
TAR RNA Mediated Folding of a Single-Arginine-Mutant HIV-1 Tat Protein within HeLa Cells Experiencing Intracellular Crowding


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Folding of Single-Arginine-Residue-Mutant Tat with TAR RNA
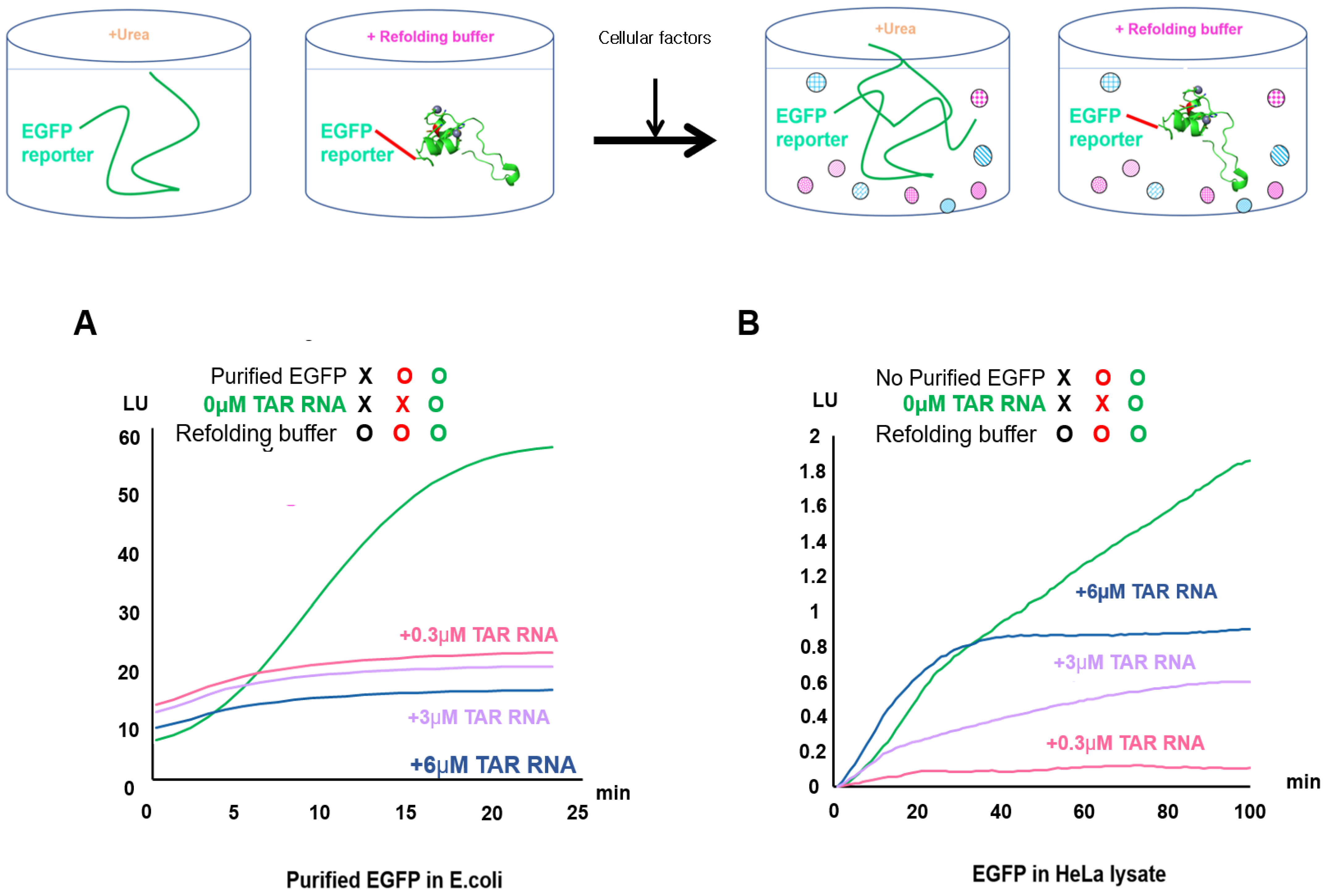
2.2. Validation of EGFP Unfolding and Refolding In Vitro and upon HeLa Cell Crowding
2.3. Effect of TAR RNA as a Helper in Refolding Mutant R52Tat-EGFP Involves Enzymes and Protein-Folding Substrates under Crowded HeLa Conditions
2.4. Increased Participation of Single-Arginine Mutants R52 and R53 in Tat-EGFP Folding Mediated by TAR RNA in Live Cells
2.5. Optimization of Biolayer Interferometry Monitoring to Analyze Arginine-Mutant Tat Refolding without EGFP
3. Discussion
4. Materials and Methods
4.1. Cloning
4.2. RNA Sequences
4.3. Cell Culture
4.4. Isolation of HIV Tat Protein from HeLa Cells
4.5. Refolding of Fluorescence EGFP Reporter In Vitro
4.6. Tat–BLI Assay In Vitro
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- van den Berg, B.; Ellis, R.J.; Dobson, C.M. Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 1999, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, B.; Wain, R.; Dobson, C.M.; Ellis, R.J. Macromolecular crowding perturbs protein refolding kinetics: Implications for folding inside the cell. EMBO J. 2000, 19, 3870–3875. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X. Influence of crowded cellular environments on protein folding, binding, and oligomerization: Biological consequences and potentials of atomistic modeling. FEBS Lett. 2013, 587, 1053–1061. [Google Scholar] [CrossRef]
- Vavouri, T.; Semple, J.I.; Garcia-Verdugo, R.; Lehner, B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 2009, 138, 198–208. [Google Scholar] [CrossRef]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Schreck, J.S.; Bridstrup, J.; Yuan, J.M. Investigating the Effects of Molecular Crowding on the Kinetics of Protein Aggregation. J. Phys. Chem. B 2020, 124, 9829–9839. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X. Protein folding and binding in confined spaces and in crowded solutions. J. Mol. Recognit. 2004, 17, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.M.; Chyan, C.L.; Zhou, H.X.; Chung, T.Y.; Peng, H.; Ping, G.; Yang, G. The effects of macromolecular crowding on the mechanical stability of protein molecules. Protein. Sci. 2008, 17, 2156–2166. [Google Scholar] [CrossRef]
- Tokuriki, N.; Kinjo, M.; Negi, S.; Hoshino, M.; Goto, Y.; Urabe, I.; Yomo, T. Protein folding by the effects of macromolecular crowding. Protein. Sci. 2004, 13, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Bolen, D.W. Efficacy of macromolecular crowding in forcing proteins to fold. Biophys. Chem. 2002, 101–102, 155–165. [Google Scholar] [CrossRef]
- Martin, J. Chaperonin function--effects of crowding and confinement. J. Mol. Recognit. 2004, 17, 465–472. [Google Scholar] [CrossRef]
- Perham, M.; Stagg, L.; Wittung-Stafshede, P. Macromolecular crowding increases structural content of folded proteins. FEBS Lett. 2007, 581, 5065–5069. [Google Scholar] [CrossRef]
- Sasahara, K.; McPhie, P.; Minton, A.P. Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol. 2003, 326, 1227–1237. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Patel, C.N.; Young, G.B.; Pielak, G.J. FlgM gains structure in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 12681–12684. [Google Scholar] [CrossRef]
- Gnutt, D.; Sistemich, L.; Ebbinghaus, S. Protein Folding Modulation in Cells Subject to Differentiation and Stress. Front Mol. Biosci. 2019, 6, 38. [Google Scholar] [CrossRef]
- Ignatova, Z.; Gierasch, L.M. Monitoring protein stability and aggregation in vivo by real-time fluorescent labeling. Proc. Nat. Acad. Sci. USA 2004, 101, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Ebbinghaus, S.; Dhar, A.; McDonald, J.D.; Gruebele, M. Protein folding stability and dynamics imaged in a living cell. Nat. Methods 2010, 7, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, J.; Mu, X.; Lang, L.; Wang, H.; Binolfi, A.; Theillet, F.X.; Bekei, B.; Logan, D.T.; Selenko, P.; Wennerstrom, H.; et al. Thermodynamics of protein destabilization in live cells. Proc. Natl. Acad. Sci. USA 2015, 112, 12402–12407. [Google Scholar] [CrossRef] [PubMed]
- Delling, U.; Reid, L.S.; Barnett, R.W.; Ma, M.Y.; Climie, S.; Sumner-Smith, M.; Sonenberg, N. Conserved nucleotides in the TAR RNA stem of human immunodeficiency virus type 1 are critical for Tat binding and trans activation: Model for TAR RNA tertiary structure. J. Virol. 1992, 66, 3018–3025. [Google Scholar] [CrossRef]
- Ulich, C.; Dunne, A.; Parry, E.; Hooker, C.W.; Gaynor, R.B.; Harrich, D. Functional domains of Tat required for efficient human immunodeficiency virus type 1 reverse transcription. J. Virol. 1999, 73, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, A.; Meredith, L.W.; Suhrbier, A.; Kiernan, R.; Harrich, D. The HIV-1 Tat protein stimulates reverse transcription in vitro. Curr. HIV Res. 2007, 5, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Schulte, A.; Vogel-Bachmayr, K.; Scheffzek, K.; Geyer, M. Structural insights into the cyclin T1-Tat-TAR RNA transcription activation complex from EIAV. Nat. Str. Mol. Biol. 2008, 15, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Schulte, A.; Fujinaga, K.; Scheffzek, K.; Geyer, M. Cyclin box structure of the P-TEFb subunit cyclin T1 derived from a fusion complex with EIAV tat. J. Mol. Biol. 2007, 370, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.V.; Salguero, C.; Khan, S.N.; Meagher, J.L.; Brown, W.C.; Humbert, N.; de Rocquigny, H.; Smith, J.L.; D’Souza, V.M. HIV-1 Tat interactions with cellular 7SK and viral TAR RNAs identifies dual structural mimicry. Nat. Communal. 2018, 9, 4266. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Rudolph, R. Suppression of protein aggregation by L-arginine. Curr. Pharm. Biotechnol. 2009, 10, 408–414. [Google Scholar] [CrossRef]
- Rocco, M.A.; Waraho-Zhmayev, D.; DeLisa, M.P. Twin-arginine translocase mutations that suppress folding quality control and permit export of misfolded substrate proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 13392–13397. [Google Scholar] [CrossRef]
- Singh, V.; Patel, K.A.; Sharma, R.K.; Patil, P.R.; Joshi, A.S.; Parihar, R.; Athilingam, T.; Sinha, N.; Ganesh, S.; Sinha, P.; et al. Discovery of Arginine Ethyl Ester as Polyglutamine Aggregation Inhibitor: Conformational Transitioning of Huntingtin N-Terminus Augments Aggregation Suppression. ACS Chem. Neurosci. 2019, 10, 3969–3985. [Google Scholar] [CrossRef]
- Ishibashi, M.; Tsumoto, K.; Tokunaga, M.; Ejima, D.; Kita, Y.; Arakawa, T. Is arginine a protein-denaturant? Prot. Expr. Purif. 2005, 42, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tsumoto, K.; Umetsu, M.; Kumagai, I.; Ejima, D.; Philo, J.S.; Arakawa, T. Role of arginine in protein refolding, solubilization, and purification. Biotechnol. Prog. 2004, 20, 1301–1308. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What macromolecular crowding can do to a protein. Int. J. Mol. Sci. 2014, 15, 23090–23140. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Choi, H.S.; Seong, B.L. The folding competence of HIV-1 Tat mediated by interaction with TAR RNA. RNA Biol. 2017, 14, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, E.N.; Popiel, H.A.; Tada, M.; Takahashi, T.; Yamane, H.; Saitoh, Y.; Takahashi, Y.; Ozawa, D.; Takeda, A.; Takeuchi, T.; et al. Arginine is a disease modifier for polyQ disease models that stabilizes polyQ protein conformation. Brain 2020, 143, 1811–1825. [Google Scholar] [CrossRef] [PubMed]
- Jarvelin, A.I.; Noerenberg, M.; Davis, I.; Castello, A. The new (dis)order in RNA regulation. Cell Commun. Signal. 2016, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Herschlag, D. RNA chaperones and the RNA folding problem. J. Biolog. Chem. 1995, 270, 20871–20874. [Google Scholar] [CrossRef]
- Weeks, K.M. Protein-facilitated RNA folding. Cur. Opin. Str. Bio. 1997, 7, 336–342. [Google Scholar] [CrossRef]
- Kudlicki, W.; Coffman, A.; Kramer, G.; Hardesty, B. Ribosomes and ribosomal RNA as chaperones for folding of proteins. Fold. Des. 1997, 2, 101–108. [Google Scholar] [CrossRef]
- Frankel, A.D.; Smith, C.A. Induced folding in RNA-protein recognition: More than a simple molecular handshake. Cell 1998, 92, 149–151. [Google Scholar] [CrossRef]
- Biro, J.C. Nucleic acid chaperons: A theory of an RNA-assisted protein folding. Bio. Med. Model. 2005, 2, 35. [Google Scholar]
- Sulijoadikusumo, I.; Horikoshi, N.; Usheva, A. Another function for the mitochondrial ribosomal RNA: Protein folding. Biochemistry 2001, 40, 11559–11564. [Google Scholar] [CrossRef]
- Clodi, E.; Semrad, K.; Schroeder, R. Assaying RNA chaperone activity in vivo using a novel RNA folding trap. EMBO J. 1999, 18, 3776–3782. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Webb, A.E.; Weeks, K.M. A collapsed state functions to self-chaperone RNA folding into a native ribonucleoprotein complex. Nat. Str. Biol. 2001, 8, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Son, A.; Choi, S.I.; Han, G.; Seong, B.L. M1 RNA is important for the in-cell solubility of its cognate C5 protein: Implications for RNA-mediated protein folding. RNA Biol. 2015, 12, 1198–1208. [Google Scholar] [CrossRef]
- Choi, S.I.; Ryu, K.; Seong, B.L. RNA-mediated chaperone type for de novo protein folding. RNA Biol. 2009, 6, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Borges, J.C.; Ramos, C.H. Protein folding assisted by chaperones. Protein Pept. Let. 2005, 12, 257–261. [Google Scholar] [CrossRef]
- Chakraborty, K.; Chatila, M.; Sinha, J.; Shi, Q.; Poschner, B.C.; Sikor, M.; Jiang, G.; Lamb, D.C.; Hartl, F.U.; Hayer-Hartl, M. Chaperonin-Catalyzed Rescue of Kinetically Trapped States in Protein Folding. Cell 2010, 142, 112–122. [Google Scholar] [CrossRef]
- Clare, D.K.; Vasishtan, D.; Stagg, S.; Quispe, J.; Farr, G.W.; Topf, M.; Horwich, A.L.; Saibil, H.R. ATP-triggered conformational changes delineate substrate-binding and -folding mechanics of the GroEL chaperonin. Cell 2012, 149, 113–123. [Google Scholar] [CrossRef]
- Horowitz, S.; Bardwell, J.C. RNAs as chaperones. RNA Biol. 2016, 13, 1228–1231. [Google Scholar] [CrossRef]
- Waldo, G.S.; Standish, B.M.; Berendzen, J.; Terwilliger, T.C. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 1999, 17, 691–695. [Google Scholar] [CrossRef]
- Sacchetti, A.; Alberti, S. Protein tags enhance GFP folding in eukaryotic cells. Nat. Biotechnol. 1999, 17, 1046. [Google Scholar] [CrossRef]
- Son, A.; Horowitz, S.; Seong, B.L. Chaperna: Linking the ancient RNA and protein worlds. RNA Biol. 2020, 18, 16–23. [Google Scholar] [CrossRef] [PubMed]
- De Marco, A.; Dans, P.D.; Knezevich, A.; Maiuri, P.; Pantano, S.; Marcello, A. Subcellular localization of the interaction between the human immunodeficiency virus transactivator Tat and the nucleosome assembly protein 1. Amino. Acids 2010, 38, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Shida, H.; Maki, M.; Hatanaka, M. Effects of a highly basic region of human immunodeficiency virus Tat protein on nucleolar localization. J. Virol. 1990, 64, 1803–1807. [Google Scholar] [CrossRef] [PubMed]
- Hautbergue, G.M.; Hung, M.L.; Golovanov, A.P.; Lian, L.Y.; Wilson, S.A. Mutually exclusive interactions drive handover of mRNA from export adaptors to TAP. Proc. Natl. Acad. Sci. USA 2008, 105, 5154–5159. [Google Scholar] [CrossRef]
- Bayer, T.S.; Booth, L.N.; Knudsen, S.M.; Ellington, A.D. Arginine-rich motifs present multiple interfaces for specific binding by RNA. RNA 2005, 11, 1848–1857. [Google Scholar] [CrossRef]
- Pastor, A.; Singh, A.K.; Fisher, M.T.; Chaudhuri, T.K. Protein folding on biosensor tips: Folding of maltodextrin glucosidase monitored by its interactions with GroEL. FEBS J. 2016, 283, 3103–3114. [Google Scholar] [CrossRef]
- Santra, S.; Jana, M. Insights into the Sensitivity of Arginine Concentration to Preserve the Folded Form of Insulin Monomer under Thermal Stress. J. Chem. Inf. Model. 2020, 60, 3105–3119. [Google Scholar] [CrossRef]
- Calnan, B.J.; Tidor, B.; Biancalana, S.; Hudson, D.; Frankel, A.D. Arginine-mediated RNA recognition: The arginine fork. Science 1991, 252, 1167–1171. [Google Scholar] [CrossRef]
- Mousseau, G.; Mediouni, S.; Valente, S.T. Targeting HIV transcription: The quest for a functional cure. Curr. Top. Microbiol. Immunol. 2015, 389, 121–145. [Google Scholar]
- Jeang, K.T.; Xiao, H.; Rich, E.A. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem. 1999, 274, 28837–28840. [Google Scholar] [CrossRef]
- Berkhout, B.; Silverman, R.H.; Jeang, K.T. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 1989, 59, 273–282. [Google Scholar] [CrossRef]
- Cullen, B. Cullen, Trans-activation of human immunodeficiency virus occurs via a bimodal mechanism. Cell 1986, 46, 973–982. [Google Scholar] [CrossRef]
- Berkhout, B.; Gatignol, A.; Rabson, A.B.; Jeang, K.T. Tar-independent activation of the hiv-1-ltr—Evidence that tat requires specific regions of the promoter. Cell 1990, 62, 757–767. [Google Scholar] [CrossRef]
- Dang, C.V.; Lee, W.M. Nuclear and nucleolar targeting sequences of c-erb-A, c-myb, N-myc, p53, HSP70, and HIV tat proteins. J. Biolo. Chem. 1989, 264, 18019–18023. [Google Scholar] [CrossRef]
- Choi, S.I.; Seong, B.L. A Conceptual Framework for Integrating Cellular Protein Folding, Misfolding and Aggregation. Life 2021, 11, 605. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular Chaperones in the Cytosol: From Nascent Chain to Folded Protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Kerner, M.J.; Naylor, D.J.; Ishihama, Y.; Maier, T.; Chang, H.-C.; Stines, A.P.; Georgopoulos, C.; Frishman, D.; Hayer-Hartl, M.; Mann, M. Proteome-wide Analysis of Chaperonin-Dependent Protein Folding in Escherichia coli. Cell 2005, 122, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Vorderwülbecke, S.; Kramer, G.; Merz, F.; Kurz, T.A.; Rauch, T.; Zachmann-Brand, B.; Bukau, B.; Deuerling, E. Low temperature or GroEL/ES overproduction permits growth of Escherichia coli cells lacking trigger factor and DnaK. FEBS Lett. 2004, 559, 181–187. [Google Scholar] [CrossRef]
- Ullers, R.S. SecB is a bona fide generalized chaperone in Escherichia coli. Proc. Natl. Acad. Sci. USA 2004, 101, 7583–7588. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Marinelli, P.; Navarro, S.; Bano-Polo, M.; Morel, B.; Grana-Montes, R.; Sabe, A.; Canals, F.; Fernandez, M.R.; Conejero-Lara, F.; Ventura, S. Global Protein Stabilization Does Not Suffice to Prevent Amyloid Fibril Formation. ACS Chem. Biol. 2018, 13, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Ciryam, P.; Tartaglia, G.G.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013, 5, 781–790. [Google Scholar] [CrossRef]
- Varela, A.E.; Lang, J.F.; Wu, Y.; Dalphin, M.D.; Stangl, A.J.; Okuno, Y.; Cavagnero, S. Kinetic Trapping of Folded Proteins Relative to Aggregates under Physiologically Relevant Conditions. J. Phys. Chem. B 2018, 122, 7682–7698. [Google Scholar] [CrossRef]
- Kuciak, M.; Gabus, C.; Ivanyi-Nagy, R.; Semrad, K.; Storchak, R.; Chaloin, O.; Muller, S.; Mely, Y.; Darlix, J.L. The HIV-1 transcriptional activator Tat has potent nucleic acid chaperoning activities in vitro. Nucleic Acids Res. 2008, 36, 3389–3400. [Google Scholar] [CrossRef]
- Connell, G.J.; Illangesekare, M.; Yarus, M. Three small ribooligonucleotides with specific arginine sites. Biochemistry 1993, 32, 5497–5502. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: Which way to go? Cell Mol. Life Sci. 2003, 60, 1852–1871. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lim, J.; Sung, J.; Cheong, Y.; Lee, E.Y.; Kim, J.; Oh, H.; Kim, Y.S.; Cho, N.H.; Choi, S.; et al. Built-in RNA-mediated chaperone (chaperna) for antigen folding tailored to immunized hosts. Biotechnol. Bioeng. 2020, 117, 1990–2007. [Google Scholar] [CrossRef]
- Lee, J.; Son, A.; Kim, P.; Kwon, S.B.; Yu, J.E.; Han, G.; Seong, B.L. RNA-dependent chaperone (chaperna) as an engineered pro-region for the folding of recombinant microbial transglutaminase. Biotechnol. Bioeng. 2019, 116, 490–502. [Google Scholar] [CrossRef]
- Yang, S.W.; Jang, Y.H.; Kwon, S.B.; Lee, Y.J.; Chae, W.; Byun, Y.H.; Kim, P.; Park, C.; Lee, Y.J.; Kim, C.K.; et al. Harnessing an RNA-mediated chaperone for the assembly of influenza hemagglutinin in an immunologically relevant conformation. FASEB J. 2018, 32, 2658–2675. [Google Scholar] [CrossRef]
- Holloway, A.F.; Occhiodoro, F.; Mittler, G.; Meisterernst, M.; Shannon, M.F. Functional interaction between the HIV transactivator Tat and the transcriptional coactivator PC4 in T cells. J. Biolog. Chem. 2000, 275, 21668–21677. [Google Scholar] [CrossRef]
- Rudolph, C.; Adam, G.; Simm, A. Determination of copy number of c-Myc protein per cell by quantitative Western blotting. Anal. Biochem. 1999, 269, 66–71. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.M.; Chun, H. TAR RNA Mediated Folding of a Single-Arginine-Mutant HIV-1 Tat Protein within HeLa Cells Experiencing Intracellular Crowding. Int. J. Mol. Sci. 2021, 22, 9998. https://doi.org/10.3390/ijms22189998
Kim JM, Chun H. TAR RNA Mediated Folding of a Single-Arginine-Mutant HIV-1 Tat Protein within HeLa Cells Experiencing Intracellular Crowding. International Journal of Molecular Sciences. 2021; 22(18):9998. https://doi.org/10.3390/ijms22189998
Chicago/Turabian StyleKim, Jung Min, and Honggu Chun. 2021. "TAR RNA Mediated Folding of a Single-Arginine-Mutant HIV-1 Tat Protein within HeLa Cells Experiencing Intracellular Crowding" International Journal of Molecular Sciences 22, no. 18: 9998. https://doi.org/10.3390/ijms22189998
APA StyleKim, J. M., & Chun, H. (2021). TAR RNA Mediated Folding of a Single-Arginine-Mutant HIV-1 Tat Protein within HeLa Cells Experiencing Intracellular Crowding. International Journal of Molecular Sciences, 22(18), 9998. https://doi.org/10.3390/ijms22189998