
Mass Spectrometric and Bio-Computational Binding Strength Analysis of Multiply Charged RNAse S Gas-Phase Complexes Obtained by Electrospray Ionization from Varying In-Solution Equilibrium Conditions

 , , ,
, , ,
Abstract
1. Introduction
2. Results
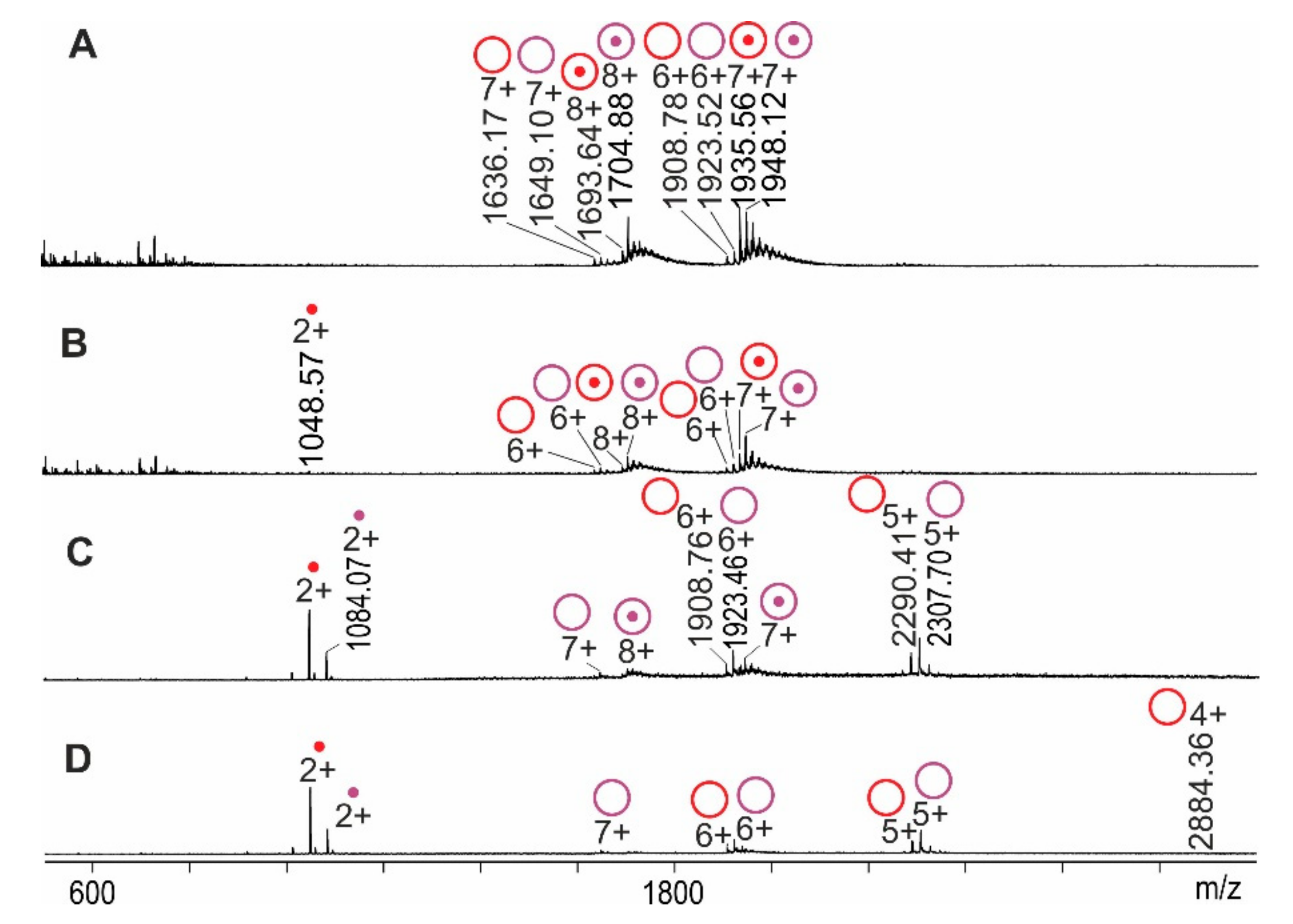
2.1. RNAse S Gas Phase Dissociation Studies upon Electrospraying Protein Solutions with Different pHs
2.2. RNAse S Gas Phase Dissociation Studies upon Electrospraying Protein Solutions with Different Content of Organic Co-Solvent
2.3. Bio-Computational Studies of RNAse S Conformations Forming during Electrospray Desolvation or in Bulk Solution
2.4. Bio-Computational Studies on the Dependence of Complex Binding Strength on S-Peptide Amino Acid Residue Position
3. Discussion
4. Conclusions and Outlook
5. Material and Methods
5.1. Preparation of NanoESI-MS Compatible RNAse S Solutions
5.2. Nanospray Needle Preparation
5.3. Off-Line NanoESI-MS Instrument Settings and Data Acquisition Conditions
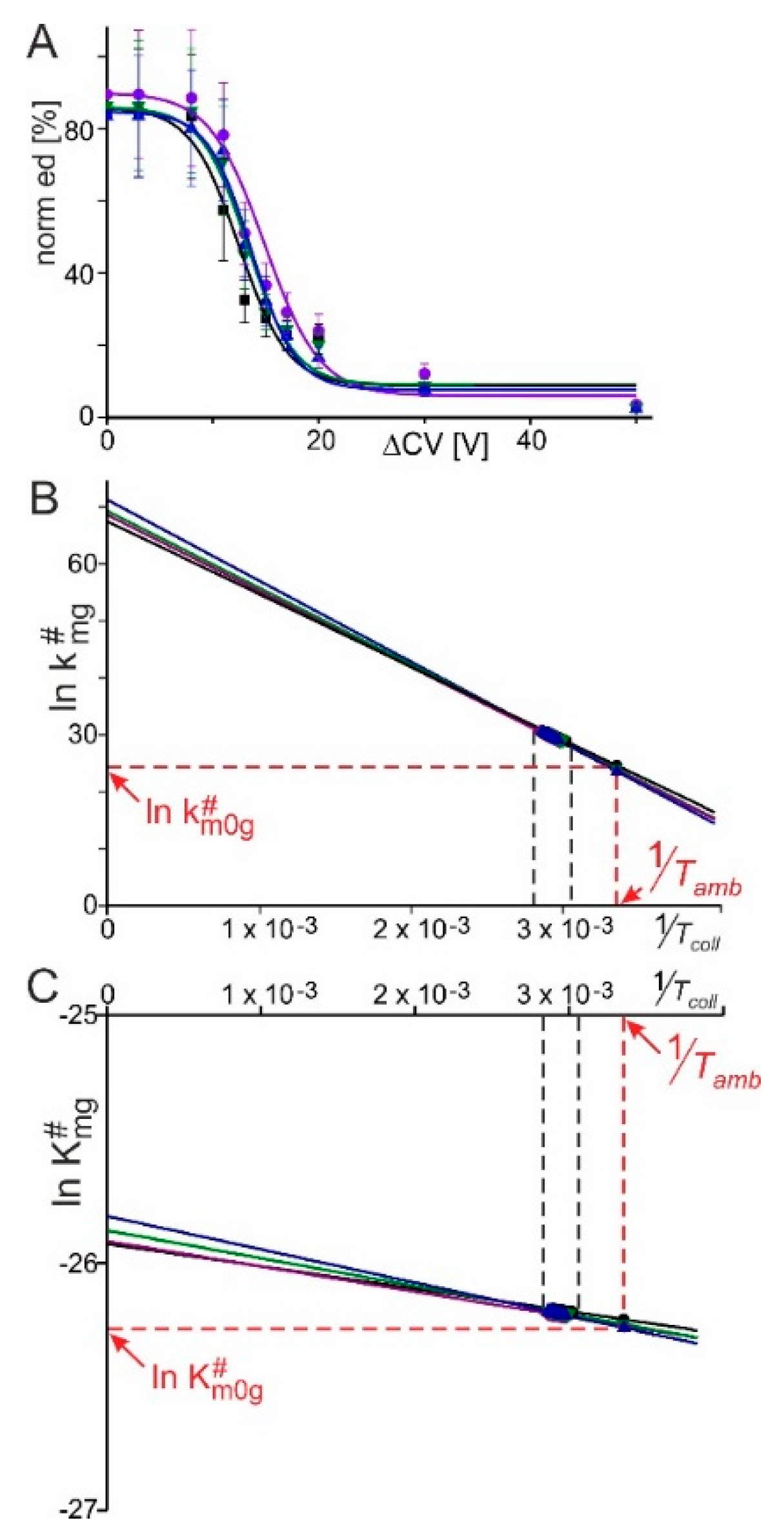
5.4. Mass Spectral Data Analysis and Calculation of Kinetic and Thermodynamic Values of RNAse S
- change of Gibbs energy of activation to transform educt ions into product ions
- heat change between educt ions and product ions
- change of disorder of educt ions and product ions(#: apparent; m: mean charge state; g: gas phase)
- change of Gibbs energy of activation
- gas constant, 8.314 J/mol·K
- educt and/or product temperature during collision in the collision cell
- gas phase thermodynamic equilibrium dissociation constant
- heat change between educt ions and product ions(#: apparent; m: mean charge state; 0: no external energy contribution; g: gas phase)
- absolute ambient temperature, 298 K.
5.5. In-Silico Simulation Methodology for Estimating RNAse S Conformational Changes upon Desolvation
5.6. Visualization of RNAse S Structure and Calculation of Inter-Residue Contacts
5.7. Calculation of Binding Energy Changes of S-Peptide Amino Acid Residue Substitutions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FAB | fast atom bombardment |
| PD | plasma desorption |
| LD | laser desorption |
| MALDI | matrix-assisted laser desorption/ionization |
| ESI | electrospray ionization |
| ITEM | intact transition epitope mapping |
| ITEM-ONE | ITEM- One-step Non-covalent force Exploration |
| ITEM-TWO | ITEM- Thermodynamic Weak-force Order |
| ITEM-THREE | ITEM- Targeted High-Energy Rupture of Extracted Epitopes |
| CID | collision induced dissociation |
| TFE | trifluoroethanol |
| TIC | total ion current |
| RNAse S | ribonuclease S |
| ASA | accessible surface area |
| PDB | protein database |
| PRIDE | proteomics identifications database |
| Q-ToF | quadrupole time-of-flight |
| ΔCV | collisional voltage difference |
| m/z | mass-to-charge ratio |
References
- Barber, M.; Bordoli, R.S.; Sedgwick, R.D.; Tyler, A.N. Fast atom bombardment of solids (F.A.B.): A new ion source for mass spectrometry. J. Chem. Soc. Chem. Commun. 1981, 325–327. [Google Scholar] [CrossRef]
- Torgerson, D.; Skowronski, R.; Macfarlane, R. New approach to the mass spectroscopy of non-volatile compounds. Biochem. Biophys. Res. Commun. 1974, 60, 616–621. [Google Scholar] [CrossRef]
- Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y. Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 151–153. [Google Scholar] [CrossRef]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, M.L.; Gall, L.N.; Krasnov, N.V.; Nikolaev, V.I.; Pavlenko, V.A.; Shkurov, V.A. Extraction of ions from solutions under atmospheric pressure as a method for mass spectrometric analysis of bioorganic compounds. Dokl. Akad. Nauk. SSSR 1984, 277, 379–383. [Google Scholar] [CrossRef]
- Yamashita, M.; Fenn, J.B. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984, 88, 4451–4459. [Google Scholar] [CrossRef]
- Katta, V.; Chait, B.T. Observation of the hemdlobin complex in native myoglobin by electrospray-ionization mass spectrometry. J. Am. Chem. Soc. 1991, 113, 8534–8535. [Google Scholar] [CrossRef]
- Light-Wahl, K.J.; Winger, B.E.; Smith, R.D. Observation of the multimeric forms of concanavalin A by electrospray ionization mass spectrometry. J. Am. Chem. Soc. 1993, 115, 5869–5870. [Google Scholar] [CrossRef]
- Przybylski, M.; Kast, J.; Glocker, M.O.; Dürr, E.; Bosshard, H.R.; Nock, S.; Sprmzl, M. Mass spectrometric approaches to molecular characterization of protein-nucleic acid interactions. Toxicol. Lett. 1995, 82, 567–575. [Google Scholar] [CrossRef]
- Glocker, M.O.; Bauer, S.H.; Kast, J.; Volz, J.; Przybylski, M. Characterization of Specific noncovalent protein complexes by UV matrix-assisted laser desorption ionization mass spectrometry. J. Mass Spectrom. 1996, 31, 1221–1227. [Google Scholar] [CrossRef]
- Rosinke, B.; Strupat, K.; Hillenkamp, F.; Rosenbusch, J.; Dencher, N.; Krüger, U.; Galla, H.-J. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) of membrane proteins and non-covalent complexes. J. Mass Spectrom. 1995, 30, 1462–1468. [Google Scholar] [CrossRef]
- Chen, F.; Gulbakan, B.; Weidmann, S.; Fagerer, S.R.; Ibanez, A.; Zenobi, R. Applying mass spectrometry to study non-covalent biomolecule complexes. Mass Spectrom. Rev. 2015, 35, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997, 16, 1–23. [Google Scholar] [CrossRef]
- Przybylski, M.; Glocker, M.O. Electrospray mass spectrometry of biomacromolecular complexes with noncovalent interactions—New analytical perspectives for supramolecular chemistry and molecular recognition processes. Angew. Chem. Int. Ed. 1996, 35, 806–826. [Google Scholar] [CrossRef]
- Goodlelt, D.R.; Loo, R.R.O.; Loo, J.A.; Wahl, J.H.; Udseth, H.R.; Smith, R.D. A study of the thermal denaturation of ribonuclease S by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 1994, 5, 614–622. [Google Scholar] [CrossRef]
- Loo, R.R.O.; Goodlett, D.R.; Smith, R.; Loo, J.A. Observation of a noncovalent ribonuclease S-protein/S-peptide complex by electrospray ionization mass spectrometry. J. Am. Chem. Soc. 1993, 115, 4391–4392. [Google Scholar] [CrossRef]
- Erba, E.B.; Petosa, C. The emerging role of native mass spectrometry in characterizing the structure and dynamics of macromolecular complexes. Protein Sci. 2015, 24, 1176–1192. [Google Scholar] [CrossRef] [PubMed]
- Sharon, M.; Robinson, C. The Role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu. Rev. Biochem. 2007, 76, 167–193. [Google Scholar] [CrossRef]
- Heck, A.J.R. Native mass spectrometry: A bridge between interactomics and structural biology. Nat. Methods 2008, 5, 927–933. [Google Scholar] [CrossRef]
- Barth, M.; Schmidt, C. Native mass spectrometry—A valuable tool in structural biology. J. Mass Spectrom. 2020, 55, e4578. [Google Scholar] [CrossRef]
- Marcoux, J.; Cianférani, S. Towards integrative structural mass spectrometry: Benefits from hybrid approaches. Methods 2015, 89, 4–12. [Google Scholar] [CrossRef]
- Yefremova, Y.; Melder, F.T.I.; Danquah, B.D.; Opuni, K.F.-M.; Koy, C.; Ehrens, A.; Frommholz, D.; Illges, H.; Koelbel, K.; Sobott, F.; et al. Apparent activation energies of protein–protein complex dissociation in the gas–phase determined by electrospray mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Chingin, K.; Barylyuk, K.; Chen, H. On the preservation of non-covalent protein complexes during electrospray ionization. Philos. Trans. R. Soc. B 2016, 374, 20150377. [Google Scholar] [CrossRef] [PubMed]
- Yefremova, Y.; Danquah, B.D.; Opuni, K.F.-M.; El-Kased, R.; Koy, C.; Glocker, O.M. Mass spectrometric characterization of protein structures and protein complexes in condensed and gas phase. Eur. J. Mass Spectrom. 2017, 23, 445–459. [Google Scholar] [CrossRef]
- Opuni, K.F.-M.; Al-Majdoub, M.; Yefremova, Y.; El-Kased, R.; Koy, C.; Glocker, M.O. Mass spectrometric epitope mapping. Mass Spectrom. Rev. 2016, 37, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Yefremova, Y.; Opuni, K.F.M.; Danquah, B.D.; Thiesen, H.-J.; Glocker, M.O. Intact transition epitope mapping (ITEM). J. Am. Soc. Mass Spectrom. 2017, 28, 1612–1622. [Google Scholar] [CrossRef]
- Danquah, B.D.; Yefremova, Y.; Opuni, K.F.-M.; Röwer, C.; Koy, C.; Glocker, M.O. Intact Transition epitope mapping-Thermodynamic weak-force order (ITEM-TWO). J. Proteom. 2019, 212, 103572. [Google Scholar] [CrossRef]
- Danquah, B.D.; Röwer, C.; Opuni, K.F.M.; El-Kased, R.; Frommholz, D.; Illges, H.; Koy, C.; Glocker, M.O. Intact transition epitope mapping—Targeted high-energy rupture of extracted epitopes (ITEM-THREE) *. Mol. Cell. Proteom. 2019, 18, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Danquah, B.D.; Opuni, K.F.M.; Roewer, C.; Koy, C.; Glocker, M.O. Mass Spectrometric analysis of antibody—Epitope peptide complex dissociation: Theoretical concept and practical procedure of binding strength characterization. Molecules 2020, 25, 4776. [Google Scholar] [CrossRef]
- Kim, E.E.; Varadarajan, R.; Wyckoff, H.W.; Richards, F.M. Refinement of the crystal structure of ribonuclease S. Comparison with and between the various ribonuclease A structures. Biochemistry 1992, 31, 12304–12314. [Google Scholar] [CrossRef]
- Jørgensen, T.J.; Delforge, D.; Remacle, J.; Bojesen, G.; Roepstorff, P. Collision-induced dissociation of noncovalent complexes between vancomycin antibiotics and peptide ligand stereoisomers: Evidence for molecular recognition in the gas phase. Int. J. Mass Spectrom. 1999, 188, 63–85. [Google Scholar] [CrossRef]
- Breuker, K.; McLafferty, F.W. Stepwise evolution of protein native structure with electrospray into the gas phase, 10–12 to 102 s. Proc. Natl. Acad. Sci. USA 2008, 105, 18145–18152. [Google Scholar] [CrossRef]
- Warnke, S.; Von Helden, G.; Pagel, K. Protein structure in the gas phase: The influence of side-chain microsolvation. J. Am. Chem. Soc. 2013, 135, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Jensen, J.H.; Gordon, M.S. On the number of water molecules necessary to stabilize the glycine zwitterion. J. Am. Chem. Soc. 1995, 117, 8159–8170. [Google Scholar] [CrossRef]
- Loo, R.R.O.; Loo, J.A. Salt Bridge Rearrangement (SaBRe) Explains the dissociation behavior of noncovalent complexes. J. Am. Soc. Mass Spectrom. 2016, 27, 975–990. [Google Scholar] [CrossRef]
- Sciuto, S.V.; Liu, J.; Konermann, L. An electrostatic charge partitioning model for the dissociation of protein complexes in the gas phase. J. Am. Soc. Mass Spectrom. 2011, 22, 1679–1689. [Google Scholar] [CrossRef][Green Version]
- Opuni, K.F.; Koy, C.; Russ, M.; Reepmeyer, M.; Danquah, B.D.; Weresow, M.; Alef, A.; Lorenz, P.; Thiesen, H.-J.; Glocker, M.O. ITEM-THREE analysis of a monoclonal anti-malaria antibody reveals its assembled epitope on the pfMSP119 antigen. J. Biol. Chem. 2020, 295, 14987–14997. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Llinares, M.B.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2018, 47, D442–D450. [Google Scholar] [CrossRef]
- Berisio, R.; Sica, F.; Lamzin, V.; Wilson, K.; Zagari, A.; Mazzarella, L. Atomic resolution structures of ribonuclease A at six pH values. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 441–450. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Li, J.; Rossetti, G.; Dreyer, J.; Raugei, S.; Ippoliti, E.; Lüscher, B.; Carloni, P. Molecular Simulation-based structural prediction of protein complexes in mass spectrometry: The human insulin dimer. PLoS Comput. Biol. 2014, 10, e1003838. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.; Demirev, P.A.; Karshikoff, A.D. Multiply-protonated protein ions in the gas phase: Calculation of the electrostatic interactions between charged sites. J. Phys. Chem. B 1997, 101, 9645–9650. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS tool for high-throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rosell, G.; Giorgino, T.; De Fabritiis, G. PlayMolecule ProteinPrepare: A Web application for protein preparation for molecular dynamics simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Opuni, K.F.-M.; Solomon, S.; Metzen, F.; Frommholz, D.; Koy, C.; Röwer, C.; Glocker, M.O.; Illges, H.; Anderson, P.C. In silico epitope mapping of glucose-6-phosphate isomerase: A Rheumatoid arthritis autoantigen. J. Proteom. Bioinform. 2017, 10, 60–72. [Google Scholar]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef]
- Linnebacher, M.; Lorenz, P.; Koy, C.; Jahnke, A.; Born, N.; Steinbeck, F.; Wollbold, J.; Latzkow, T.; Thiesen, H.-J.; Glocker, M.O. Clonality characterization of natural epitope-specific antibodies against the tumor-related antigen topoisomerase IIa by peptide chip and proteome analysis: A pilot study with colorectal carcinoma patient samples. Anal. Bioanal. Chem. 2012, 403, 227–238. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH/MeOH [-] (a)/[vol. %] | Mean Charge ± Std. Dev. (a) | Initial Educt Rate [%] (b) | Final Educt Rate [%] (b) | ∆CV50 [V] | dx [V] | Slope [%/V] |
|---|---|---|---|---|---|---|
| 4.5/5 | 7.3 ± 0.1 | 63.00 | 0.84 | 21.10 | 6.59 | −2.36 |
| 7/5 | 6.9 ± 0.1 | 100.00 | 5.60 | 13.01 | 2.37 | −9.94 |
| 7/10 | 6.4 ± 0.2 | 86.00 | 8.77 | 12.50 | 2.30 | −8.39 |
| 7/20 | 6.1 ± 0.6 | 90.00 | 5.99 | 14.90 | 2.60 | −8.08 |
| 7/30 | 6.4 ± 0.2 | 86.00 | 9.04 | 13.30 | 2.20 | −8.75 |
| 7/40 | 6.5 ± 0.2 | 84.75 | 7.65 | 13.66 | 2.08 | −9.28 |
| pH/MeOH [-] (a,b)/[vol. %] | [1/s] | ||||
|---|---|---|---|---|---|
| 4.5/5 | 6.90 × 1011 | 4.39 × 10−12 | 64.79 | −1.83 | −66.54 |
| 7/5 | 1.03 × 1010 | 3.71 × 10−12 | 65.21 | +1.28 | −63.83 |
| 7/10 | 5.00 × 1010 | 3.97 × 10−12 | 65.04 | +0.76 | −64.22 |
| 7/20 | 2.09 × 1010 | 3.83 × 10−12 | 65.13 | +0.84 | −64.20 |
| 7/30 | 2.80 × 1010 | 3.87 × 10−12 | 65.10 | +0.94 | −64.09 |
| 7/40 | 1.71 × 1010 | 3.79 × 10−12 | 65.15 | +1.12 | −63.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koy, C.; Opuni, K.F.M.; Danquah, B.D.; Neamtu, A.; Glocker, M.O. Mass Spectrometric and Bio-Computational Binding Strength Analysis of Multiply Charged RNAse S Gas-Phase Complexes Obtained by Electrospray Ionization from Varying In-Solution Equilibrium Conditions. Int. J. Mol. Sci. 2021, 22, 10183. https://doi.org/10.3390/ijms221910183
Koy C, Opuni KFM, Danquah BD, Neamtu A, Glocker MO. Mass Spectrometric and Bio-Computational Binding Strength Analysis of Multiply Charged RNAse S Gas-Phase Complexes Obtained by Electrospray Ionization from Varying In-Solution Equilibrium Conditions. International Journal of Molecular Sciences. 2021; 22(19):10183. https://doi.org/10.3390/ijms221910183
Chicago/Turabian StyleKoy, Cornelia, Kwabena F. M. Opuni, Bright D. Danquah, Andrei Neamtu, and Michael O. Glocker. 2021. "Mass Spectrometric and Bio-Computational Binding Strength Analysis of Multiply Charged RNAse S Gas-Phase Complexes Obtained by Electrospray Ionization from Varying In-Solution Equilibrium Conditions" International Journal of Molecular Sciences 22, no. 19: 10183. https://doi.org/10.3390/ijms221910183
APA StyleKoy, C., Opuni, K. F. M., Danquah, B. D., Neamtu, A., & Glocker, M. O. (2021). Mass Spectrometric and Bio-Computational Binding Strength Analysis of Multiply Charged RNAse S Gas-Phase Complexes Obtained by Electrospray Ionization from Varying In-Solution Equilibrium Conditions. International Journal of Molecular Sciences, 22(19), 10183. https://doi.org/10.3390/ijms221910183