
Insights into the Genetic Variations of Human Cytochrome P450 2C9: Structural Analysis, Characterization and Comparison

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
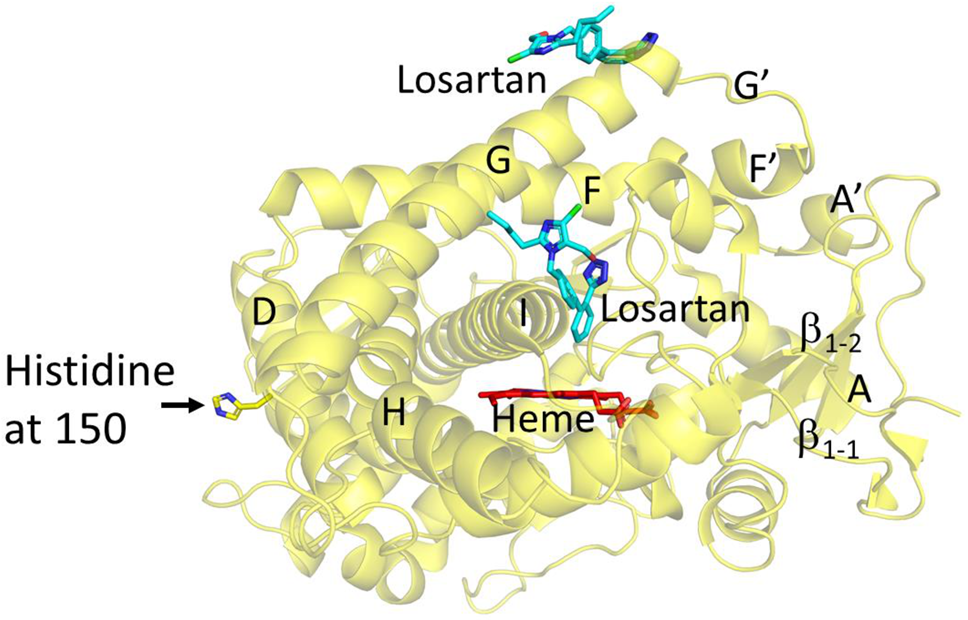
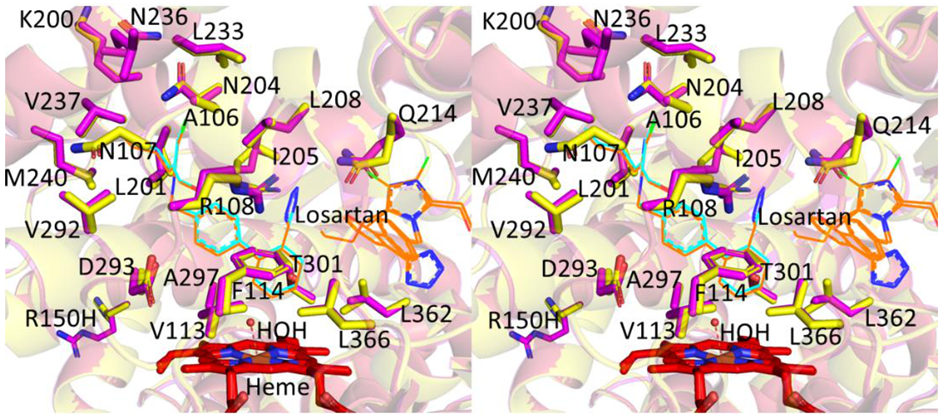
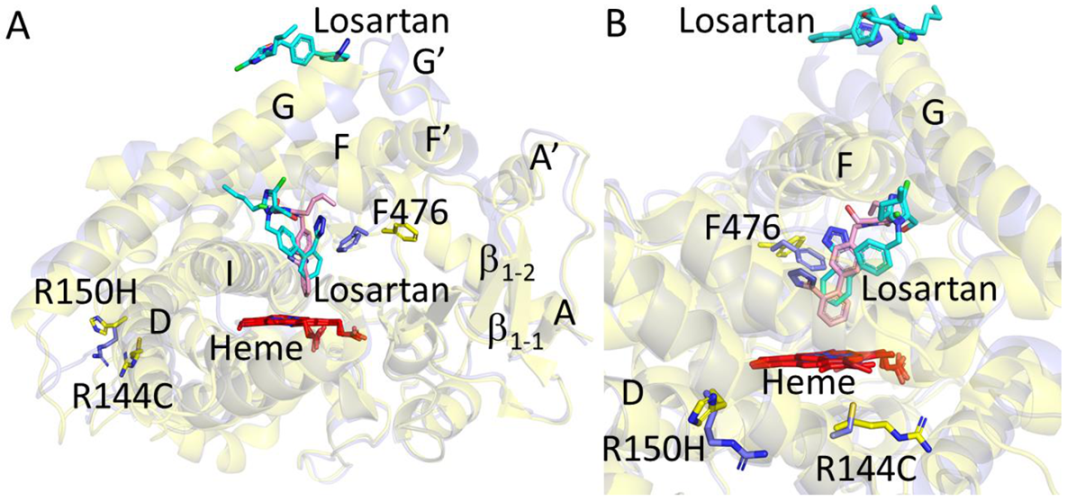
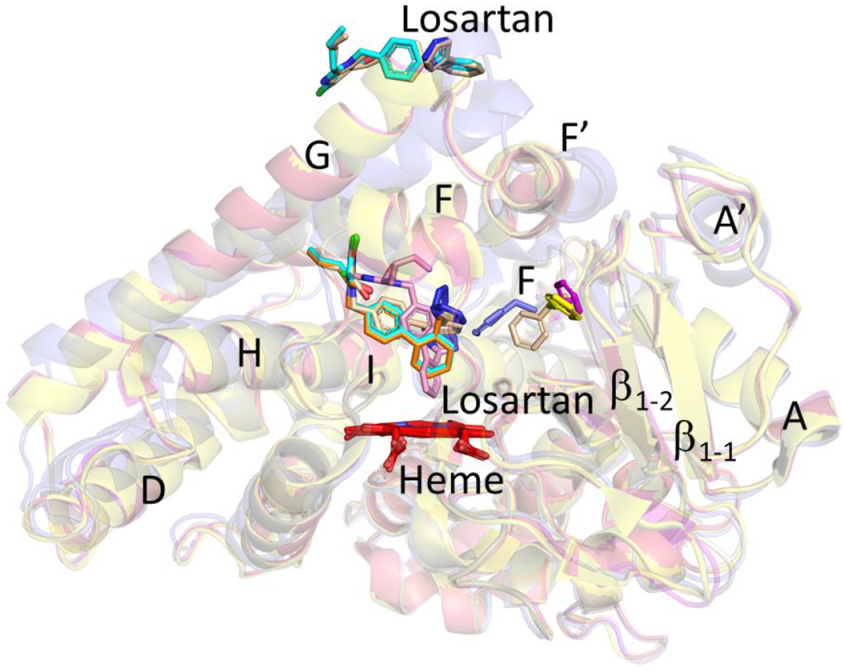
2.1. Structure Determination and Comparison of CYP2C9*8 in Complex with Losartan
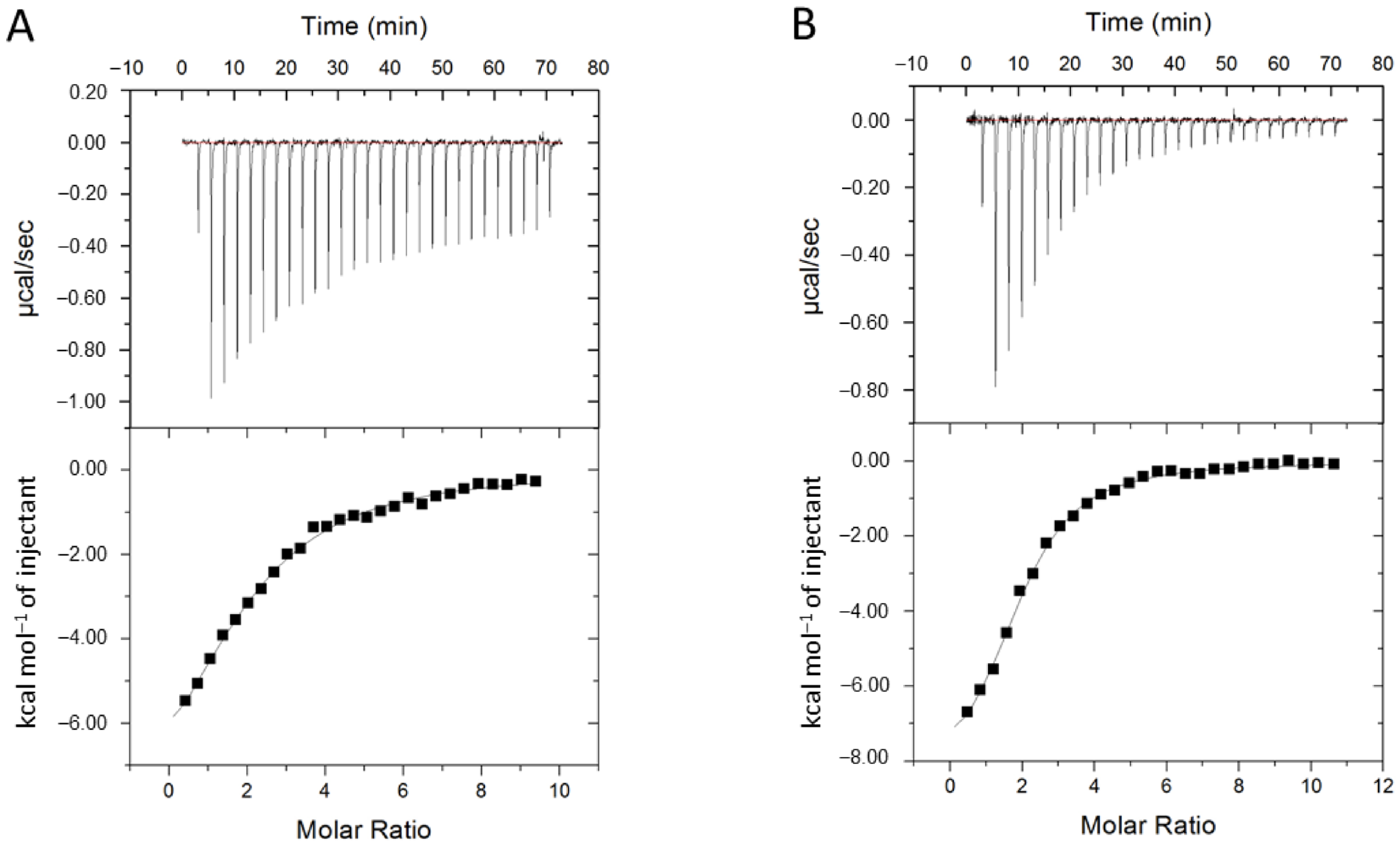
2.2. Functional Characterization of Losartan Binding to CYP2C9 WT and Variants
3. Materials and Methods
3.1. Materials
3.2. Protein Expression and Purification
3.3. Crystallization
3.4. Structure Determination and Refinement
3.5. NADPH Consumption Assay
3.6. Thermal Stability Assays
3.7. Isothermal Titration Calorimetry
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CYP | Cytochrome P450 |
| ALA | 5-aminolevulinic acid hydrochloride |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| PDB | Protein Data Bank |
| EDTA | Ethylenediaminetetraacetic acid |
| IPTG | Isopropyl β-F-1-thiogalactopyranoside |
| CYMAL-5 | 5-Cymal-hexylpentyl-β-D-maltoside |
| CHAPS | (2-2-Cholamidopropyldimethylammonio)-1-propanesulfonate |
| TCEP | Tris (2-carboxyethyl) phosphine hydrochloride |
| RMSD | Root mean square deviation |
| PMSF | Phenylmethylsulfonyl fluoride |
| ITC | Isothermal titration calorimetry. |
References
- Gray, I.C.; Nobile, C.; Muresu, R.; Ford, S.; Spurr, N.K. A 2.4-megabase physical map spanning the CYP2C gene cluster on chromosome 10q24. Genomics 1995, 28, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Birkett, D.J. Cytochrome P4502C9: An enzyme of major importance in human drug metabolism. Br. J. Clin. Pharmacol. 1998, 45, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, T.H.; Jung, F.; Griffin, K.J.; Wester, M.; Raucy, J.L.; Kemper, B.; Bornheim, L.M.; Hassett, C.; Omiecinski, C.J.; Johnson, E.F. A universal approach to the expression of human and rabbit cytochrome P450s of the 2C subfamily in Escherichia coli. Arch. Biochem. Biophys. 1995, 323, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Rettie, A.E.; Korzekwa, K.R.; Kunze, K.L.; Lawrence, R.F.; Eddy, A.C.; Aoyama, T.; Gelboin, H.V.; Gonzalez, F.J.; Trager, W.F. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: A role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem. Res. Toxicol. 1992, 5, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Pieper, J.A.; Frye, R.F.; Hinderliter, A.L.; Blaisdell, J.A.; Goldstein, J.A. Tolbutamide, flurbiprofen, and losartan as probes of CYP2C9 activity in humans. J. Clin. Pharmacol. 2003, 43, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Yasumori, T.; Chen, L.S.; Li, Q.H.; Ueda, M.; Tsuzuki, T.; Goldstein, J.A.; Kato, R.; Yamazoe, Y. Human CYP2C-mediated stereoselective phenytoin hydroxylation in Japanese: Difference in chiral preference of CYP2C9 and CYP2C19. Biochem. Pharmacol. 1999, 57, 1297–1303. [Google Scholar] [CrossRef]
- Klose, T.S.; Ibeanu, G.C.; Ghanayem, B.I.; Pedersen, L.G.; Li, L.; Hall, S.D.; Goldstein, J.A. Identification of residues 286 and 289 as critical for conferring substrate specificity of human CYP2C9 for diclofenac and ibuprofen. Arch. Biochem. Biophys. 1998, 357, 240–248. [Google Scholar] [CrossRef]
- Daly, A.K.; Rettie, A.E.; Fowler, D.M.; Miners, J.O. Pharmacogenomics of CYP2C9: Functional and Clinical Considerations. J. Pers. Med. 2017, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Rettie, A.E.; Jones, J.P. Clinical and toxicological relevance of CYP2C9: Drug-drug interactions and pharmacogenetics. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 477–494. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.R.; Pieper, J.A.; Hinderliter, A.L.; Blaisdell, J.A.; Goldstein, J.A. Losartan and E3174 pharmacokinetics in cytochrome P450 2C9*1/*1, *1/*2, and *1/*3 individuals. Pharmacotherapy 2003, 23, 720–725. [Google Scholar] [CrossRef]
- Yamazaki, H.; Inoue, K.; Chiba, K.; Ozawa, N.; Kawai, T.; Suzuki, Y.; Goldstein, J.A.; Guengerich, F.P.; Shimada, T. Comparative studies on the catalytic roles of cytochrome P450 2C9 and its Cys- and Leu-variants in the oxidation of warfarin, flurbiprofen, and diclofenac by human liver microsomes. Biochem. Pharmacol. 1998, 56, 243–251. [Google Scholar] [CrossRef]
- Allabi, A.C.; Gala, J.L.; Horsmans, Y. CYP2C9, CYP2C19, ABCB1 (MDR1) genetic polymorphisms and phenytoin metabolism in a Black Beninese population. Pharm. Genom. 2005, 15, 779–786. [Google Scholar] [CrossRef]
- Kidd, R.S.; Curry, T.B.; Gallagher, S.; Edeki, T.; Blaisdell, J.; Goldstein, J.A. Identification of a null allele of CYP2C9 in an African-American exhibiting toxicity to phenytoin. Pharmacogenetics 2001, 11, 803–808. [Google Scholar] [CrossRef]
- Allabi, A.C.; Gala, J.L.; Horsmans, Y.; Babaoglu, M.O.; Bozkurt, A.; Heusterspreute, M.; Yasar, U. Functional impact of CYP2C95, CYP2C96, CYP2C98, and CYP2C911 in vivo among black Africans. Clin. Pharmacol. Ther. 2004, 76, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Blaisdell, J.; Jorge-Nebert, L.F.; Coulter, S.; Ferguson, S.S.; Lee, S.J.; Chanas, B.; Xi, T.; Mohrenweiser, H.; Ghanayem, B.; Goldstein, J.A. Discovery of new potentially defective alleles of human CYP2C9. Pharmacogenetics 2004, 14, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Langaee, T.Y.; Wong, S.S.; Cavallari, L.H. Pyrosequencing of the CYP2C9-1766T>C polymorphism as a means of detecting the CYP2C9*8 allele. Pharmacogenomics 2014, 15, 1717–1722. [Google Scholar] [CrossRef]
- Cavallari, L.H.; Vaynshteyn, D.; Freeman, K.M.; Wang, D.; Perera, M.A.; Takahashi, H.; Drozda, K.; Patel, S.R.; Jeong, H. CYP2C9 promoter region single-nucleotide polymorphisms linked to the R150H polymorphism are functional suggesting their role in CYP2C9*8-mediated effects. Pharm. Genom. 2013, 23, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.A.; Jaremko, M.; Lubitz, S.A.; Kornreich, R.; Halperin, J.L.; Desnick, R.J. CYP2C9*8 is prevalent among African-Americans: Implications for pharmacogenetic dosing. Pharmacogenomics 2009, 10, 1243–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jeong, H.; Takahashi, H.; Drozda, K.; Patel, S.R.; Shapiro, N.L.; Nutescu, E.A.; Cavallari, L.H. Decreased warfarin clearance associated with the CYP2C9 R150H (*8) polymorphism. Clin. Pharmacol. Ther. 2012, 91, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, V.M.; Cavallari, L.H.; Del Tredici, A.L.; Hachad, H.; Ji, Y.; Moyer, A.M.; Scott, S.A.; Whirl-Carrillo, M.; Weck, K.E. Recommendations for Clinical CYP2C9 Genotyping Allele Selection: A Joint Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2019, 21, 746–755. [Google Scholar] [CrossRef]
- Gyulkhandanyan, A.; Rezaie, A.R.; Roumenina, L.; Lagarde, N.; Fremeaux-Bacchi, V.; Miteva, M.A.; Villoutreix, B.O. Analysis of protein missense alterations by combining sequence- and structure-based methods. Mol. Genet. Genomic. Med. 2020, 8, e1166. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.C.; Ingelman-Sundberg, M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: A peer-reviewed database of CYP variants and their associated effects. Hum. Genom. 2010, 4, 278–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.W.; Goldberg, M.R.; McCrea, J.B.; Lu, H.; Furtek, C.I.; Bjornsson, T.D. Pharmacokinetics of losartan, an angiotensin II receptor antagonist, and its active metabolite EXP3174 in humans. Clin. Pharmacol. Ther. 1995, 58, 641–649. [Google Scholar] [CrossRef]
- Sica, D.A.; Lo, M.W.; Shaw, W.C.; Keane, W.F.; Gehr, T.W.; Halstenson, C.E.; Lipschutz, K.; Furtek, C.I.; Ritter, M.A.; Shahinfar, S. The pharmacokinetics of losartan in renal insufficiency. J. Hypertens. Suppl. 1995, 13, S49–S52. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, K.; Adachi, M.; Matsuzawa, Y.; Zhang, Q.; Kuroki, R.; Saito, Y.; Shah, M.B. Structural Basis of Single-Nucleotide Polymorphisms in Cytochrome P450 2C9. Biochemistry 2017, 56, 5476–5480. [Google Scholar] [CrossRef]
- Schoch, G.A.; Yano, J.K.; Wester, M.R.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J. Biol. Chem. 2004, 279, 9497–9503. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef]
- Parikh, S.J.; Evans, C.M.; Obi, J.O.; Zhang, Q.; Maekawa, K.; Glass, K.C.; Shah, M.B. Structure of Cytochrome P450 2C9*2 in Complex with Losartan: Insights into the Effect of Genetic Polymorphism. Mol. Pharmacol. 2020, 98, 529–539. [Google Scholar] [CrossRef]
- Bumpus, N.N.; Hollenberg, P.F. Cross-linking of human cytochrome P450 2B6 to NADPH-cytochrome P450 reductase: Identification of a potential site of interaction. J. Inorg. Biochem. 2010, 104, 485–488. [Google Scholar] [CrossRef] [Green Version]
- Whirl-Carrillo, M.; Huddart, R.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Whaley, R.; Klein, T.E. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2021, 110, 563–572. [Google Scholar] [CrossRef]
- Lee, S.C.; Bennett, B.C.; Hong, W.X.; Fu, Y.; Baker, K.A.; Marcoux, J.; Robinson, C.V.; Ward, A.B.; Halpert, J.R.; Stevens, R.C.; et al. Steroid-based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E1203–E1211. [Google Scholar] [CrossRef] [Green Version]
- Soltis, S.M.; Cohen, A.E.; Deacon, A.; Eriksson, T.; Gonzalez, A.; McPhillips, S.; Chui, H.; Dunten, P.; Hollenbeck, M.; Mathews, I.; et al. New paradigm for macromolecular crystallography experiments at SSRL: Automated crystal screening and remote data collection. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 1210–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-Ice and the Distributed Control System: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. [Google Scholar] [CrossRef]
- Cohen, A.E.; Ellis, P.J.; Miller, M.D.; Deacon, A.M.; Phizackerley, R.P. An automated system to mount cryo-cooled protein crystals on a synchrotron beam line, using compact sample cassettes and a small-scale robot. J. Appl. Crystallogr. 2002, 35, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Leslie, A.G. Integration of macromolecular diffraction data. Acta. Crystallogr. D Biol. Crystallogr. 1999, 55, 1696–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta. Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Potterton, E.; Briggs, P.; Turkenburg, M.; Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1131–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Vagin, A.A.; Young, P.; Murshudov, G.N. BALBES: A molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bart, A.G.; Scott, E.E. Structural and functional effects of cytochrome b5 interactions with human cytochrome P450 enzymes. J. Biol. Chem. 2017, 292, 20818–20833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilderman, P.R.; Shah, M.B.; Jang, H.H.; Stout, C.D.; Halpert, J.R. Structural and thermodynamic basis of (+)-alpha-pinene binding to human cytochrome P450 2B6. J. Am. Chem. Soc. 2013, 135, 10433–10440. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytochrome P450 | Amino Acid Change |
|---|---|
| CYP2C9 WT (wild-type) | None |
| CYP2C9*2 | R144C |
| CYP2C9*3 | I359L |
| CYP2C9*5 | D360E |
| CYP2C9*6 | Frameshift |
| CYP2C9*8 | R150H |
| CYP2C9*11 | R335W |
| CYP2C9*27 | R150L |
| CYP2C9*30 | A477T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parikh, S.J.; Kamat, S.; Phillips, M.; Boyson, S.P.; Yarbrough, T.; Davie, D.; Zhang, Q.; Glass, K.C.; Shah, M.B. Insights into the Genetic Variations of Human Cytochrome P450 2C9: Structural Analysis, Characterization and Comparison. Int. J. Mol. Sci. 2021, 22, 10206. https://doi.org/10.3390/ijms221910206
Parikh SJ, Kamat S, Phillips M, Boyson SP, Yarbrough T, Davie D, Zhang Q, Glass KC, Shah MB. Insights into the Genetic Variations of Human Cytochrome P450 2C9: Structural Analysis, Characterization and Comparison. International Journal of Molecular Sciences. 2021; 22(19):10206. https://doi.org/10.3390/ijms221910206
Chicago/Turabian StyleParikh, Sonia J., Sumit Kamat, Margaret Phillips, Samuel P. Boyson, Thomas Yarbrough, Dylan Davie, Qinghai Zhang, Karen C. Glass, and Manish B. Shah. 2021. "Insights into the Genetic Variations of Human Cytochrome P450 2C9: Structural Analysis, Characterization and Comparison" International Journal of Molecular Sciences 22, no. 19: 10206. https://doi.org/10.3390/ijms221910206
APA StyleParikh, S. J., Kamat, S., Phillips, M., Boyson, S. P., Yarbrough, T., Davie, D., Zhang, Q., Glass, K. C., & Shah, M. B. (2021). Insights into the Genetic Variations of Human Cytochrome P450 2C9: Structural Analysis, Characterization and Comparison. International Journal of Molecular Sciences, 22(19), 10206. https://doi.org/10.3390/ijms221910206