
Personalised Medicine for Colorectal Cancer Using Mechanism-Based Machine Learning Models


Abstract
1. Introduction
2. Results
2.1. Determining Significant Genes and Proteins in CRC: A Kaplan–Meier Survival Analysis
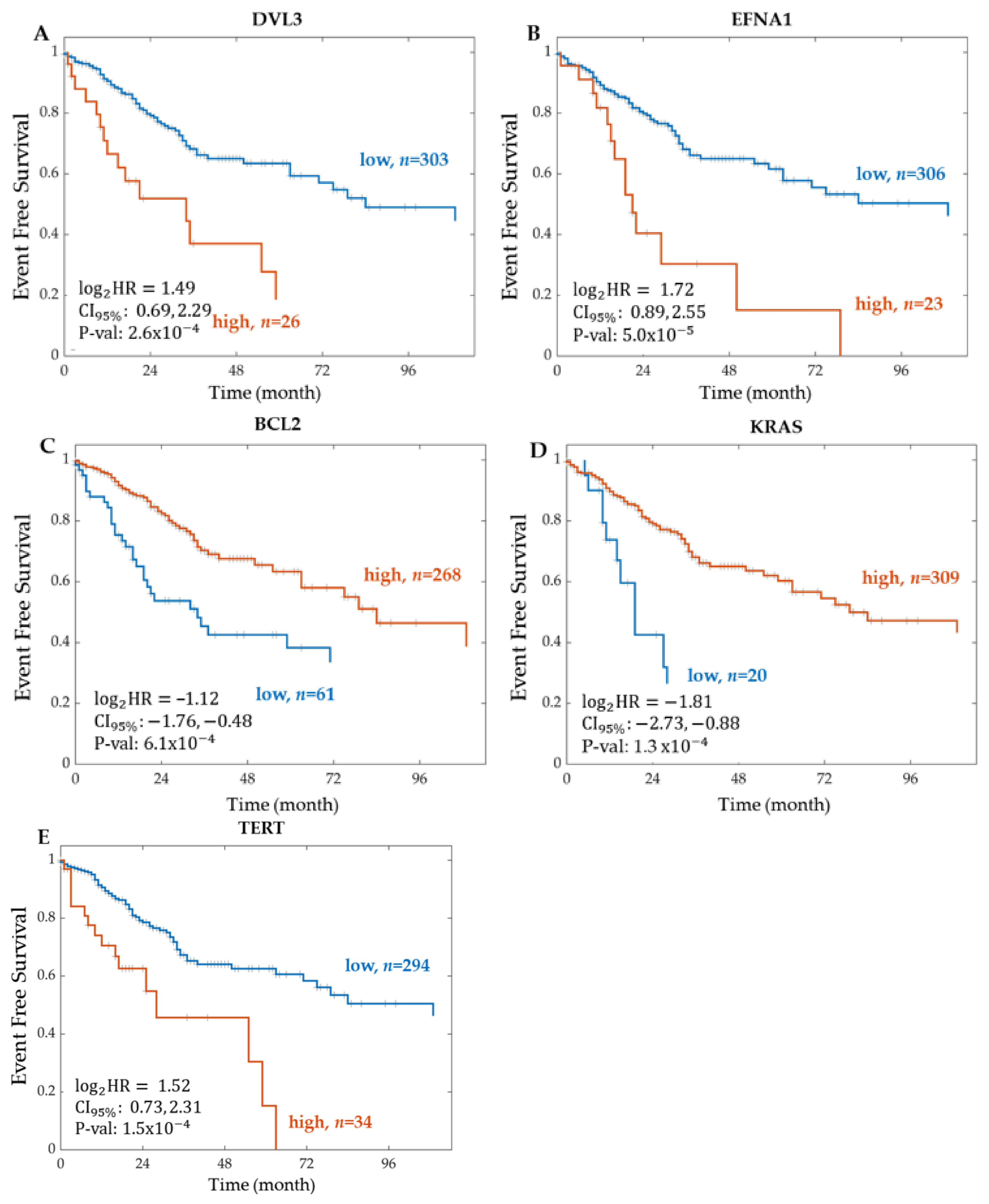
2.1.1. Significant Genes from RNA Sequencing
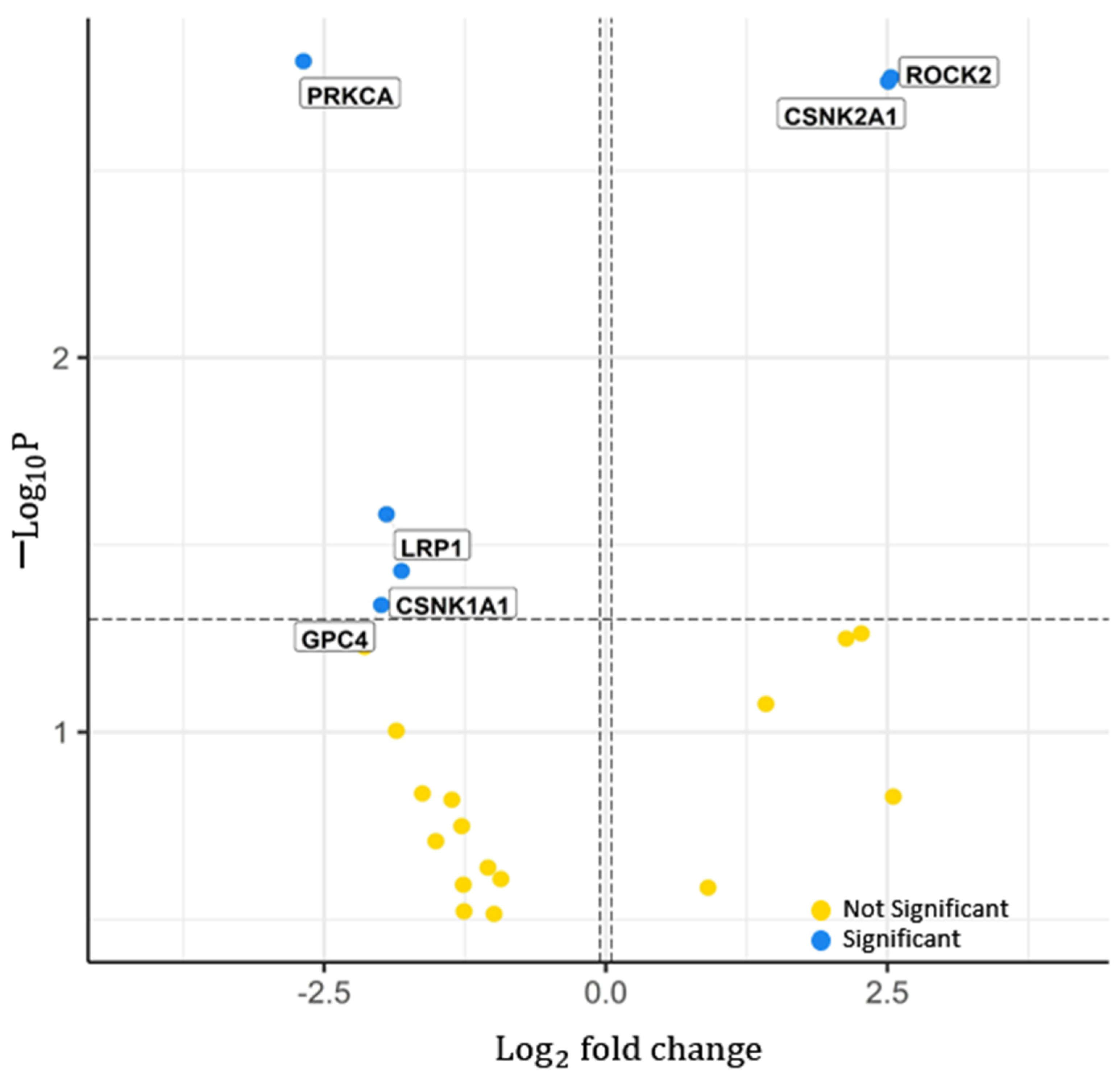
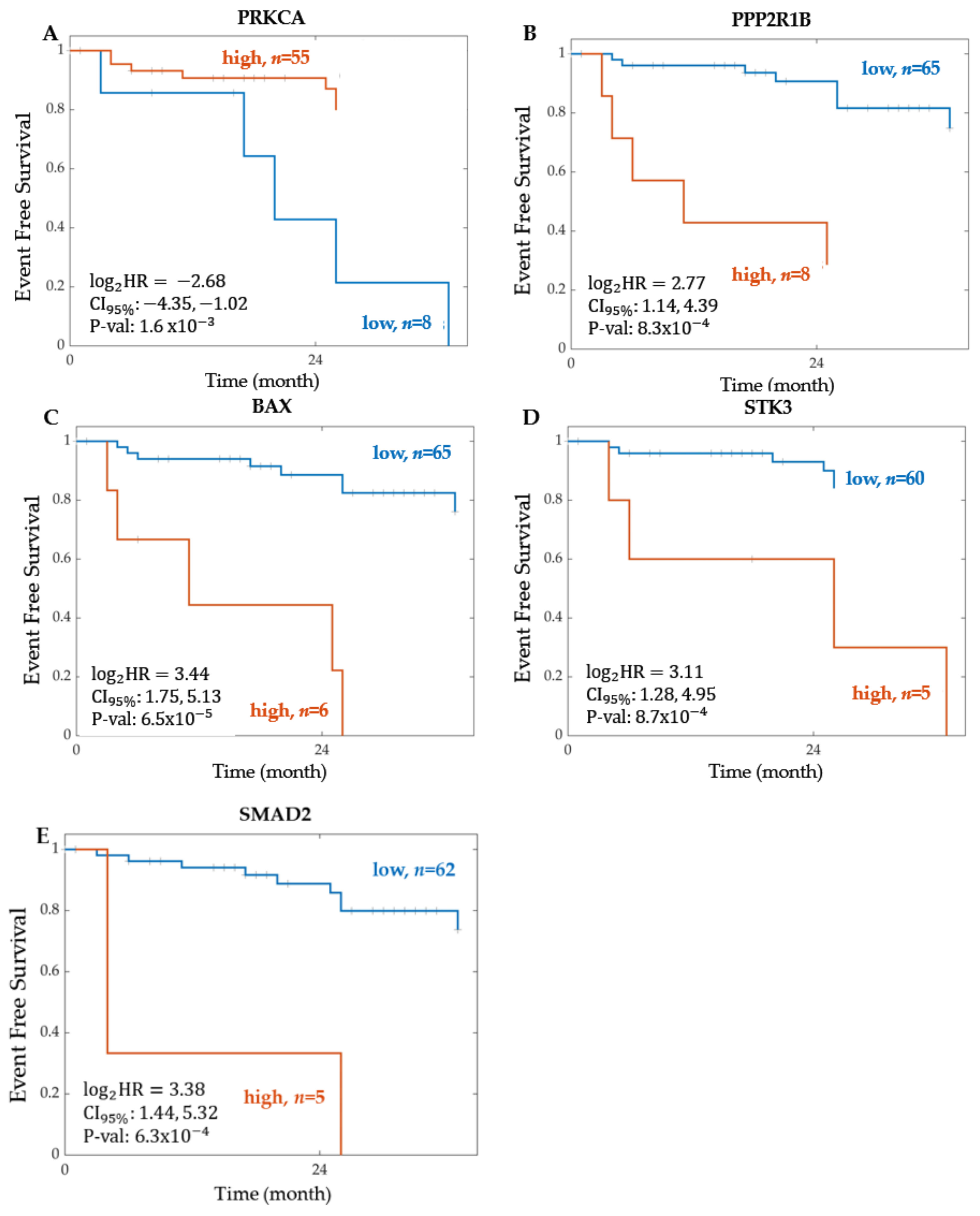
2.1.2. Significant Proteins from Proteomics
2.1.3. Summary of the Event-Free Survival Analysis Results
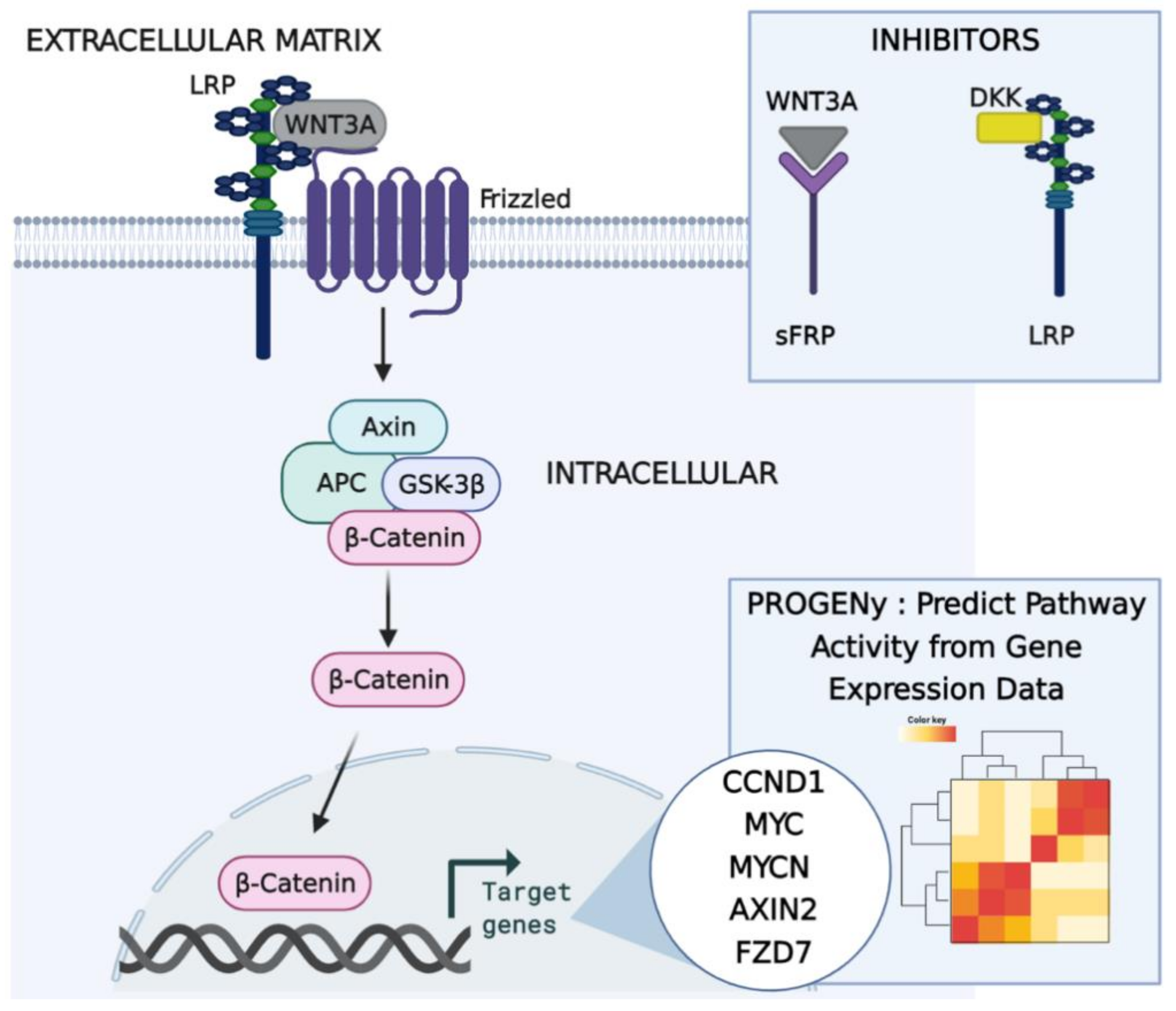
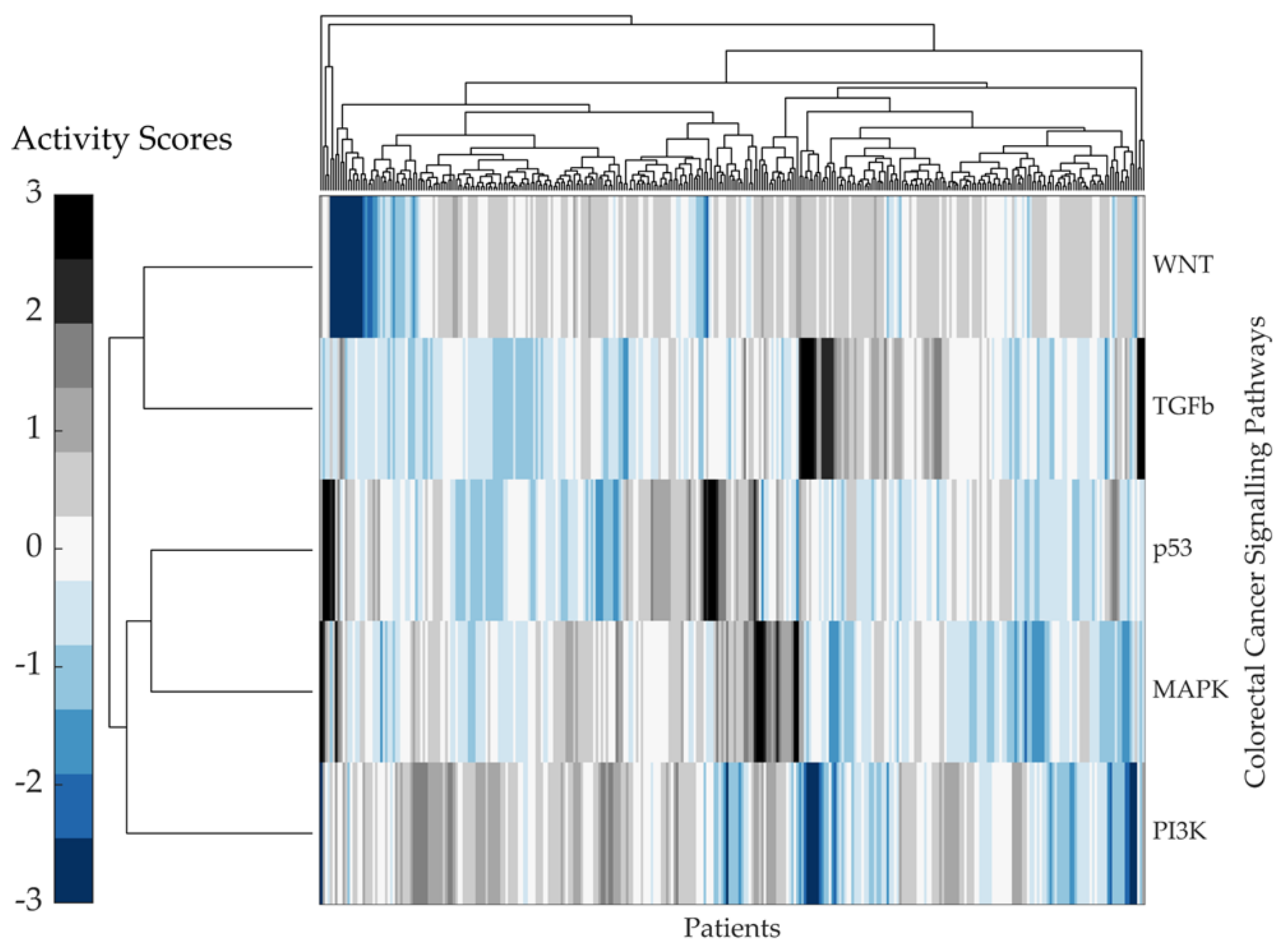
2.2. Predicting Pathway Responsive Genes for Activity Interference from Gene Expression: A PROGENy Analysis
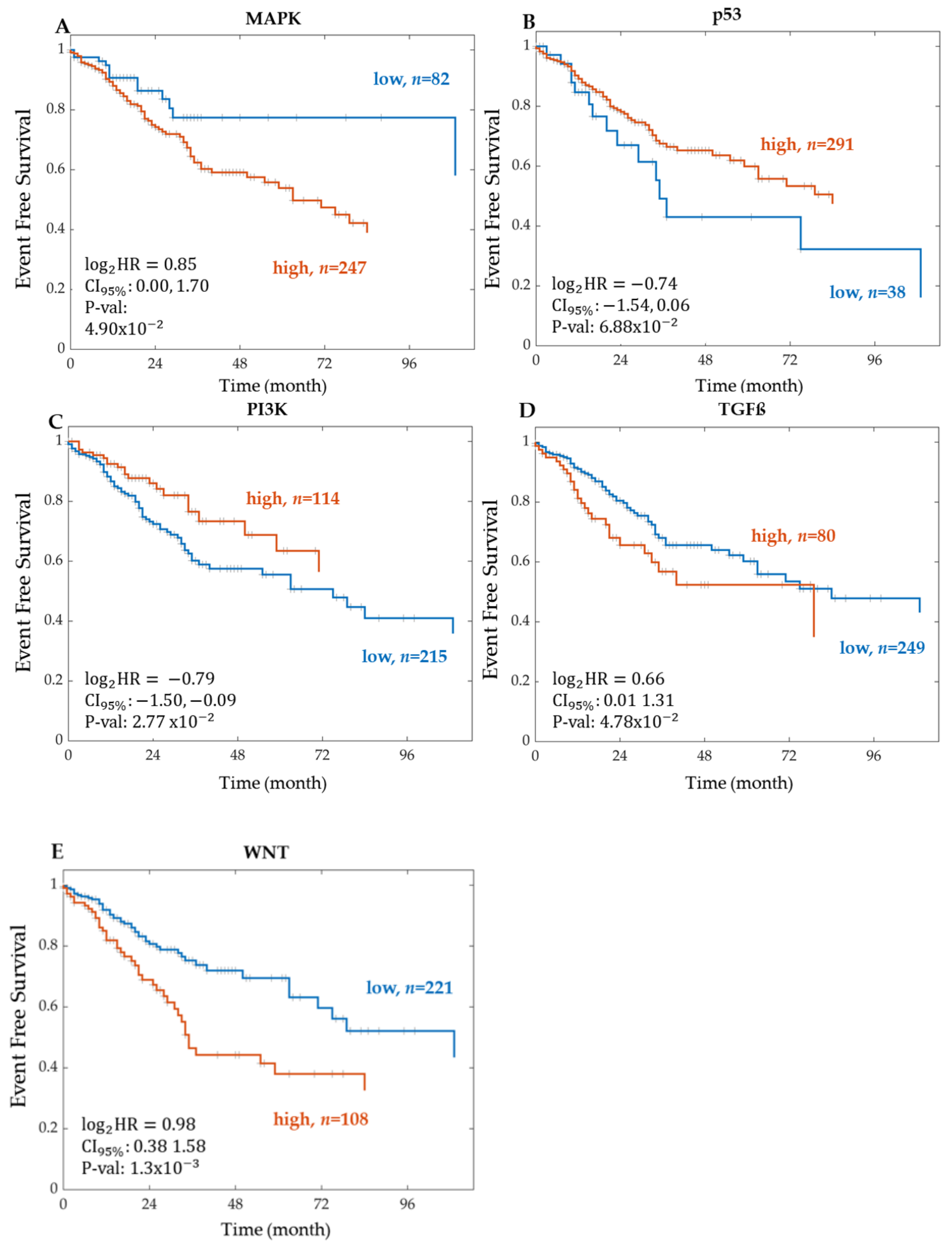
Kaplan–Meier Survival Analysis on PROGENy Activity Scores
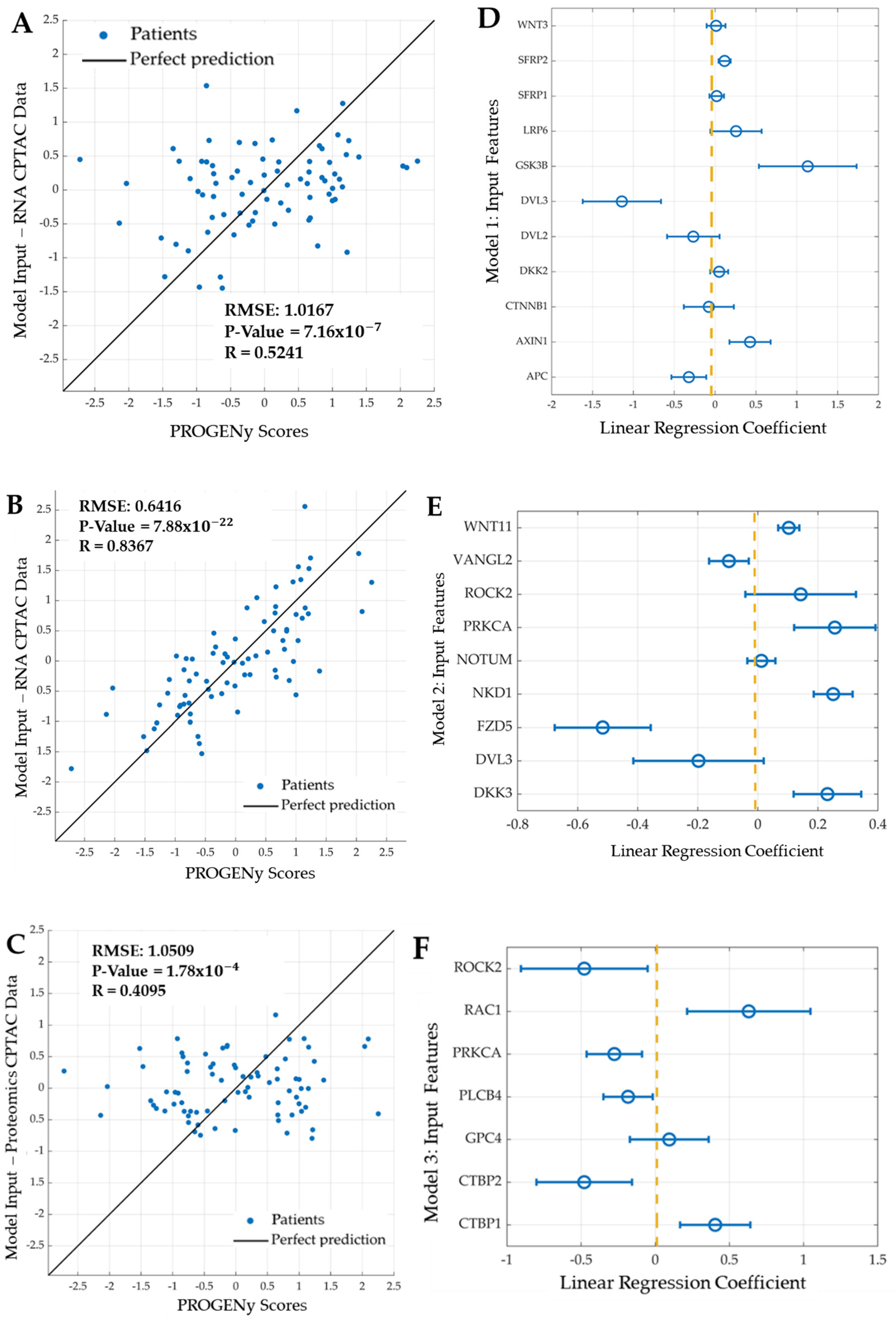
2.3. Developing Linear Regression Machine Learning Models
- Model 1: Features are eleven genes taken from the most recent WNT mechanistic model developed by Kogan et al.: APC, AXIN1, CTNNB1, DKK2, DVL3, GSK3B, LRP6, SFRP1, SFRP2, and WNT3.
- Model 2: Features are the mRNA expression of nine genes: seven features selected using LASSO and the two most significant genes (DVL3, VANGL2): DKK3, FZD5, NKD1, NOTUM (notum, palmitoleoyl-protein carboxylesterase), WNT11, PRKCA, and ROCK2.
- Model 3: Features are the protein expression of seven proteins; five identified using LASSO and the two most significant proteins (PRKCA, ROCK2): CTBP1 (c-terminal binding protein 1), CTBP2 (c-terminal binding protein 2), GPC4, PLCB4 (phospholipase c beta 4), and RAC1 (ras-related C3 botulinum toxin substrate 1).
2.4. Summary
3. Discussion
3.1. Event-Free Survival Analysis
3.2. A PROGENy Analysis
3.3. Development of Linear Regression Models
4. Conclusions
5. Materials and Methods
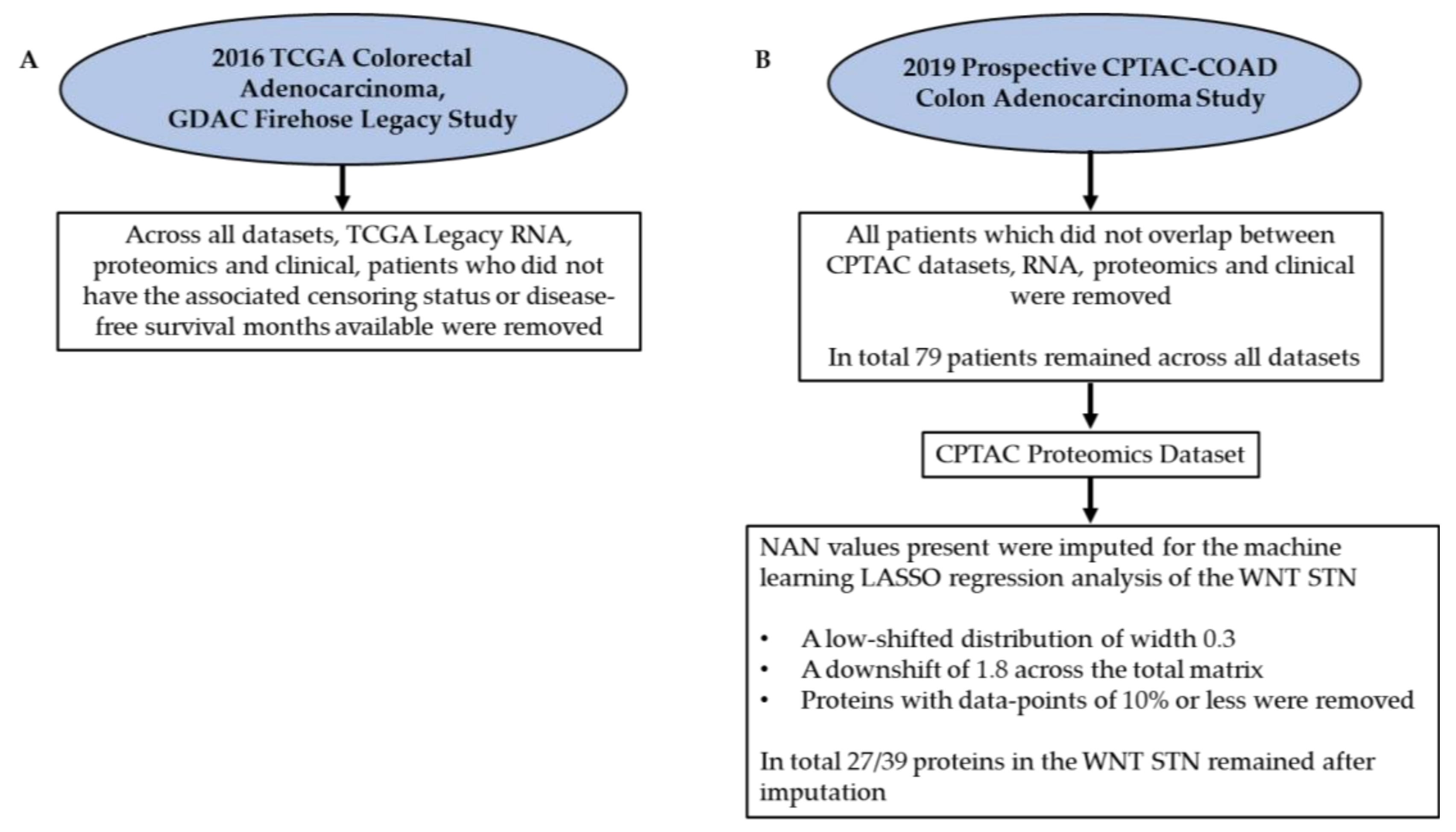
5.1. Data Acquisition from the Literature
5.2. Kaplan–Meier Survival Analysis
5.3. PROGENy Analysis
5.4. Developing Linear Regression Machine Learning Models
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. In Signal Transduction and Targeted Therapy; Springer Nature: Basingstoke, UK, 2020; Volume 5, pp. 1–30. [Google Scholar] [CrossRef]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Stintzing, S.; Wirapati, P.; Lenz, H.-J.; Neureiter, D.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Kaiser, F.; Al-Batran, S.; Heintges, T.; et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann. Oncol. 2019, 30, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef] [PubMed]
- Mooi, J.; Wirapati, P.; Asher, R.; Lee, C.; Savas, P.S.; Price, T.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef]
- Roelands, J.; Kuppen, P.J.K.; Vermeulen, L.; Maccalli, C.; Decock, J.; Wang, E.; Marincola, F.M.; Bedognetti, D.; Hendrickx, W. Immunogenomic classification of colorectal cancer and therapeutic implications. Int. J. Mol. Sci. 2017, 18, 2229. [Google Scholar] [CrossRef]
- Kolch, W.; Fey, D. Personalized computational models as biomarkers. J. Pers. Med. 2017, 7, 9. [Google Scholar] [CrossRef]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- McDonald, G.T.; Sullivan, R.; Paré, G.C.; Graham, C.H. Inhibition of phosphatidylinositol 3-kinase promotes tumor cell resistance to chemotherapeutic agents via a mechanism involving delay in cell cycle progression. Exp. Cell Res. 2010, 316, 3197–3206. [Google Scholar] [CrossRef]
- Temiz, T.K.; Altun, A.; Turgut, N.H.; Balcı, E.; Turgut, H.; Balcı, E. Investigation of the effects of drugs effective on PI3K-AKT signaling pathway in colorectal cancer alone and in combination Kolorektal kanserde PI3K-AKT sinyal yolağı üzerinden etki gösteren ilaçların tek başlarına ve kombinasyonlarının etkilerinin araştırılması. Cumhur. Med. J. 2014, 36, 167–177. [Google Scholar] [CrossRef][Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Shi, Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007, 17, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Cell Cycle Pathway|Aviva Systems Biology—Bio-Connect. Available online: https://www.bio-connect.nl/cell-cycle-pathway/cnt/page/4814 (accessed on 9 April 2021).
- Wang, X.W.; Zhang, Y.J. Targeting mTOR network in colorectal cancer therapy. World J. Gastroenterol. 2014, 20, 4178–4188. [Google Scholar] [CrossRef]
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; Peymani, M.; Forootan, F.S.; Esfahani, M.H.N.; Ghaedi, K. Signaling pathways involved in colorectal cancer progression. In Cell and Bioscience; BioMed Central Ltd.: London, UK, 2019; Volume 9. [Google Scholar] [CrossRef]
- Previs, R.A.; Coleman, R.L.; Harris, A.L.; Sood, A.K. Molecular Pathways: Translational and Therapeutic Implications of the Notch Signaling Pathway in Cancer. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Li, G.; You, Y.; Wan, H.; Wu, Q.; Wang, C.; Lv, N. Antitumor activity of Notch-1 inhibition in human colorectal carcinoma cells. Oncol. Rep. 2018, 39, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.K.S. KEGG: Kyoto Encyclopedia of Genes and Genomes. In Nucleic Acids Research; Oxford University Press: Oxford, UK, 2000; Volume 28, pp. 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. Thed KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef]
- Iurii, M. Genetics of colorectal cancer. J. Med. Life 2014, 7, 507–511. [Google Scholar]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. Wnt Signaling and Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kogan, Y.; Halevi-Tobias, K.E.; Hochman, G.; Baczmanska, A.K.; Leyns, L.; Agur, Z. A new validated mathematical model of the Wnt signalling pathway predicts effective combinational therapy by sFRP and Dkk. Biochem. J. 2012, 444, 115–125. [Google Scholar] [CrossRef][Green Version]
- Wolf, B.K.J. Mathematical modelling of Wnt/β-catenin signalling. Biochem. Soc. Trans. 2010, 38, 1281–1285. [Google Scholar] [CrossRef][Green Version]
- Lee, E.; Salic, A.; Krüger, R.; Heinrich, R.; Kirschner, M.W. The roles of APC and axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003, 1, e10. [Google Scholar] [CrossRef]
- Lee, M.; Chen, G.T.; Puttock, E.; Wang, K.; Edwards, R.A.; Waterman, M.L.; Lowengrub, J. Mathematical modeling links Wnt signaling to emergent patterns of metabolism in colon cancer. Mol. Syst. Biol. 2017, 13, 912. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Singh, T.R. Network-based approach to understand dynamic behaviour of Wnt signaling pathway regulatory elements in colorectal cancer. Netw. Modeling Anal. Health Inform. Bioinform. 2018, 7, ra130. [Google Scholar] [CrossRef]
- Heinrich, R. Mathematical modelling of the Wnt-pathway. Syst. Biol. 2005, 13, 259–275. [Google Scholar] [CrossRef]
- Fey, D.; Halasz, M.; Dreidax, D.; Kennedy, S.P.; Hastings, J.F.; Rauch, N.; Munoz, A.G.; Pilkington, R.; Fischer, M.; Westermann, F.; et al. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci. Signal. 2015, 8, ra130. [Google Scholar] [CrossRef]
- Golumbeanu, M.; Yang, G.; Camponovo, F.; Stuckey, E.M.; Hamon, N.; Mondy, M.; Rees, S.; Chitnis, N.; Cameron, E.; Penny, M.A. Combining machine learning and mathematical models of disease dynamics to guide development of novel disease interventions. medRxiv 2021. [Google Scholar] [CrossRef]
- Schubert, M.; Klinger, B.; Klünemann, M.; Sieber, A.; Uhlitz, F.; Sauer, S.; Garnett, M.J.; Blüthgen, N.; Saez-Rodriguez, J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018, 9, 20. [Google Scholar] [CrossRef]
- Holland, C.H.; Tanevski, J.; Perales-Patón, J.; Gleixner, J.; Kumar, M.P.; Mereu, E.; Joughin, B.A.; Stegle, O.; Lauffenburger, D.A.; Heyn, H.; et al. Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 2020, 21, 1–19. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. In Protein Science; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2019; Volume 28, pp. 1947–1951. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. 2014, 4, 13. [Google Scholar] [CrossRef]
- Darras, S.; Fritzenwanker, J.H.; Uhlinger, K.R.; Farrelly, E.; Pani, A.M.; Hurley, I.A.; Norris, R.P.; Osovitz, M.; Terasaki, M.; Wu, M.; et al. Anteroposterior axis patterning by early canonical Wnt signaling during hemichordate development. PLoS Biol. 2018, 16, e2003698. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chen, Y.G. Dishevelled: The hub of Wnt signaling. Cell. Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef]
- DVL3 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000161202-DVL3 (accessed on 12 April 2021).
- Zhao, Q.; Zhuang, K.; Han, K.; Tang, H.; Wang, Y.; Si, W.; Yang, Z. Silencing DVL3 defeats MTX resistance and attenuates stemness via Notch Signaling Pathway in colorectal cancer. Pathol. Res. Pract. 2020, 216, 152964. [Google Scholar] [CrossRef] [PubMed]
- Shafer, B.; Onishi, K.; Lo, C.; Colakoglu, G.; Zou, Y. Vangl2 Promotes Wnt/Planar Cell Polarity-like Signaling by Antagonizing Dvl1-Mediated Feedback Inhibition in Growth Cone Guidance. Dev. Cell 2011, 20, 177–191. [Google Scholar] [CrossRef]
- Dupasquier, S.; Blache, P.; Lasorsa, L.P.; Zhao, H.; Abraham, J.-D.; Haigh, J.J.; Ychou, M.; Prevostel, C. Modulating PKCα activity to target Wnt/β-catenin signaling in colon cancer. Cancers 2019, 11, 693. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Holland, C.H.; Szalai, B.; Saez-Rodriguez, J. Transfer of regulatory knowledge from human to mouse for functional genomics analysis. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2019, 1863, 194431. [Google Scholar] [CrossRef] [PubMed]
- LinkedOmics: Data Download. Datasets for Colon Adenocarcinoma (Prospective CPTAC-COAD). Available online: http://linkedomics.org/cptac-colon/ (accessed on 8 April 2021).
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. In Nature Methods; Nature Publishing Group: Berlin, Germany, 2016; Volume 13, pp. 731–740. [Google Scholar] [CrossRef]
- Nicioli da Silva, G.; Hanspers, K.; Pico, A.; Waagmeester, A. TP53 Network (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP1742 (accessed on 8 April 2021).
- Hanspers, K.; Riutta, A.; Willighagen, E. PI3K-Akt Signaling Pathway (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP4172 (accessed on 8 April 2021).
- Pandey, A.; Hansper, K.; Pico, A.; Salomonis, N. TGF-beta Signaling Pathway (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP366 (accessed on 8 April 2021).
- Lieberman, M.; Hanspers, K.; Pico, A.; Kelder, T. Wnt Signaling (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP428 (accessed on 8 April 2021).
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Kelder, T.; Pico, A.R.; Hanspers, K.; van Iersel, M.P.; Evelo, C. Mining Biological Pathways Using WikiPathways Web Services. PLoS ONE 2009, 4, 6447. [Google Scholar] [CrossRef]
- Team Reactome; Miller, R. MTOR Signalling (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP3318 (accessed on 8 April 2021).
- Zambon, A.C.; Alexander, P.; Coort, S. Apoptosis (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP254 (accessed on 8 April 2021).
- Conklin, B.; Sach, I.C.; Coort, S.; Salomonis, N. Cell Cycle (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP179 (accessed on 8 April 2021).
- Burel, S.; Hanspers, K.; Pico, A. MAPK Signaling Pathway (Homo Sapiens)—WikiPathways. Available online: https://www.wikipathways.org/index.php/Pathway:WP382 (accessed on 8 April 2021).
- Giuseppe, C. Logrank—File Exchange—MATLAB Central. 2021. Available online: https://uk.mathworks.com/matlabcentral/fileexchange/22317-logrank (accessed on 7 April 2021).
- Regression Learner App—MATLAB & Simulink—MathWorks United Kingdom. Available online: https://uk.mathworks.com/help/stats/regression-learner-app.html (accessed on 9 April 2021).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRC Signalling Pathways | Features | Key Functions in Colorectal Cancer |
|---|---|---|
| WNT | 119 | Function: Normal activation leads to tumour growth in advanced CRC. This function depends on the amount of B-catenin in the cytoplasm Cellular Activities: Cell fate specification, proliferation, migration, and asymmetric cell division Activated: WNT signal or APC mutation |
| PI3K/Akt | 340 | Function: Oncogenic role in the initiation and progression of CRC Inhibition: Reduction in CRC cell growth and increase in apoptosis Cellular Activities: Cell growth, proliferation, differentiation, and migration Activated: EGFR (epidermal growth factor receptor) Signalling
|
| MAPK | 252 | Function: Oncogenic role in CRC associated with tumour growth and disease progression Cellular Activities: Cell growth, differentiation, and survival |
| TGFß | 135 | Function: Reduces colon epithelial cells proliferation and induces apoptosis and differentiation Cellular Activities: Cell proliferation, differentiation, migration, apoptosis, and adhesion. Activated: Binding of TGF-β ligands to type II TGF-β receptors
|
| TP53 | 38 | Function: Regulation of the cell cycle, DNA replication and apoptosis |
| Apoptosis | 87 | Function: Apoptotic cell death induction by two main pathways, intrinsic and extrinsic signalling Resulting in the formation of a death-inducing signalling complex and an apoptosome |
| Cell Cycle | 122 | Function: Controls cell division |
| mTOR | 51 | Function: Regulation of cell growth and division Cellular Activities: Cell growth, proliferation, and survival |
| Notch | 48 | Function: Promotes CRC through regulating the cell cycle and cell apoptosis by regulation of p21 and PUMA (p53 upregulated modulator of apoptosis) genes Cellular Activities: Normal cell development, differentiation, proliferation, and apoptosis |
| Genes | Hazard Ratio | 95% Confidence Interval | p-Value | mafdr | Standard Error | Patient | Significant |
|---|---|---|---|---|---|---|---|
| (1) WNT | 53/119 | ||||||
| DVL3 | 2.8158 | (1.6159–4.9066) | 2.58 | 1.37 | 0.2833 | 329 | |
| VANGL2 | 2.7692 | (1.5039–5.0992) | 1.08 | 1.40 | 0.3115 | 329 | |
| (2) PI3K-Akt | 159/340 | ||||||
| EFNA1 | 3.2860 | (1.8496–5.8377) | 4.96 | 9.92 | 0.2932 | 329 | |
| KRAS | 0.2853 | (0.1502–0.5418) | 1.27 | 1.26 | 0.3272 | 329 | |
| (3) TP53 | 11/38 | ||||||
| BCL2 | 0.4610 | (0.2960–0.7179) | 6.12 | 0.0012 | 0.2260 | 329 | |
| CDKN2A | 1.8156 | (1.1931–2.7629) | 5.36 | 0.0054 | 0.2142 | 329 | |
| (4) MAPK | 113/252 | ||||||
| KRAS | 0.2853 | (0.1502–0.5418) | 1.27 | 2.09 | 0.3272 | 329 | |
| CACNA1I | 2.7685 | (1.6159–4.7432) | 2.10 | 2.09 | 0.2747 | 313 | |
| (5) TGF-Beta | 53/135 | ||||||
| TERT | 2.8734 | (1.6639–4.96201) | 1.53 | 3.05 | 0.2787 | 328 | |
| TGFB1I1 | 2.2381 | (1.3807–3.6281) | 1.08 | 0.0011 | 0.2465 | 329 | |
| Genes | Hazard Ratio | 95% Confidence Intervals | p-Value | mafdr | Standard Error | Patient | Significant |
|---|---|---|---|---|---|---|---|
| (1) WNT | 6/27/119 | ||||||
| PRKCA | 0.1557 | (0.0490–0.4947) | 1.61 | 0.0036 | 0.5898 | 63 | |
| ROCK2 | 5.7810 | (1.9229–17.3789) | 1.78 | 0.0036 | 0.5616 | 71 | |
| (2) PI3K-Akt | 29/100/340 | ||||||
| PPP2R1B | 6.8050 | (2.2113–20.9407) | 8.26 | 8.32 | 0.5735 | 73 | |
| EIF4E | 7.6285 | (2.3168–25.1176) | 8.32 | 8.32 | 0.6080 | 73 | |
| (3) TP53 | 2/5/38 | ||||||
| BAX | 10.8648 | (3.3684–35.0445) | 6.54 | 1.30 | 0.5975 | 71 | |
| BID | 0.2650 | (0.0814–0.8616) | 2.73 | 0.0273 | 0.6016 | 67 | |
| (4) MAPK | 20/80/252 | ||||||
| STK3 | 8.6543 | (2.4297–30.8245) | 8.69 | 0.0092 | 0.6481 | 65 | |
| PRKCA | 0.1557 | (0.0490–0.4947) | 1.61 | 0.0092 | 0.5898 | 63 | |
| (5) TGF-Beta | 15/71/135 | ||||||
| SMAD2 | 10.4343 | (2.7196–40.0330) | 6.30 | 0.0013 | 0.6860 | 67 | |
| SPTBN1 | 5.8318 | (1.9510–17.4318) | 1.60 | 0.0016 | 0.5587 | 73 | |
| A | Genes | Hazard Ratio | p-Value | mafdr | Patient | Proteomics |
|---|---|---|---|---|---|---|
| 1 | DVL3 | 2.8158 | 2.58 | 1.37 | 329 | |
| 2 | VANGL2 | 2.7693 | 1.08 | 1.40 | 329 | |
| 3 | WNT6 | 2.3175 | 1.34 | 1.40 | 299 | |
| 4 | TCF7L1 | 2.0324 | 1.35 | 1.40 | 329 | |
| 5 | CER1 | 2.8871 | 1.42 | 1.40 | 147 | |
| 6 | SOX17 | 2.1048 | 1.59 | 1.40 | 329 | |
| 7 | NKD2 | 2.8598 | 1.85 | 1.40 | 329 | |
| 8 | DKK1 | 1.9732 | 2.16 | 1.43 | 309 | |
| 9 | SFRP2 | 1.8539 | 3.94 | 2.09 | 327 | Y |
| 10 | PLCB1 | 2.9436 | 4.32 | 2.09 | 329 | |
| 11 | PRICKLE1 | 1.8229 | 5.01 | 2.09 | 329 | |
| 12 | MAPK8 | 0.5491 | 5.19 | 2.09 | 329 | Y |
| 13 | PRICKLE2 | 1.8104 | 5.38 | 2.09 | 329 | |
| 14 | MYC | 0.5535 | 5.53 | 2.09 | 329 | |
| 15 | WIF1 | 2.4639 | 6.46 | 2.20 | 246 | |
| 16 | CAMK2B | 1.9981 | 6.66 | 2.20 | 302 | |
| 17 | WNT3A | 2.3137 | 7.09 | 2.21 | 243 | |
| 18 | LEF1 | 1.9751 | 8.65 | 2.52 | 329 | |
| 19 | RHOA | 0.4792 | 9.47 | 2.52 | 329 | Y |
| 20 | MAPK10 | 1.7294 | 9.88 | 2.52 | 329 | |
| 21 | CAMK2D | 0.5514 | 1.05 | 2.52 | 329 | Y |
| 22 | WNT3 | 1.8141 | 1.06 | 2.52 | 328 | |
| 23 | FZD8 | 1.8913 | 1.09 | 2.52 | 329 | |
| 24 | SERPINF1 | 1.7116 | 1.14 | 2.52 | 329 | Y |
| 25 | FZD1 | 1.7251 | 1.24 | 2.55 | 329 | |
| 26 | NKD1 | 1.8480 | 1.25 | 2.55 | 329 | |
| 27 | CXXC4 | 1.8290 | 2.62 | 1.34 | 318 | |
| 28 | PPP3CA | 0.4335 | 2.62 | 1.39 | 329 | Y |
| 29 | PLCB3 | 0.5772 | 2.74 | 1.50 | 329 | |
| 30 | CSNK1A1L | 0.4710 | 2.74 | 1.55 | 321 | |
| 31 | PLCB2 | 1.6955 | 3.46 | 2.10 | 329 | |
| 32 | ROR2 | 1.6511 | 3.46 | 2.14 | 329 | |
| 33 | PRKCB | 0.5888 | 3.46 | 2.15 | 329 | |
| 34 | WNT2 | 0.5479 | 3.52 | 2.26 | 329 | |
| 35 | SFRP1 | 1.6386 | 3.53 | 2.33 | 322 | |
| 36 | WNT10A | 2.1170 | 3.69 | 2.56 | 328 | |
| 37 | GSK3B | 1.6397 | 3.69 | 2.61 | 329 | Y |
| 38 | FRAT1 | 0.6144 | 3.69 | 2.65 | 329 | |
| 39 | SENP2 | 2.2535 | 3.84 | 2.92 | 329 | |
| 40 | DKK4 | 1.7497 | 3.84 | 2.96 | 301 | |
| 41 | CTNNBIP1 | 1.5838 | 3.84 | 2.97 | 329 | |
| 42 | FZD7 | 2.0424 | 4.18 | 3.40 | 329 | |
| 43 | WNT1 | 1.7533 | 4.18 | 3.46 | 244 | |
| 44 | DKK2 | 1.8152 | 4.18 | 3.59 | 326 | |
| 45 | WNT16 | 2.0367 | 4.18 | 3.62 | 276 | |
| 46 | WNT2B | 0.6417 | 4.18 | 3.68 | 329 | |
| 47 | NFATC4 | 1.5715 | 4.18 | 3.70 | 329 | |
| 48 | NLK | 1.7784 | 4.23 | 3.83 | 329 | |
| 49 | NOTUM | 1.5754 | 4.34 | 4.08 | 329 | |
| 50 | CSNK2A3 | 0.4868 | 4.34 | 4.09 | 329 | |
| 51 | WNT4 | 0.6060 | 4.82 | 4.64 | 329 | |
| 52 | FZD5 | 0.6579 | 4.82 | 4.81 | 329 | |
| 53 | DAAM1 | 1.7076 | 4.82 | 4.82 | 329 | |
| B | Proteins | Hazard Ratio | p-Value | mafdr | Patient | |
| 1 | PRKCA | 0.1557 | 1.62 | 3.66 | 63 | |
| 2 | ROCK2 | 5.7810 | 1.78 | 3.66 | 71 | |
| 3 | CSNK2A1 | 5.6824 | 1.83 | 3.66 | 73 | |
| 4 | LRP1 | 0.2594 | 2.62 | 3.93 | 72 | |
| 5 | CSNK1A1 | 0.2846 | 3.71 | 4.46 | 72 | |
| 6 | GPC4 | 0.2512 | 4.58 | 4.58 | 50 |
| Pathway | Hazard Ratio | p-Value |
|---|---|---|
| WNT | 1.9731 | 0.0013 |
| PI3K | 0.5775 | 0.0276 |
| TGFβ | 1.5792 | 0.0477 |
| MAPK | 1.8058 | 0.0489 |
| p53 | 0.5972 | 0.0687 |
| (A) Top Six Prognostic Genes and Proteins | Features |
|---|---|
| mRNA Sequencing TCGA Legacy | DVL3 |
| VANGL2 | |
| WNT6 | |
| TCF7L1 | |
| CER1 | |
| SOX17 | |
| Proteomics TCGA Legacy | PRKCA |
| ROCK2 | |
| CSNK2A1 | |
| LRP1 | |
| CSNK1A1 | |
| GPC4 | |
| (B) Linear Regression Models | Features |
| Model 1: Mechanistic Model Model Input: CPTAC RNA RMSE: 1.0167 11 Features | APC |
| AXIN1 | |
| CTNNB1 | |
| DKK2 | |
| DVL2 | |
| DVL3 | |
| GSK3B | |
| LRP6 | |
| SFRP1 | |
| SFRP2 | |
| WNT3 | |
| Model 2: Canonical WNT model with top two prognostic genes and proteins Model Input: CPTAC RNA RMSE: 0.6416 9 Features | DKK3 |
| FZD5 | |
| NKD1 | |
| NOTUM | |
| WNT11 | |
| DVL3 | |
| PRKCA | |
| ROCK2 | |
| VANGL2 | |
| Model 3: Canonical WNT model with top two prognostic proteins Model Input: CPTAC proteomics RMSE: 1.0509 7 Features | CTBP1 |
| CTBP2 | |
| GPC4 | |
| PLCB4 | |
| PRKCA | |
| RAC1 | |
| ROCK2 |
| Study | Datasets | Analyses |
|---|---|---|
| 2016 TCGA Colorectal Adenocarcinoma, GDAC Firehose Legacy |
20,532 genes Cohort of 329 patients
5562 proteins Cohort of 74 patients
Event Free Survival |
|
| 2019 Prospective CPTAC-COAD Colon Adenocarcinoma |
RNA Expression (RSEM-UQ, Log2(Val+1)) 13,482 genes Cohort of 106 patients
Protein Expression (TMT, Log2ratio) 6422 proteins Cohort of 96 patients
Cohort of 110 patients 79 patients analysed Event Free Survival |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nwaokorie, A.; Fey, D. Personalised Medicine for Colorectal Cancer Using Mechanism-Based Machine Learning Models. Int. J. Mol. Sci. 2021, 22, 9970. https://doi.org/10.3390/ijms22189970
Nwaokorie A, Fey D. Personalised Medicine for Colorectal Cancer Using Mechanism-Based Machine Learning Models. International Journal of Molecular Sciences. 2021; 22(18):9970. https://doi.org/10.3390/ijms22189970
Chicago/Turabian StyleNwaokorie, Annabelle, and Dirk Fey. 2021. "Personalised Medicine for Colorectal Cancer Using Mechanism-Based Machine Learning Models" International Journal of Molecular Sciences 22, no. 18: 9970. https://doi.org/10.3390/ijms22189970
APA StyleNwaokorie, A., & Fey, D. (2021). Personalised Medicine for Colorectal Cancer Using Mechanism-Based Machine Learning Models. International Journal of Molecular Sciences, 22(18), 9970. https://doi.org/10.3390/ijms22189970