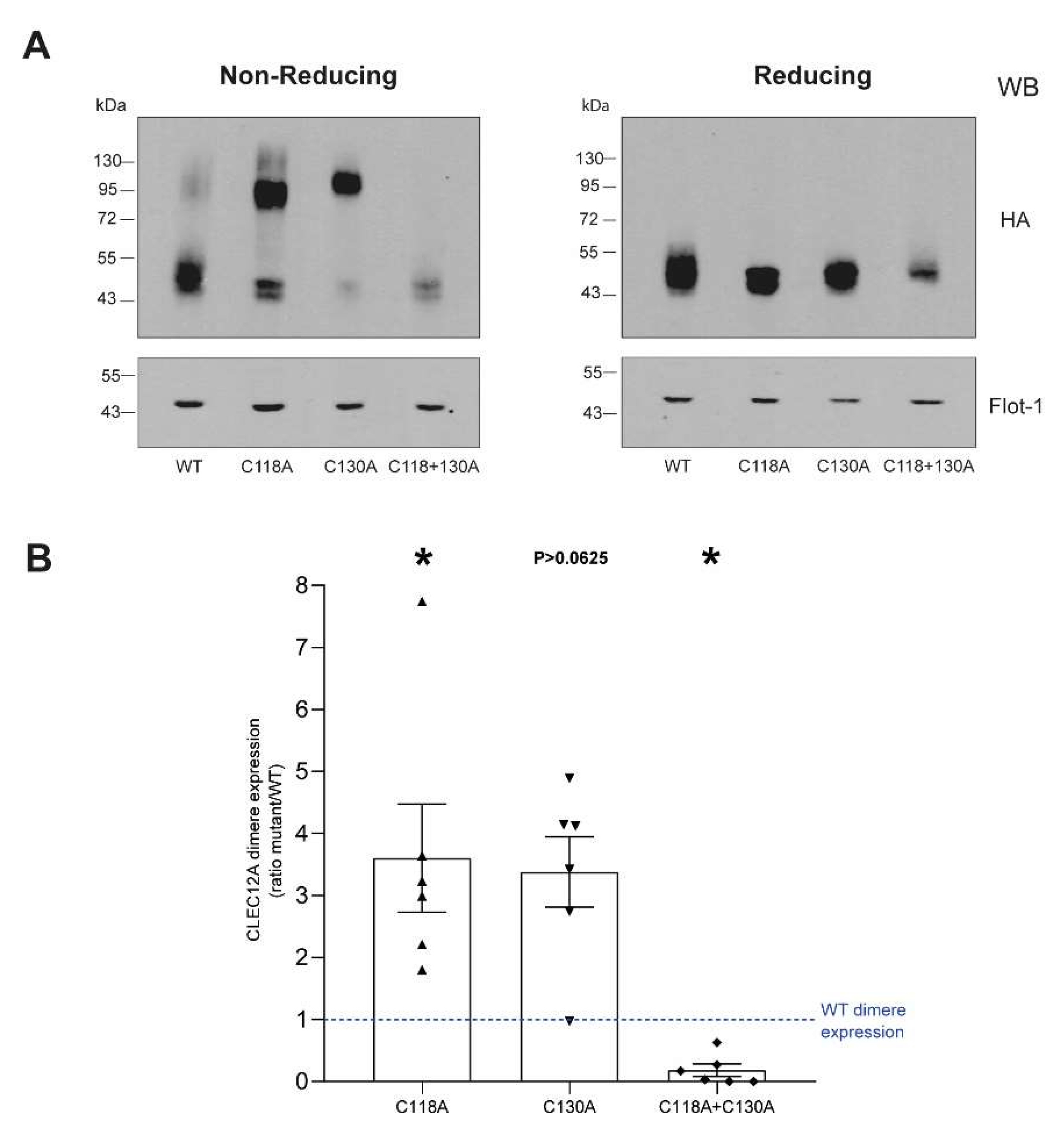
CLR stalk cysteine residues are mostly known for their role in receptor oligomerisation through the formation of disulfide bonds [
24]. CLEC12A’s stalk cysteines are conserved with CLEC-1, a CLR that dimerizes through the formation of disulfide linkages in its stalk domain [
25]. Similarly, the CLR LOX-1 that shares a high degree of homology with CLEC12A’s CTLD also forms homodimers through a disulfide bond formed by Cys140 within its stalk domain [
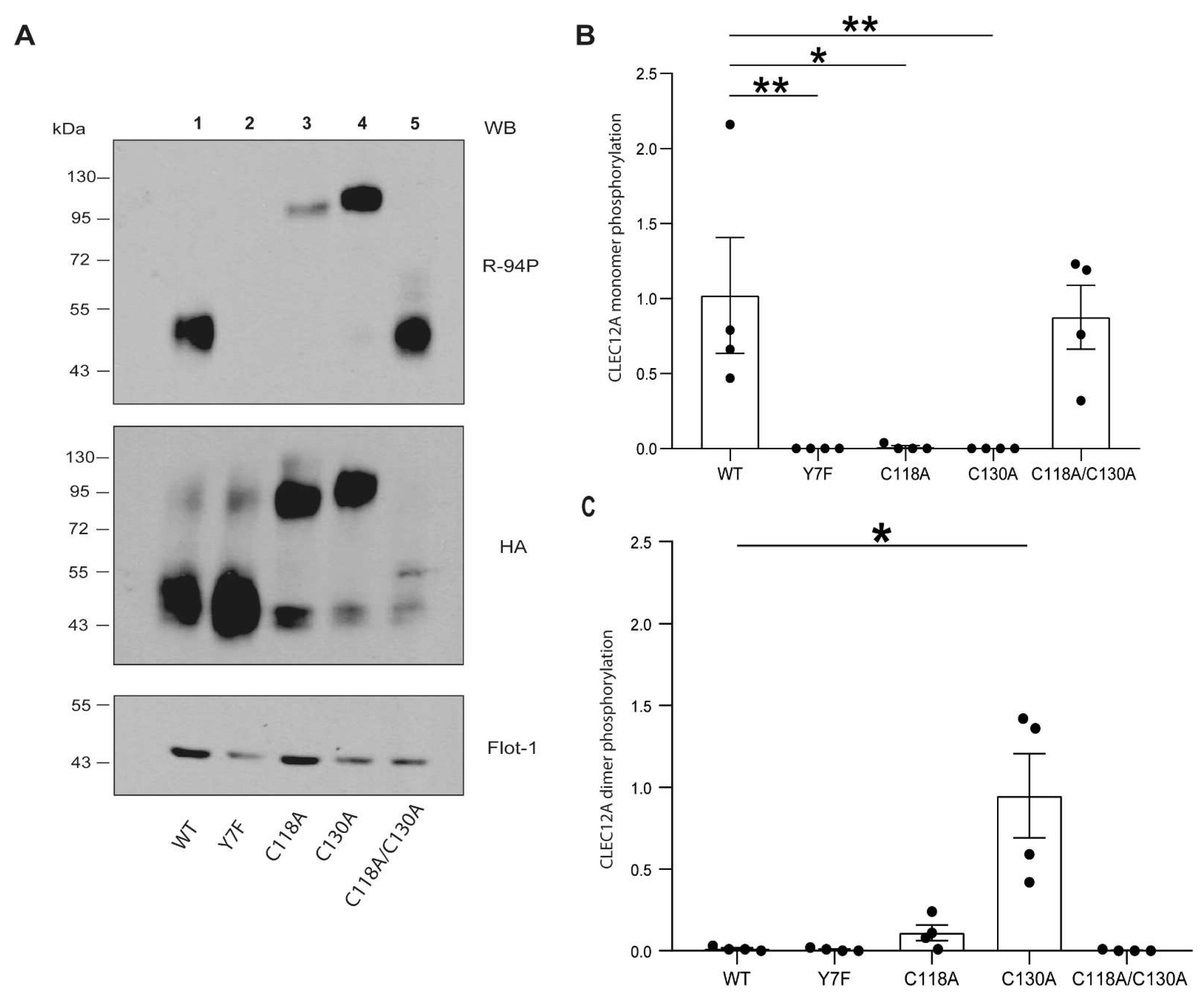
26]. We provide direct evidence that C118 and C130 are also involved in disulfide bond formation as the dimerisation of the CLEC12A combinatorial mutant of both cysteines is significantly compromised. In contrast, the single C130A mutant enhances CLEC12A dimerisation and phosphorylation, and recruits SHP-2 to the flotillin-rich membrane, cellular fraction. We interpret this ligand-independent induced oligomerisation as a gain-of-function phenotype. A search of the the gnomAD database revealed a naturally occurring polymorphism that substitutes C130 with a tyrosine residue. Our data suggest that individuals with this polymorphism will have a constitutively active CLEC12A and a significant down-regulation of myeloid cell activation. Since CLR oligomerisation increases ligand binding affinity, the formation of CLEC12A oligomers due to the loss of C130 may further potentiate CLEC12A function through enhanced interactions with ligands. The C130Y polymorphism is more frequent in Asian than European populations suggestive that environmental and/or epigenetic factors may favor the retention of this polymorphism in a greater proportion of the Asian than the European population.
Cysteine residues ensure protein quality control through proper protein folding in the tightly controlled redox environment of the ER [
19]. Naturally occurring mutations in membrane and secretory proteins causing ER retention and loss of protein function are common in genetic diseases including Pelizaeus–Merzbacher disease and von Willebrand’s disease [
19]. While we provide evidence that loss of C118 disrupts CLEC12A’s transit through the secretory pathway significantly diminishing its cell-surface expression, naturally occurring polymorphisms at this residue have not been reported. It is highly likely that there is considerable selection pressure to avoid changes at this amino acid due to its crucial role in CLEC12A expression. To our knowledge, a role for stalk cysteine residues in CLR, cell-surface expression has not been previously reported.
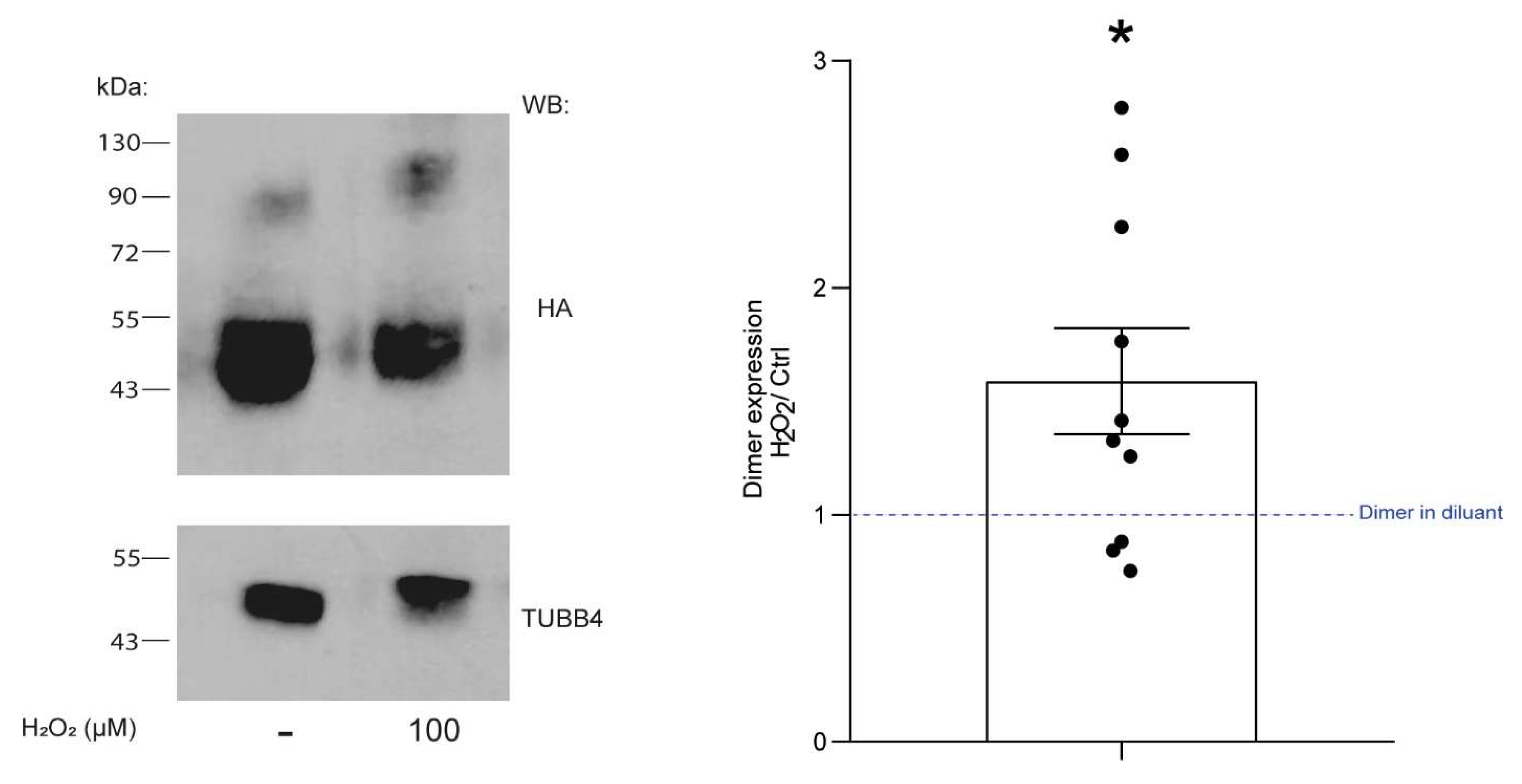
In addition to naturally occurring polymorphisms, the redox status of the surrounding environment could modify CLEC12A function by affecting C118 and C130 formation of disulfide bonds due to changes in their thiol reactivity. Cysteines are highly reactive residues and they also play a role as redox molecular switches in addition to their oxidative protein folding properties in several proteins such as HMGB1 [
19]. The function of this DNA-binding nuclear protein changes depending on the redox state of its cysteines. When fully reduced, HGMB1 promotes inflammation by activating cell migration and stimulating cytokine secretion. In contrast, sulphonylation inactivates HMGB1. Moroever, cysteines within the same protein may play differential roles in protein trafficking, dimerisation and function as reported for the HDL receptor, SR-B1 [
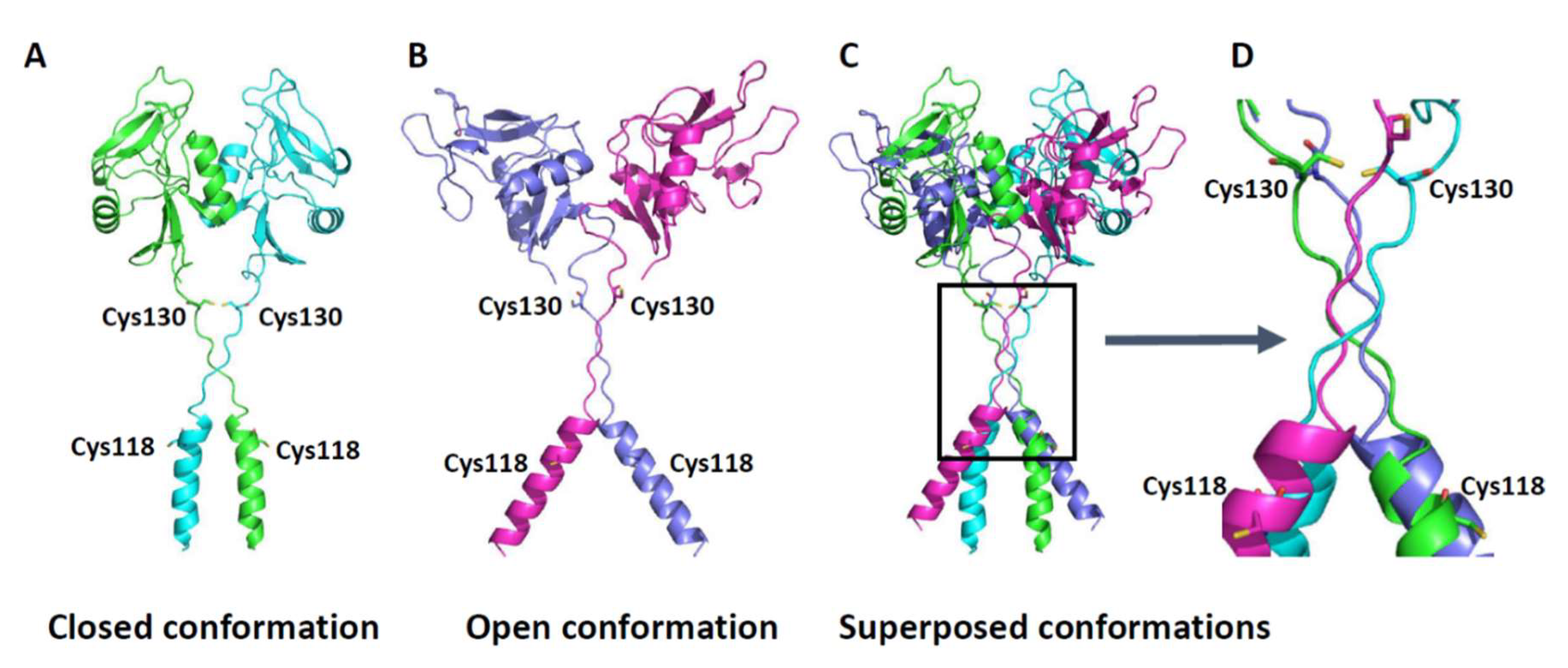
27]. Our findings have implications for the role of CLEC12A in inflammation as we also show that hydrogen peroxide enhances CLEC12A oligomerisation. Whether C118 and C130 are equally reactive towards oxygen radicals remains unknown. The preferential oxidation of C118 would result in it forming a disulfide bond as shown in
Figure 6 (closed conformation) and a gain-of-function phenotype. In contrast, oxidation of C130 would favor disulfide bonding at this residue as shown in
Figure 6 and result in a diminution in receptor function. These two cysteines thus have distinct and counter-regulatory roles to ensure the appropriate post-translational processing and function of CLEC12A. While the presence of C118 ensures that CLEC12A’s transport from the ER to the plasma membrane is not hindered, the presence of C130 is necessary to regulate CLEC12A oligomerisation. Our data strongly suggest that CLEC12A stalk cysteines function as regulatory switches of CLEC12A cell-surface expression, oligomerisation and signaling. This regulatory function is highly likely influenced by oxidative environments.
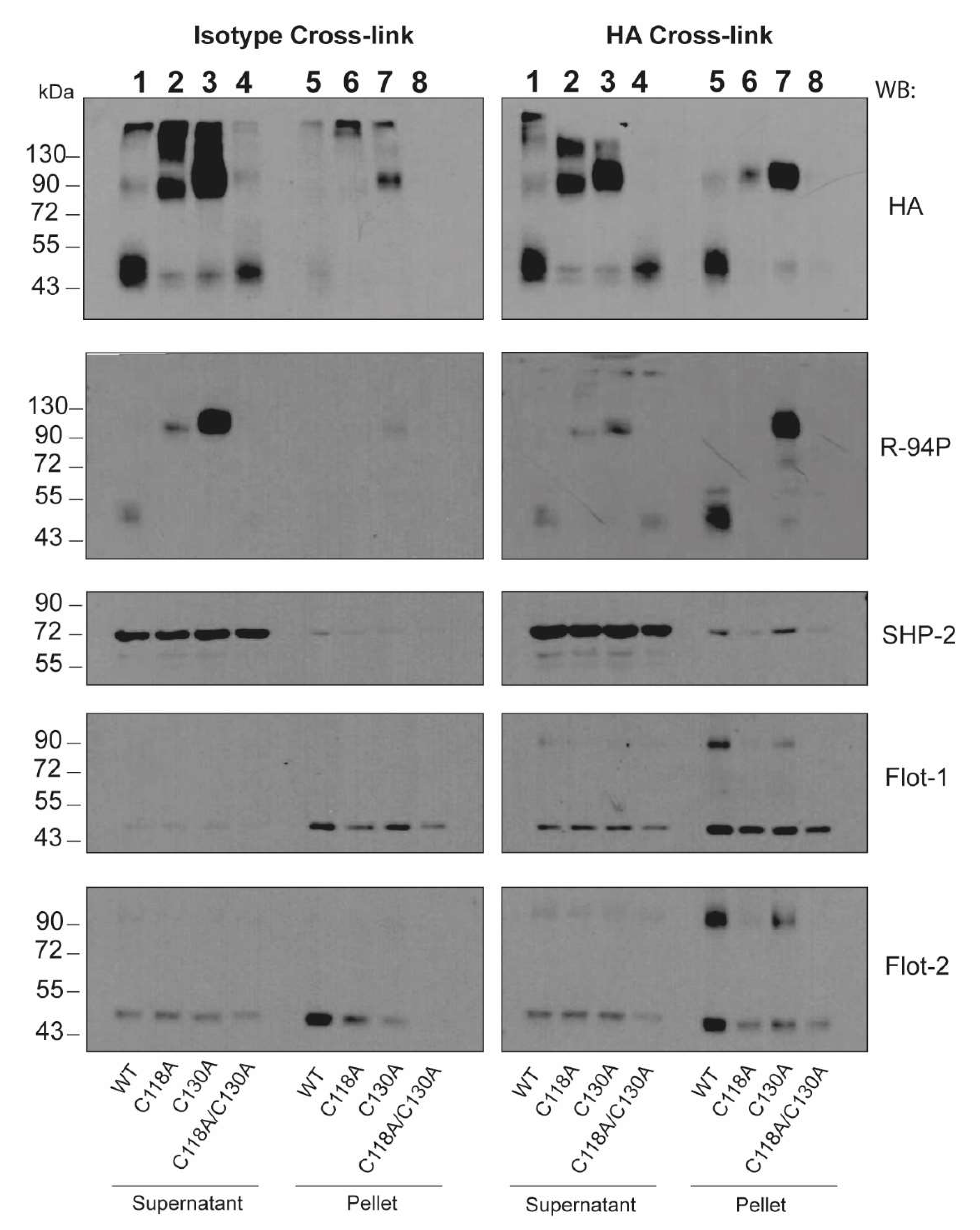
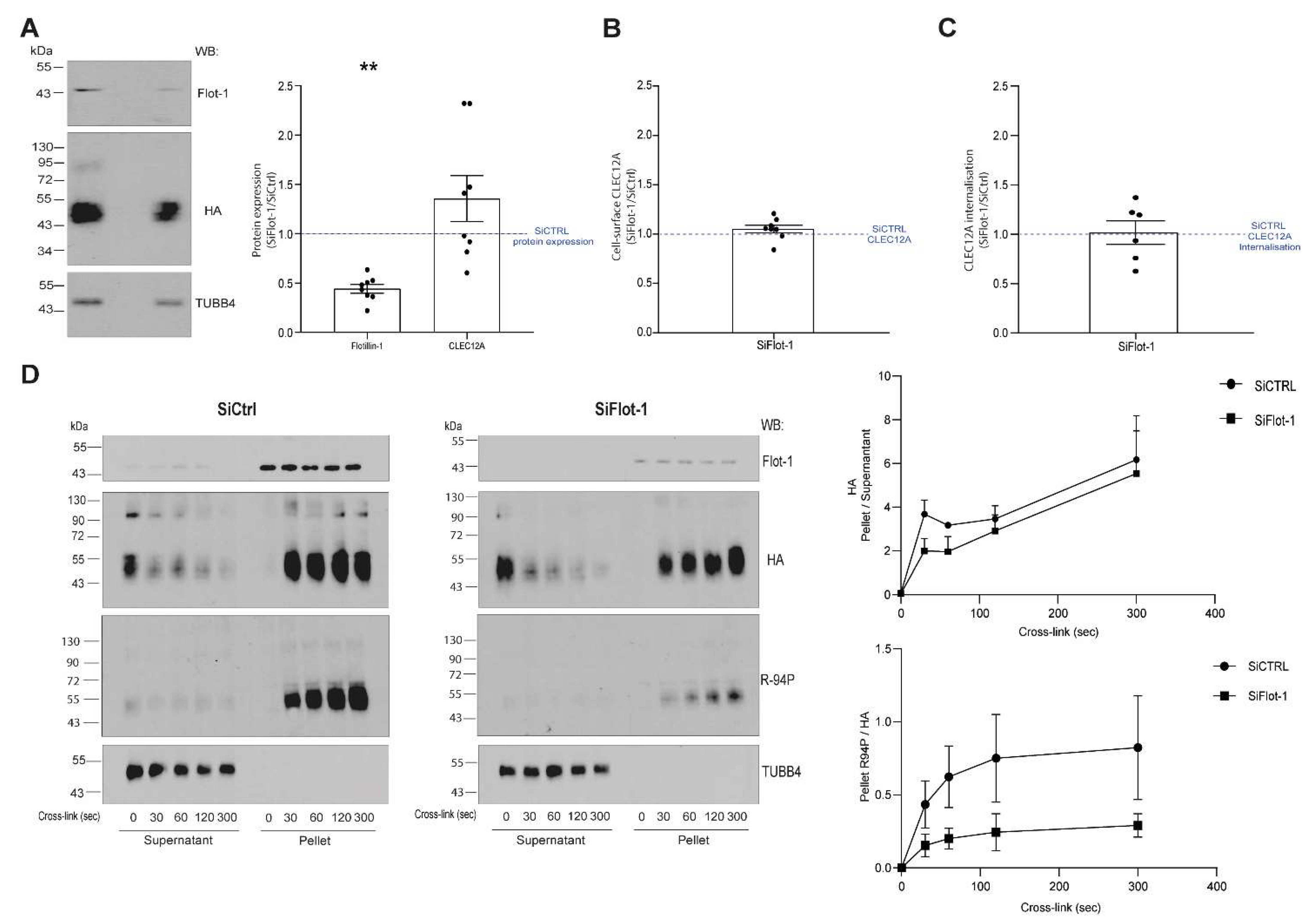
We previously reported that CLEC12A co-localizes with flotillin in detergent-resistant membranes after antibody-induced cross-linking [
17]. The current study shows that antibody-induced cross-linking of CLEC12A also induces flotillin-1 and flotillin-2 oligomerisation in the detergent-resistant membrane fraction. This is in line with previous reports that demonstrated the requirement of flotillin oligomerisation for its recruitment to these plasma membrane domains [
28]. Consistent with the signaling promoting properties of flotillin-enriched membrane domains, a knock-down of flotillin-1 expression significantly diminished the phosphorylation of the CLEC12A ITIM. These observations underscore the crucial role of flotillin membrane domains in CLEC12A signaling. Flotillin interacts with a variety of receptors and signaling proteins explaining its involvement in a myriad of cellular processes including cell adhesion, endocytosis, phagocytosis and cell signaling [
23,
28]. Whether CLEC12A can also, in turn, regulate any of the diverse roles of flotillin through hetero-oligomerisation and/or altering flotillin phosphorylation remains to be determined.
While we provide evidence for a regulatory the role for Cys118 and Cys130 in CLEC12A expression, oligomerisaton and phosphorylation, the role of these residues in downstream CLEC12A signaling and function remains to be determined. Further experimentation will also reveal how flotillin and CLEC12A interact to regulate myeloid cell function.
In summary, our observations significantly further our understanding of CLEC12A function by identifying a crucial role for non-CTLD cysteines in regulating CLEC12A expression and signaling. Additionally, our data suggest that these cysteines act as redox-regulatory switches of CLEC12A signaling. Insight into how different inflammatory environments modulate CLEC12A expression and function through the stalk cysteines, will reveal how this myeloid inhibitory receptor contributes to the pathogenesis of autoimmune and inflammatory diseases.
4. Materials and Methods
4.1. Antibodies
Two different antibodies against the HA-tag were used, namely, the anti-HA.11 (mouse monoclonal 16B12; no. 90150) from BioLegend (Pacific Heights Blvd, San Diego, CA) and the rabbit polyclonal anti-HA (NB600-363B) from Novus biologicals (Oakville, ON, Canada). The former was used to cross-link CLEC12A-HA on HEK-293T cells and the latter, for immunoblotting. A mouse IgG1 isotype antibody (no. IM0571) was obtained from Beckman Coulter (Mississauga, ON, Canada) and used as a negative control.
The mouse IgG2a isotype control (no. 401502) antibody was obtained from BioLegend (Pacific Heights Blvd, San Diego, CA, USA). The affiniPure F(ab’)
2 fragment goat anti-mouse IgG F(ab’)
2 fragment specific (no. 115-006-072), the horseradish peroxidase-labeled donkey anti-rabbit IgG (no. 711-035-152), the horseradish peroxidase-labeled donkey anti-mouse IgG (no. 715-035-150) and fluorescein (FITC)-AffiniPure F(ab’)
2 fragment goat anti-mouse IgG, Fcγ fragment specific (no. 115-096-071) antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The monoclonal anti-Flotillin-1 (no. 610820) and anti-Flotillin-2 (no. 610383) antibodies were purchased from BD Transduction Laboratories (Mississauga, ON, Canada) and phalloidin (no. A12381) from Thermofischer (Pacific Heights Blvd, San Diego, CA, USA). The mouse monoclonal anti-GAPDH (NBP1-47339) was obtained from Novus biologicals (Oakville, ON, Canada). Goat anti-mouse IgG (H + L) conjugated AlexaFluor 488 nm (A-11011) and the goat anti-rabbit IgG (H + L) conjugated AlexaFluor 594 nm (A-10012) antibodies were purchased from Invitrogen, Thermo Fisher Scientific (Waltham, MA, USA). The polyclonal anti-SHP-2 (sc-280) antibody was obtained from Santa Cruz (Dallas, TX, USA) and the mouse anti-human tubulin 4 (anti-TUBB4) (TUBB2C #WH0010383M2) antibody from Sigma-Aldrich Canada (Oakville, ON, Canada). The anti-phospho CLEC12A ITIM antibody (R-94P) was generated in-house and characterized in Paré et al. [
20].
4.2. Reagents
Sodium orthovanadate (Na3VO4), trypsin inhibitor, PMSF, Nonidet P-40, Triton X-100, formalin 10%, aprotinin and leupeptin were obtained from Sigma-Aldrich Canada (Oakville, ON, Canada) and the Western Lightning Chemiluminescence Plus from PerkinElmer (Guelph, ON, Canada). Fetal bovine serum (FBS) and Dulbeco’s modified Eagle’s medium (DMEM) were purchased from Wisent Bioproducts (St-Bruno, QC, Canada). And Tween 20 as well as hydrogen peroxide (30%) from Fischer Scientific (Ottawa, ON, Canada). Polyethylenimine (PEI) was obtained from VWR (Mississauga, ON, Canada) and slowFadeTM Gold antifade reagent from Thermo Fisher Scientific (Waltham, MA, USA).
4.3. Plasmid Constructs and SiRNA StealthTM RNAi
The wild type, CLEC12A coding sequence used for our constructs corresponds to the CLEC12A isoform 2 sequence (Q5QGZ9-2 Uniprot)
. The generation of the HA-tagged CLEC12A wild-type (WT) and CLEC12A-HA-Y7F construct with a mutated tyrosine in the ITIM motif (Y7F) were previously described in Paré [
20]. To generate the CLEC12A-HA-C118A mutant construct with a mutated C118A, the open reading frame of CLEC12A-HA-wt was amplified with the forward; 5′ CAATAGCCACCAAATTAGCTCGTGAGCTATATAGC 3′ and reverse primer 5′ GCTATATAGCTCACGAGCTAATTTGGTGGCTATTG 3′. The PCR product was ligated to the pCRII plasmid using the same strategy as for CLEC12A-HA-wt. To generate the CLEC12A-HA-C130A construct with a mutated C130A, we used an overlap extension polymerase chain reaction (OE-PCR). The open reading frame of CLEC12A was amplified in two different PCR reactions with different pairs of primers. For the first PCR reaction for CLEC12A-HA-C130A we used the forward primer: 5′ AAGAGCACAAAGCTAAGCCTTGTC 3′ and reverse primer: 5′ TCTAGATGCATGCTCGAGCGGCCGCTTA 3′. For the second PCR reaction for CLEC12A-HA-C130A, we used the forward primer: 5′ CGCCAGTGTGCTGGAATTCTTTACATATT 3′ and reverse primer: 5′ GACAAGGCTTAGCTTTGTGCTCTT 3′. The two PCR products, one extending “upstream” and the other “downstream” of the desired mutation were mixed and hybridized while performing a third PCR using the oligonucleotides that targeted the 5′ and 3′ ends the hybrid PCR product, namely, the forward primer: 5′ CGCCAGTGTGCTGGAATTCTTTACATATT 3′ and the reverse primer: 5′ TCTAGATGCATGCTCGAGCGGCCGCTTA 3′. The PCR product was ligated to the pCRII plasmid using the same strategy as for CLEC12A-HA-wt. To generate the CLEC12A-HA-C118A/C130A double mutant, the open reading frame of CLEC12A-HA-C130A was amplified with the same primer and cloning strategy used to generate CLEC12A-HA-C118A. The SiRNA control (SiCTRL) (452001) and the SiRNA Flotillin-1 (FLOT1HSS115567) were obtained from Invitrogen.
4.4. Cell Culture and Transfection
HEK-293T and HeLa cells were maintained in Dulbeco’s modified Eagle’s medium (DMEM) containing 4 mM L-glutamine, 1 mM sodium pyruvate and 10% heat-inactivated fetal bovine serum. No antibiotics were used to culture these cell lines. Cells were seeded at a density of 0.3 × 106 cells/well (HEK 293T or Hela) in 6-well plates the day prior to transient transfection of the CLEC12A-HA-WT or mutant plasmids (2 µg of DNA per well) with PEI as per the manufacturer’s instructions. The cells were harvested 48 h post-transfection with 10 mM EDTA in PBS prior to analysis.
4.5. Co-Transfection of Cell Lines with CLEC12A-HA-WT and the SiRNA StealthTM RNAi
Cells were seeded at a density of 0.2 × 106 cells/well (HEK 293T) in 6-well plates the day prior to transient transfection of the CLEC12A-HA-WT plasmid (2 µg of DNA per well) and 20 mM SiRNA (SiCtrl or SiFlot-1) with PEI as per the manufacturer’s instructions. The cells were harvested 72 h post-transfection with 10 mM EDTA in PBS prior to analysis. To confirm flotillin-1 expression was downregulated, we performed a western blot for each experiment (western blot and flow cytometer) where flotillin-1 expression in SiFlot-1 was compared to SiCtrl to assess the SiRNA flotillin-1 downregulation.
4.6. Lysis of Transiently Transfected Hek 293T Cells Prior to Immunoblotting
HEK-293T transfected with CLEC12A mutant construct were harvested (106 cells/100 µL PBS) and resuspended in the same volume of 2X modified Laemmli’s sample buffer reducing (composition of 1X: 62.5 mM Tris-HCl (pH 6.8), 4% (w/v) SDS, 8.5% (v/v) glycerol, 2.5 mM orthovanadate, 0.025% bromophenol blue, 10 μg/mL leupeptin, 10 μg/mL aprotinin, 5% (v/v) β-mercaptoethanol) or non-reducing (5% (v/v) β-mercaptoethanol replaced by water) and heated 95 °C for 7 min. When indicated cells were treated with pervanadate. Pervanadate was freshly prepared before use by mixing 1 mM orthovanadate, 0.03% hydrogen peroxide in H2O and then incubatingthe solution at room temperature for 15 min in the dark prior to the addition to cells 1 in 9, vol:vol dilution). Cells were incubated for 10 min at 37 °C in the dark prior to lysis in the same volume of 2X modified Laemmli’s sample buffer (see above) and immunoblotting.
When indicated, the CLEC12A-HA receptorswere cross-linked with an anti-HA, mouse monoclonal or the isotype antibody (3 µg/106 cells) for 5 min at 37 °C and centrifuged prior to incubation with the goat anti-mouse F(ab’)2 anti F(ab’)2 antibody (3 µg/106 cells) for 5 min at 37 °C. Cross-linking was stopped on ice and the cells centrifuged at 1000× g for 1 min. The cells were resuspended (106 cells/100 µL PBS) and lysed with the same volume of 2X modified, reducing or non-reducing Laemmli’s sample buffer (see above). To prepare cell-free supernatants and pellets, cells were lysed in cold 1% NP40 and the cell pellet resuspended in cold 1% NP-40 lysis buffer (10 mM Tris-HCL pH 7.3, 137.2 mM NaCl, 1 mM EDTA, 10 µg/mL aprotinin, 10 µg/mL leupeptin, 2 mM sodium orthovanadate, 50 µg/mL trypsin inhibitor, 1 mM PMSF, 1% NP-40) and incubated for 10 min on ice prior to centrifuging at 15,000× g for 10 min at 4 °C. An aliquot of the supernatant (SN) was incubated at 95 °C for 7 min in the same volume of non-reducing or reducing, modified 2X Laemmli’s sample buffer. The pellets were washed with cold, 1% NP-40 lysis buffer and centrifuged at 15,000× g for 5 min at 4 °C. Pellets were sonicated with an ultrasonic pulse for 3 s prior to the addition of non-reducing or reducing, modified 2X Laemmli’s sample buffer and incubating at 95 °C for 7 min.
4.7. Electrophoresis and Immunoblotting
Proteins (20–25 µg/sample) were separated by SDS-PAGE on 10% acrylamide gels and transferred to PVDF membranes. Blocking agents and antibodies were diluted in a TBS-Tween solution (25 mm Tris-HCl, pH 7.8, 190 mm NaCl, 0.15% (v/v) Tween 20). Non-fat milk solution (5% w/v) was used to block nonspecific sites prior to immunoblotting with the anti-flotillin-1, anti-flotillin-2, anti-HA, anti-phospho CLEC12A (R-94P), anti-GAPDH and anti-SHP-2 antibodies. Anti-flotillin-1 [0.125 µg/mL], anti-flotillin-2 [0.125 µg/mL], anti-SHP-2 [0.2 µg/mL], anti-GAPDH [1 µg/mL], anti-TUBB4 (Tubullin 4) [0.5 µg/mL], rabbit anti-HA [1 µg/mL] and were diluted in TBS-Tween (0.15%). The anti-phospho CLEC12A ITIM antibody (R-94-P) was diluted at 0.4–0.8 µg/mL in TBS-Tween + BSA (5% w/v). Horseradish peroxidase-labeled donkey anti-rabbit IgG and horseradish peroxidase-labeled donkey anti-mouse IgG antibody were diluted at 50 ng/mL in TBS-Tween solution. Chemiluminescence reagents were used to detect antibodies within a maximal exposure time of 5 min. Equal protein loading was verified by immunoblotting against flotillin-1 or GAPDH.
4.8. Confocal Microscopy
Cells were transfected with the CLEC12A constructs 24 h after being seeded on coverslips (0.22 µm). Forty-eight hours post-transfection, cells were fixed with 9% formalin for 10 min at RT and washed with PBS prior to permeabilisation with 0.05% Triton X-100 in PBS for 10 min at room temperature. This was followed by an incubation for 20 min at room temperature in blocking solution (0.05% Triton X-100 in PBS supplemented with 4% FBS) and staining with the mouse anti-HA antibody [3 µg/mL for intracellular staining] and phalloidin AlexaFluor 594 nm in blocking buffer 30 min at 37 °C in dark. For extracellular staining, cells were incubated with a rabbit anti-HA antibody 7.5 µg/mL before fixation with formalin. Cells were then washed for 30 min at 37 °C and incubated with 5 µg/mL of secondary antibody against HA (anti-mouse AlexaFluor 488 nm) for 30 min at 37 °C in dark with blocking solution. After a wash in 0.05% PBS Triton X-100 for 30 min at 37 °C and water for 2 min followed by 1 min in 90% ethanol and 1 min in 99% ethanol, prior to mounting cells with SlowFadeTM Gold antifade reagent. No non-specific binding of the secondary antibodies was observed (data not shown).
Images were acquired at 63X with a Z-stack spacing of 0.05 µm with the Quorum WAVFX spinning disc system (Quorum Technologies, Guelph, ON, Canada) and analyzed with the Volocity quantitation module. Briefly, confocal images were deconvoluted and the point spread function calculated for the GFP and Texas Red channel was applied using the velocity module for iterative restoration.
4.9. Quantitation of Antibody-Induced CLEC12A Internalisation by Flow Cytometry
HEK-293T cells transfected with the CLEC12A constructs were harvested (106 cells/100 µL PBS) and incubated with anti-HA, mouse monoclonal or the isotype antibody (3 µg/106 cells) for 5 min at 37 °C and centrifuged prior to cross-linking with the goat anti-mouse F(ab’)2 anti F(ab’)2 antibody (3 µg/106 cells) for 5 min at 37 °C or incubated in buffer (no cross-linking). Cross-linking was stopped on ice and the cells centrifuged at 1000× g for 1 min. The cells were resuspended (106 cells/100 µL PBS) prior to a 30 min incubation at 4 °C in the dark with an anti-mouse Fc-FITC conjugated (13 µg/mL) antibody. The cells were washed and resuspended in PBS and CLEC12A surface expression determined by flow cytometer.
4.10. H2O2 Treatment
HEK-293T transfected with CLEC12A WT were harvested with EDTA (106 cells/100 µL PBS) and incubated in 100 µM H2O2 for 3 min or the same v:v with diluent (H2O). The reaction was stop by adding the same volume of 2X modified Laemmli’s sample buffer (see above) without beta-mercaptoethanol and boiled for 7 min at 100 °C.
4.11. Three Dimensional Modeling of CLEC12A
The 3D homology model (105–253 amino acid) of CLEC12A (Uniprot ID: Q5QGZ9) was constructed by the modeling software Modeller (Webb and Sali, 2014), using as templates from PDB database, 3g8l_A (105–125 amino acid) and 1yxk_B (126–253 amino acid). The homodimer was obtained by Galaxy homomer software (10.1093/nar/gkx246) (
http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=HOMOMER) (accessed on 1 February 2016). Coiled-coil of CLEC12A (68–88 amino acid) was determined by PCOILS (Gruber et al., 2006) and was added to dimer. Model quality was assessed by Ramachandran plot analysis through PROCHECK (Laskowski et al., 1993). Structure images were generated using PyMOL (
http://www.pymol.org) (accessed on 1 February 2016).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







