
A Novel Mutation in GP1BB Reveals the Role of the Cytoplasmic Domain of GPIbβ in the Pathophysiology of Bernard-Soulier Syndrome and GPIb-IX Complex Assembly

 ,
,
Abstract
:1. Introduction
2. Results
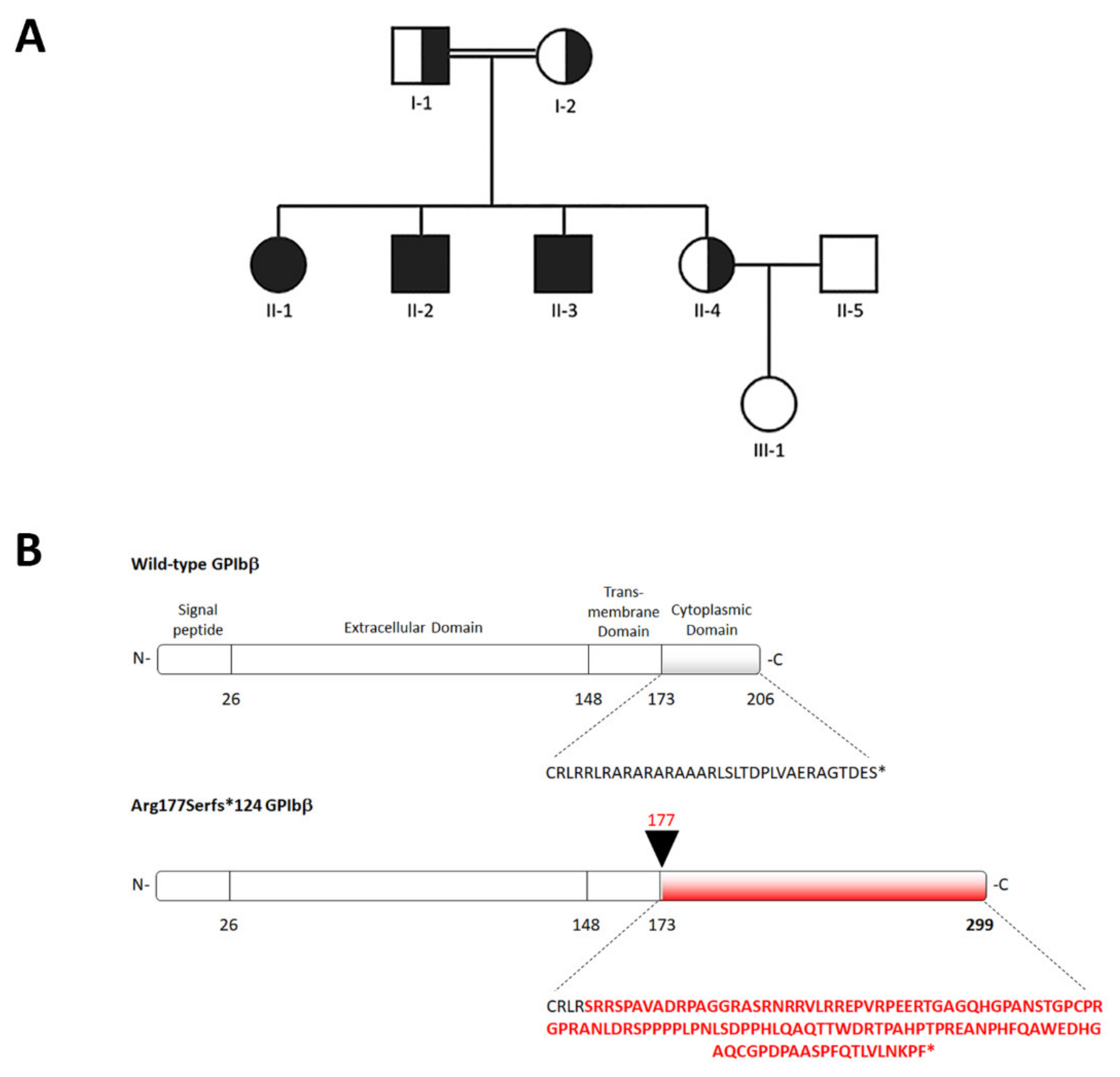
2.1. Clinical Picture of the Family Members
2.2. Identification of a Novel GP1BB Variant
2.3. In Vitro Platelet Aggregation
2.4. Flow Cytometry of Platelet Glycoproteins
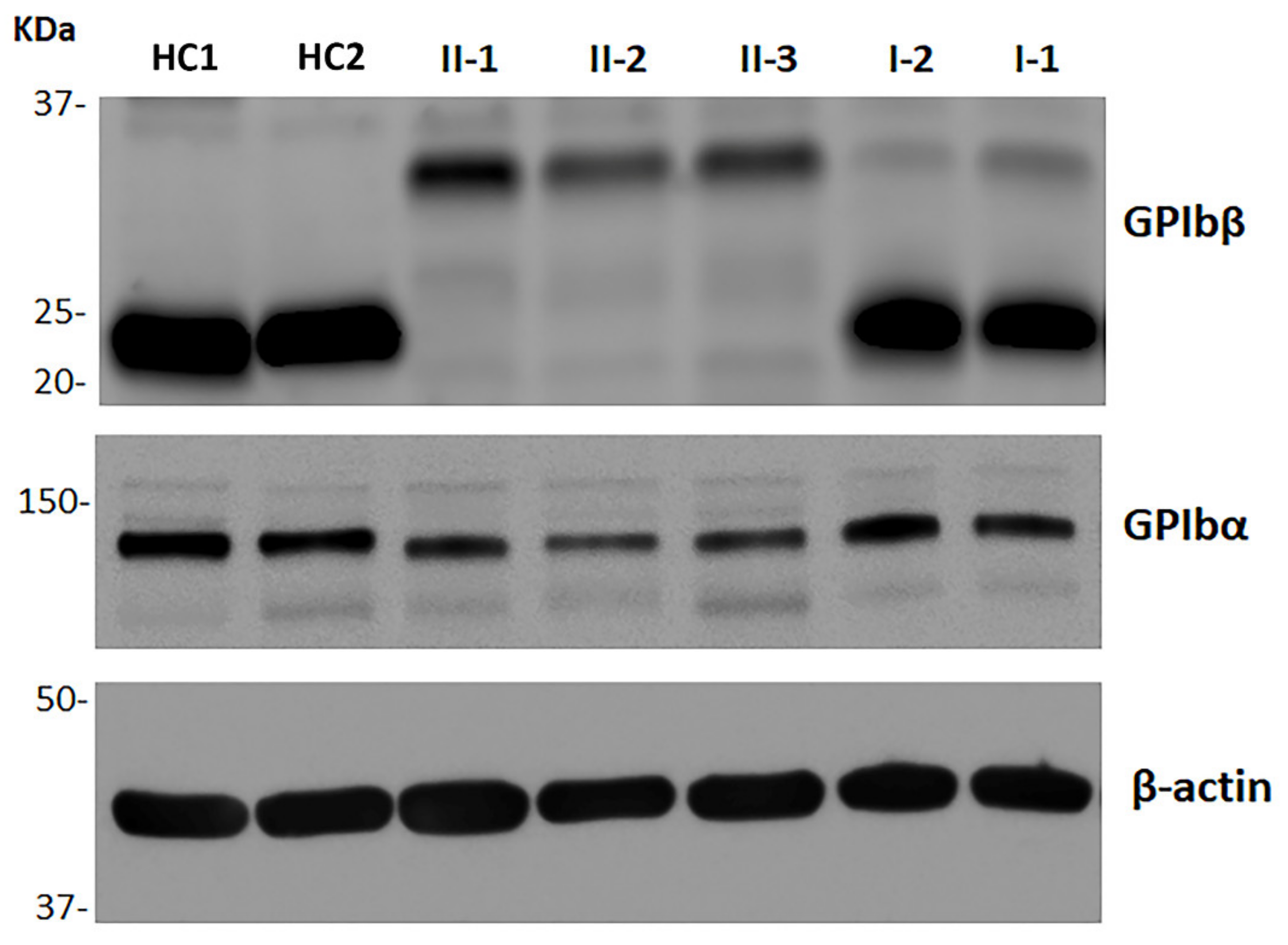
2.5. Immunoblotting Analysis of GPIbβ and GPIbα
2.6. Identification of a Second Family Carrying the c.528_550del Variant
3. Discussion
4. Patients and Methods
4.1. Patients
4.2. Platelet Aggregation
4.3. Flow Cytometry of Platelet Surface Glycoproteins
4.4. Immunoblotting Assay
4.5. Mutation Screening
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andrews, R.K.; Berndt, M.C. Bernard-Soulier Syndrome: An update. Semin. Thromb. Hemost. 2013, 39, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Savoia, A.; Kunishima, S.; De Rocco, D.; Zieger, B.; Rand, M.L.; Pujol-Moix, N.; Caliskan, U.; Tokgoz, H.; Pecci, A.; Noris, P.; et al. Spectrum of the mutations in Bernard-Soulier Syndrome. Hum. Mutat. 2014, 35, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Balduini, C.L. Inherited thrombocytopenias: An updated guide for clinicians. Blood Rev. 2021, 48, 100784. [Google Scholar] [CrossRef] [PubMed]
- Quach, M.E.; Li, R. Structure-function of platelet glycoprotein Ib-IX. J. Thromb. Haemost. 2020, 18, 3131–3141. [Google Scholar] [CrossRef]
- Savoia, A.; Pastore, A.; de Rocco, D.; Civaschi, E.; di Stazio, M.; Bottega, R.; Melazzini, F.; Bozzi, V.; Pecci, A.; Magrin, S.; et al. Clinical and genetic aspects of Bernard-Soulier Syndrome: Searching for genotype/phenotype correlations. Haematologica 2011, 96, 417–423. [Google Scholar] [CrossRef]
- Li, R.; Emsley, J. The organizing principle of the platelet glycoprotein Ib-IX-V complex. J. Thromb. Haemost. 2013, 11, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.A.; Leung, B.; Reynolds, C.C.; Li, C.Q.; Fox, J.E.B. Efficient plasma membrane expression of a functional platelet glycoprotein Ib-IX complex requires the presence of its three subunits. J. Biol. Chem. 1992, 267, 12851–12859. [Google Scholar] [CrossRef]
- Ulsemer, P.; Strassel, C.; Baas, M.J.; Salamero, J.; Chasserot-Golaz, S.; Cazenave, J.P.; De la Salle, C.; Lanza, F. Biosynthesis and intracellular post-translational processing of normal and mutant platelet glycoprotein GPIb-IX. Biochem. J. 2001, 358, 295–303. [Google Scholar] [CrossRef]
- Yamamoto, N.; Akamatsu, N.; Sakuraba, H.; Matsuno, K.; Hosoya, R.; Nogami, H.; Kasahara, K.; Mitsuyama, S.; Arai, M. Novel Bernard-Soulier Syndrome variants caused by compound heterozygous mutations (Case I) or a cytoplasmic tail truncation (Case II) of GPIbα. Thromb. Res. 2013, 131, e160–e167. [Google Scholar] [CrossRef]
- Mo, X.; Luo, S.Z.; López, J.A.; Li, R. Juxtamembrane basic residues in glycoprotein Ibβ cytoplasmic domain are required for assembly and surface expression of glycoprotein Ib-IX complex. FEBS Lett. 2008, 582, 3270–3274. [Google Scholar] [CrossRef] [Green Version]
- Strassel, C.; Nonne, C.; Eckly, A.; David, T.; Leon, C.; Freund, M.; Cazenave, J.P.; Gachet, C.; Lanza, F. Decreased thrombotic tendency in mouse models of the Bernard-Soulier Syndrome. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Klersy, C.; Gresele, P.; Giona, F.; Giordano, P.; Minuz, P.; Loffredo, G.; Pecci, A.; Melazzini, F.; Civaschi, E.; et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: A multicentric, real life study. Br. J. Haematol. 2013, 162, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Biino, G.; Pecci, A.; Civaschi, E.; Savoia, A.; Seri, M.; Melazzini, F.; Loffredo, G.; Russo, G.; Bozzi, V.; et al. Platelet diameters in inherited thrombocytopenias: Analysis of 376 Patients with all known disorders. Blood 2014, 124, e4–e10. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: A communication from the platelet physiology SSC. J. Thromb. Haemost. 2020, 18, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Sumitha, E.; Jayandharan, G.R.; David, S.; Jacob, R.R.; Sankari Devi, G.; Bargavi, B.; Shenbagapriya, S.; Nair, S.C.; Abraham, A.; George, B.; et al. Molecular basis of Bernard-Soulier Syndrome in 27 patients from India. J. Thromb. Haemost. 2011, 9, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Bragadottir, G.; Birgisdottir, E.R.; Gudmundsdottir, B.R.; Hilmarsdottir, B.; Vidarsson, B.; Magnusson, M.K.; Larsen, O.H.; Sorensen, B.; Ingerslev, J.; Onundarson, P.T. Clinical phenotype in heterozygote and biallelic Bernard-Soulier Syndrome-A Case Control Study. Am. J. Hematol. 2015, 90, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.; De Thoré, C.; Randrianaivo-Ranjatoelina, H.; Baas, M.J.; Jacquemont, M.L.; Dreyfus, M.; Lavenu-Bombled, C.; Li, R.; Gachet, C.; Dupuis, A.; et al. High prevalence of the natural Asn89Asp mutation in the GP1BB gene associated with Bernard–Soulier Syndrome in French patients from the genetic isolate of Reunion Island. Br. J. Haematol. 2020, 189, e67–e71. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Gan, C.; Zhang, X.; Liu, L.; Zakas, P.M.; Doering, C.B.; Mo, X.; Li, R. Unaccompanied mechanosensory domain mediates low expression of glycoprotein Ibα: Implications for Bernard-Soulier Syndrome. J. Thromb. Haemost. 2020, 18, 510–517. [Google Scholar] [CrossRef]
- McEwan, P.A.; Yang, W.; Carr, K.H.; Mo, X.; Zheng, X.; Li, R.; Emsley, J. Quaternary organization of GPIb-IX complex and insights into Bernard-Soulier Syndrome revealed by the structures of GPIβ and a GPIβ/GPIX chimera. Blood 2011, 118, 5292–5301. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.F.; Li, C.Q.; Sae-Tung, G.; Hyun, W.; Afshar-Kharghan, V.; López, J.A. The cytoplasmic domain of glycoprotein (GP) Ibα constrains the lateral diffusion of the GP Ib-IX complex and modulates von Willebrand factor binding. Biochemistry 1997, 36, 12421–12427. [Google Scholar] [CrossRef]
- Mistry, N.; Cranmer, S.L.; Yuan, Y.; Mangin, P.; Dopheide, S.M.; Harper, I.; Giuliano, S.; Dunstan, D.E.; Lanza, F.; Salem, H.H.; et al. Cytoskeletal regulation of the platelet glycoprotein Ib/V/IX-von Willebrand Factor interaction. Blood 2000, 96, 3480–3489. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ruggeri, Z.M.; Du, X. 14-3-3 Proteins in Platelet Biology and Glycoprotein Ib-IX Signaling. Blood 2018, 131, 2436–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, K.; Bodnar, R.; Berndt, M.C.; Du, X. A Critical Role for 14-3-3ζ Protein in Regulating the VWF Binding Function of Platelet Glycoprotein Ib-IX and Its Therapeutic Implications. Blood 2005, 106, 1975–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, R.J.; Xi, X.; Li, Z.; Berndt, M.C.; Du, X. Regulation of Glycoprotein Ib-IX-von Willebrand Factor Interaction by CAMP-Dependent Protein Kinase-Mediated Phosphorylation at Ser166 of Glycoprotein Ibβ. J. Biol. Chem. 2002, 277, 47080–47087. [Google Scholar] [CrossRef] [Green Version]
- David, T.; Strassel, C.; Eckly, A.; Cazenave, J.P.; Gachet, C.; Lanza, F. The Platelet Glycoprotein GPIbβ Intracellular Domain Participates in von Willebrand Factor Induced-Filopodia Formation Independently of the Ser 166 Phosphorylation Site. J. Thromb. Haemost. 2010, 8, 1077–1087. [Google Scholar] [CrossRef]
- Sivapalaratnam, S.; Westbury, S.K.; Stephens, J.C.; Greene, D.; Downes, K.; Kelly, A.M.; Lentaigne, C.; Astle, W.J.; Huizinga, E.G.; Nurden, P.; et al. Rare variants in GP1BB are responsible for autosomal dominant macrothrombocytopenia. Blood 2017, 129, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunstan-Harrison, C.; Morison, I.M.; Ledgerwood, E.C. A novel frameshift GP1BB mutation causes autosomal dominant macrothrombocytopenia with decreased VWF receptor expression but normal platelet aggregation. Platelets 2021, 1–4. [Google Scholar] [CrossRef]
- Noris, P.; Perrotta, S.; Bottega, R.; Pecci, A.; Melazzini, F.; Civaschi, E.; Russo, S.; Magrin, S.; Loffredo, G.; Salvo, V.D.; et al. Clinical and laboratory features of 103 patients from 42 italian families with inherited thrombocytopenia derived from the monoallelic Ala156Val mutation of Gpibα (Bolzano Mutation). Haematologica 2012, 97, 82–88. [Google Scholar] [CrossRef]
- Noris, P.; Guidetti, G.F.; Conti, V.; Ceresa, I.F.; Di Dumpo, M.; Pecci, A.; Torti, M.; Savoia, A.; Balduini, C.L. Autosomal dominant thrombocytopenias with reduced expression of glycoprotein Ia. Thromb. Haemost. 2006, 95, 483–489. [Google Scholar] [CrossRef]
- Barozzi, S.; Di Buduo, C.A.; Marconi, C.; Bozzi, V.; Seri, M.; Romano, F.; Balduini, A.; Pecci, A. Pathogenetic and clinical study of a patient with thrombocytopenia due to the p.E527K gain-of-function variant of SRC. Haematologica 2021, 106, 918–922. [Google Scholar] [CrossRef]
- Necchi, V.; Balduini, A.; Noris, P.; Barozzi, S.; Sommi, P.; di Buduo, C.; Balduini, C.L.; Solcia, E.; Pecci, A. Ubiquitin/Proteasome-Rich Particulate Cytoplasmic Structures (PaCSs) in the platelets and megakaryocytes of ANKRD26-related thrombocytopenia. Thromb. Haemost. 2013, 109, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Faleschini, M.; Melazzini, F.; Marconi, C.; Giangregorio, T.; Pippucci, T.; Cigalini, E.; Pecci, A.; Bottega, R.; Ramenghi, U.; Siitonen, T.; et al. ACTN1 mutations lead to a benign form of platelet macrocytosis not always associated with thrombocytopenia. Br. J. Haematol. 2018, 183, 276–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicchia, E.; Giordano, P.; Greco, C.; De Rocco, D.; Savoia, A. Molecular diagnosis of thrombocytopenia-absent radius syndrome using next-generation sequencing. Int. J. Lab. Hematol. 2016, 38, 412–418. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Subject | Gender/ Age (Years) | Automated Platelet Count, × 109/L 1 | Microscopic Platelet Count, × 109/L 2 | MPV, Fl 3 | Mean Platelet Diameter, µm 4 | Giant Platelets 5 | ISTH BAT Score 6 | Bleeding Symptoms |
|---|---|---|---|---|---|---|---|---|
| I-1 | M/59 | 107 | 129 | 14.1 | 3.07 | No | 0 | None |
| I-2 | F/55 | 175 | 197 | 14.5 | 2.95 | No | 0 | None |
| II-1 | F/34 | 31 | 56 | 20.4 | 4.05 | Yes | 1 | Mild menorrhagia |
| II-2 | M/33 | 22 | 45 | 18.9 | 4.45 | Yes | 0 | None |
| II-3 | M/31 | 17 | 55 | 20.2 | 4.61 | Yes | 0 | None |
| II-4 | F/30 | 103 | 110 | 14.5 | 3.01 | No | 0 | None |
| Subject | GPIbα (SZ2), % of Controls | GPIbα (MB45), % of Controls | GPIb-IX (SZ1), % of Controls | GPIIb (P2) % of Controls | GPIIIa (VIPL2), % of Controls |
|---|---|---|---|---|---|
| II-1 | 36.7 ± 2.1 | 31.1 ± 2.2 | 31.5 ± 2.9 | 231.8 ± 3.1 | 205.2 ± 4.3 |
| II-2 | 37.2 ± 1.1 | 29.3 ± 1.4 | 28.9 ± 1.3 | 252.2 ± 14.4 | 215.3 ± 10.2 |
| II-3 | 29.7 ± 3.2 | 31.4 ± 1.5 | 28.5 ± 1.8 | 267.3 ± 8.7 | 199.5 ± 12 |
| I-1 | 62.8 ± 2.9 | 87.3 ± 3.9 | 60.2 ± 4.3 | 155.6 ± 10.4 | 161.7 ± 14.2 |
| I-2 | 63.7 ± 2.5 | 69.7 ± 5.4 | 67.7 ± 5.6 | 139.0 ± 8.5 | 145.3 ± 7.3 |
| II-4 | 70.1 ± 6.9 | 65.3 ± 4.3 | 67.3 ± 3.7 | 145.5 ± 3.8 | 155.3 ± 6.9 |
| Scheme | GPIbβ, % of Controls | GPIbα, % of Controls | |
|---|---|---|---|
| 22 kDa | 32 kDa | ||
| II-1 | 0 | 27.1 ± 0.3 | 58.4 ± 7.1 |
| II-2 | 0 | 27.1 ± 2.6 | 49.9 ± 7.5 |
| II-3 | 0 | 29.7 ± 2.5 | 55.2 ± 6.3 |
| I-1 | 67.4 ± 7.5 | 7.5 ± 1.7 | 73.5 ± 9.5 |
| I-2 | 66.9 ± 3.9 | 3.4 ± 0.5 | 82.0 ± 9.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barozzi, S.; Bozzi, V.; De Rocco, D.; Giangregorio, T.; Noris, P.; Savoia, A.; Pecci, A. A Novel Mutation in GP1BB Reveals the Role of the Cytoplasmic Domain of GPIbβ in the Pathophysiology of Bernard-Soulier Syndrome and GPIb-IX Complex Assembly. Int. J. Mol. Sci. 2021, 22, 10190. https://doi.org/10.3390/ijms221910190
Barozzi S, Bozzi V, De Rocco D, Giangregorio T, Noris P, Savoia A, Pecci A. A Novel Mutation in GP1BB Reveals the Role of the Cytoplasmic Domain of GPIbβ in the Pathophysiology of Bernard-Soulier Syndrome and GPIb-IX Complex Assembly. International Journal of Molecular Sciences. 2021; 22(19):10190. https://doi.org/10.3390/ijms221910190
Chicago/Turabian StyleBarozzi, Serena, Valeria Bozzi, Daniela De Rocco, Tania Giangregorio, Patrizia Noris, Anna Savoia, and Alessandro Pecci. 2021. "A Novel Mutation in GP1BB Reveals the Role of the Cytoplasmic Domain of GPIbβ in the Pathophysiology of Bernard-Soulier Syndrome and GPIb-IX Complex Assembly" International Journal of Molecular Sciences 22, no. 19: 10190. https://doi.org/10.3390/ijms221910190
APA StyleBarozzi, S., Bozzi, V., De Rocco, D., Giangregorio, T., Noris, P., Savoia, A., & Pecci, A. (2021). A Novel Mutation in GP1BB Reveals the Role of the Cytoplasmic Domain of GPIbβ in the Pathophysiology of Bernard-Soulier Syndrome and GPIb-IX Complex Assembly. International Journal of Molecular Sciences, 22(19), 10190. https://doi.org/10.3390/ijms221910190