
Identification of FadT as a Novel Quorum Quenching Enzyme for the Degradation of Diffusible Signal Factor in Cupriavidus pinatubonensis Strain HN-2

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Media
2.2. Whole-Genome Sequencing and Assembly
2.3. Genome Component Prediction and Gene Function
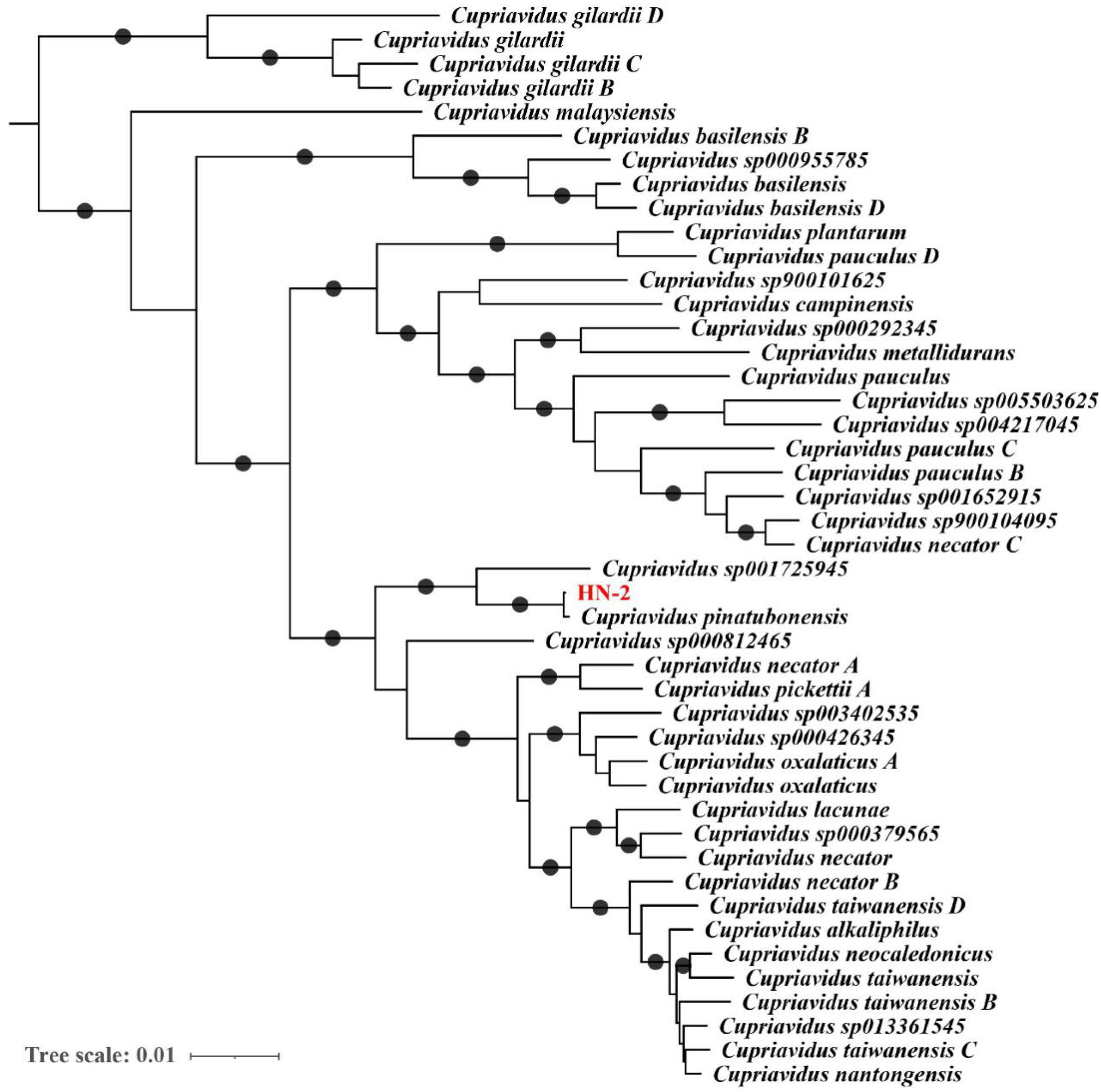
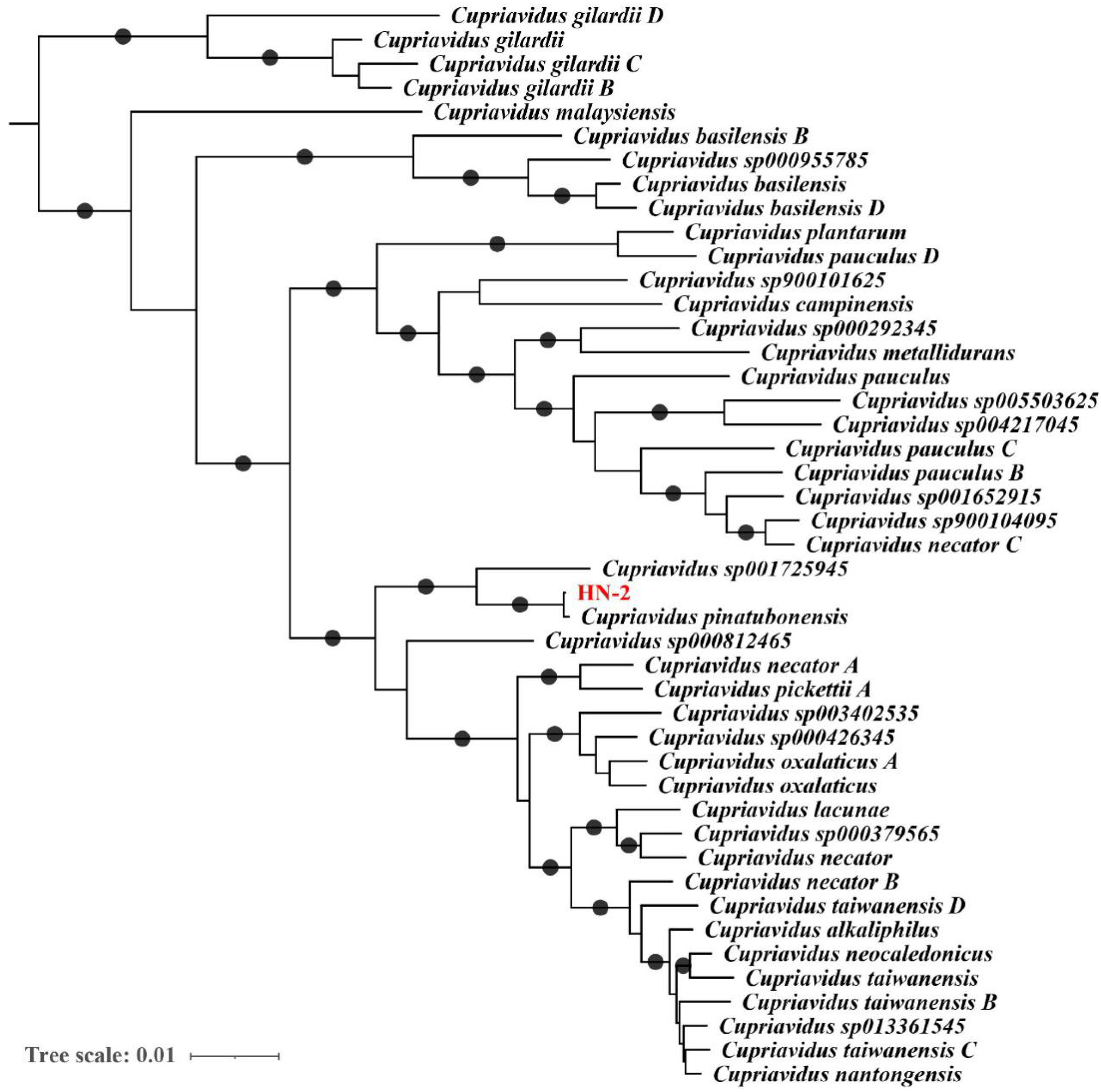
2.4. Genome-Based Taxonomic Classification Analysis
2.5. Construction of an In-Frame Deletion Mutant and Complementation
2.6. Purification of FadT
2.7. Enzyme Activity Assay
2.8. Virulence Tests
2.9. GenBank Accession Number
3. Results
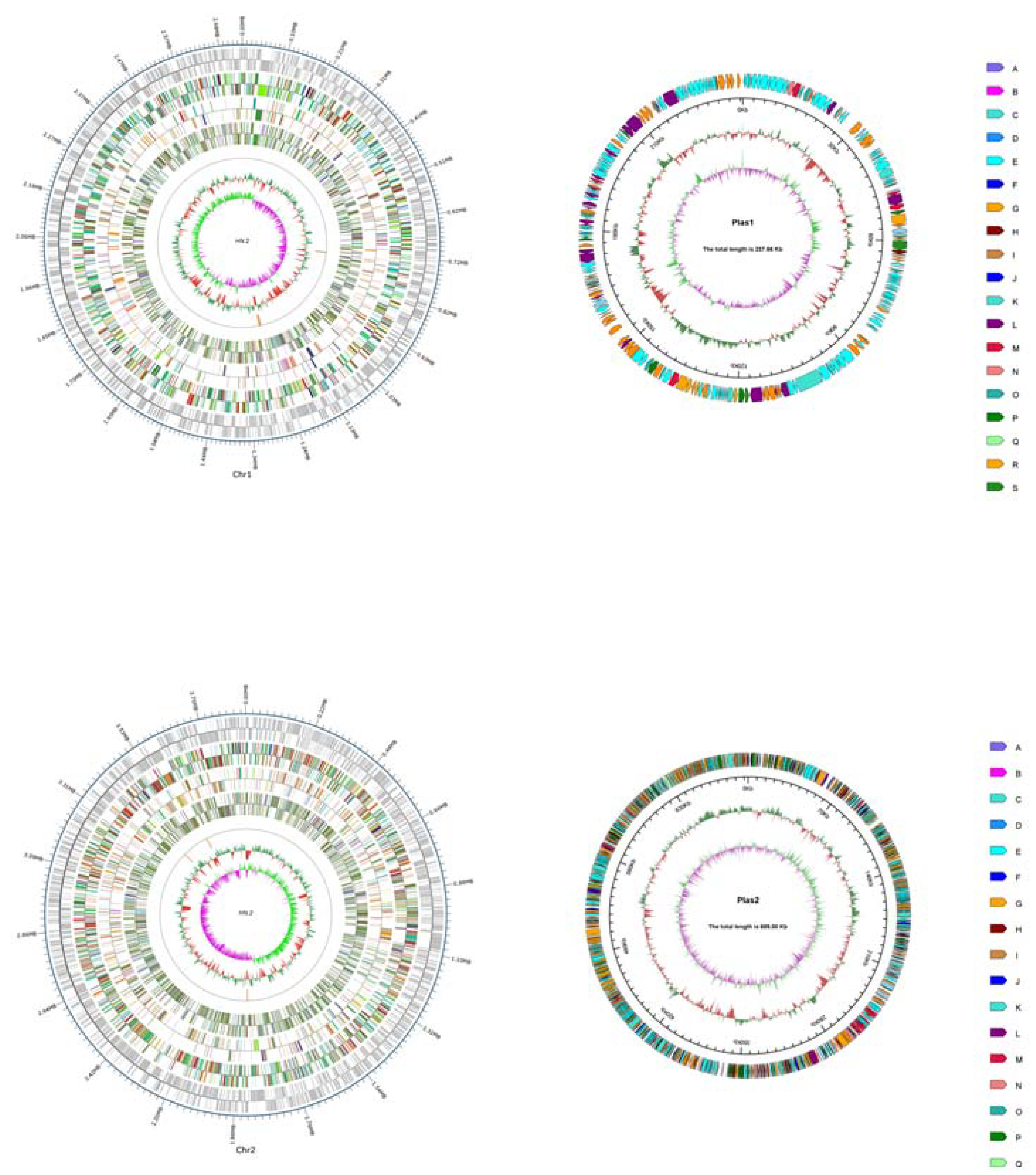
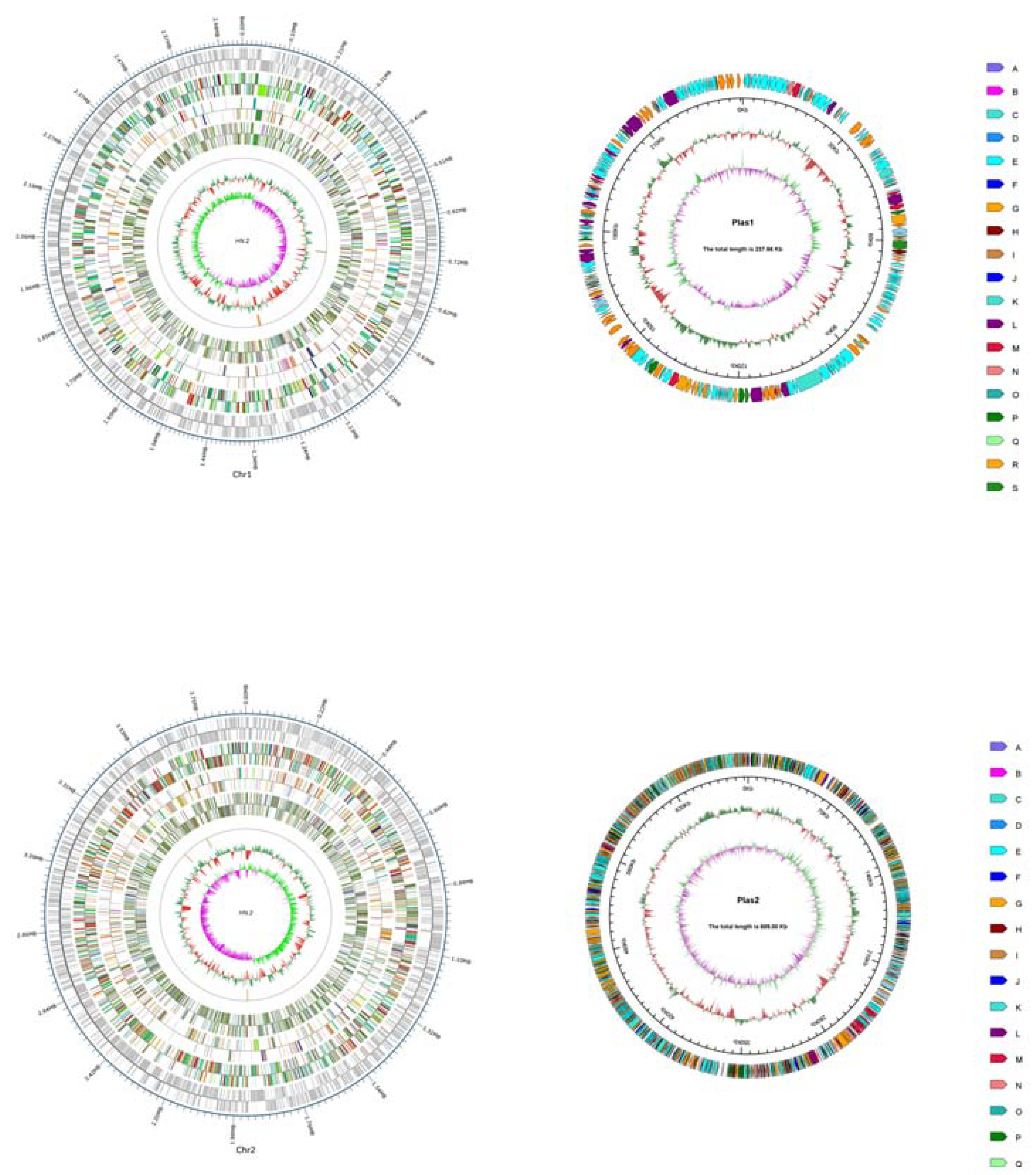
3.1. Sequencing and Analysis of the C. pinatubonensis HN-2 Genome
3.2. Identification and Cloning of the Gene Responsible for the DSF Degradation
3.3. Codon Optimization, Expression, and Purification of FadT
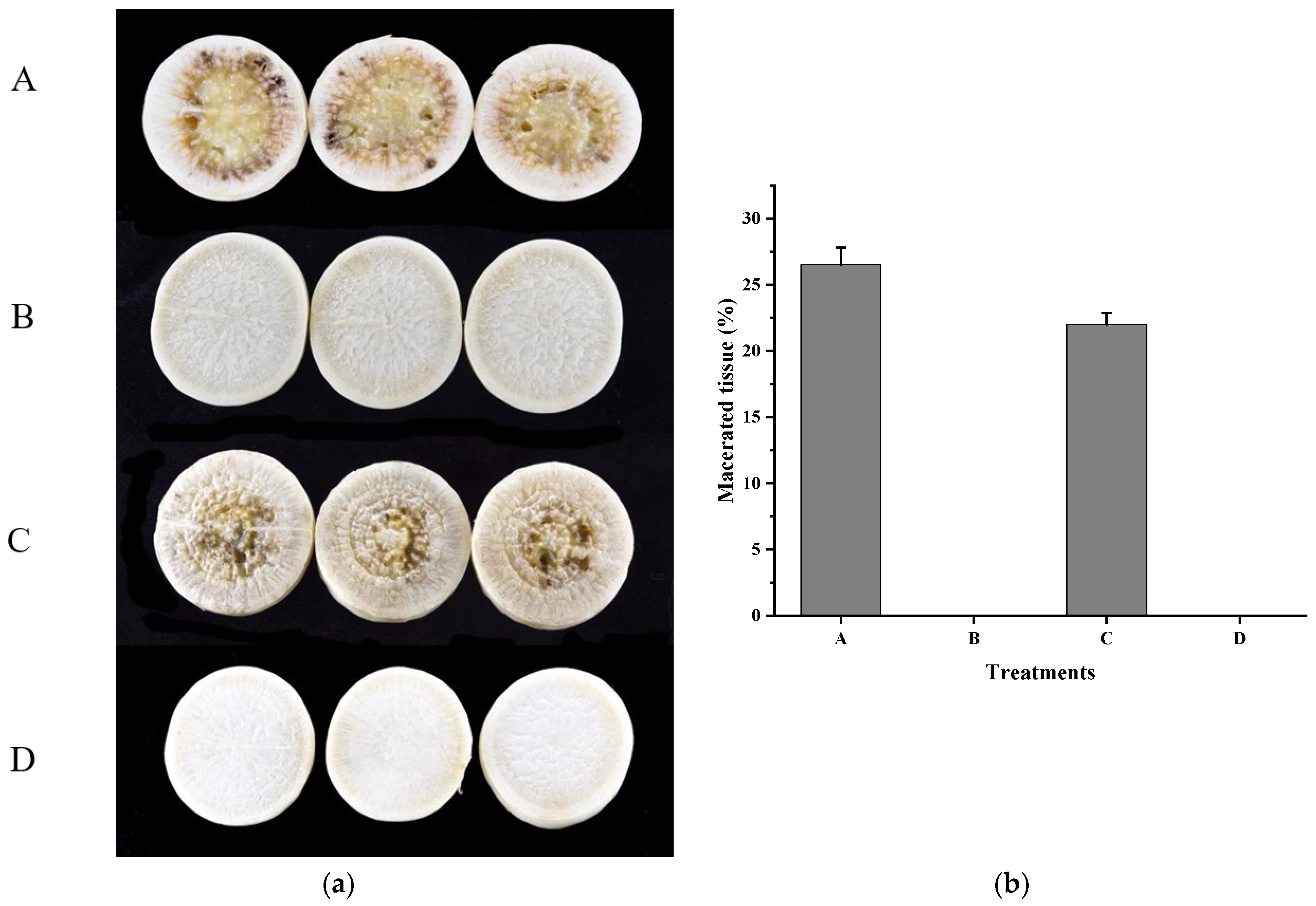
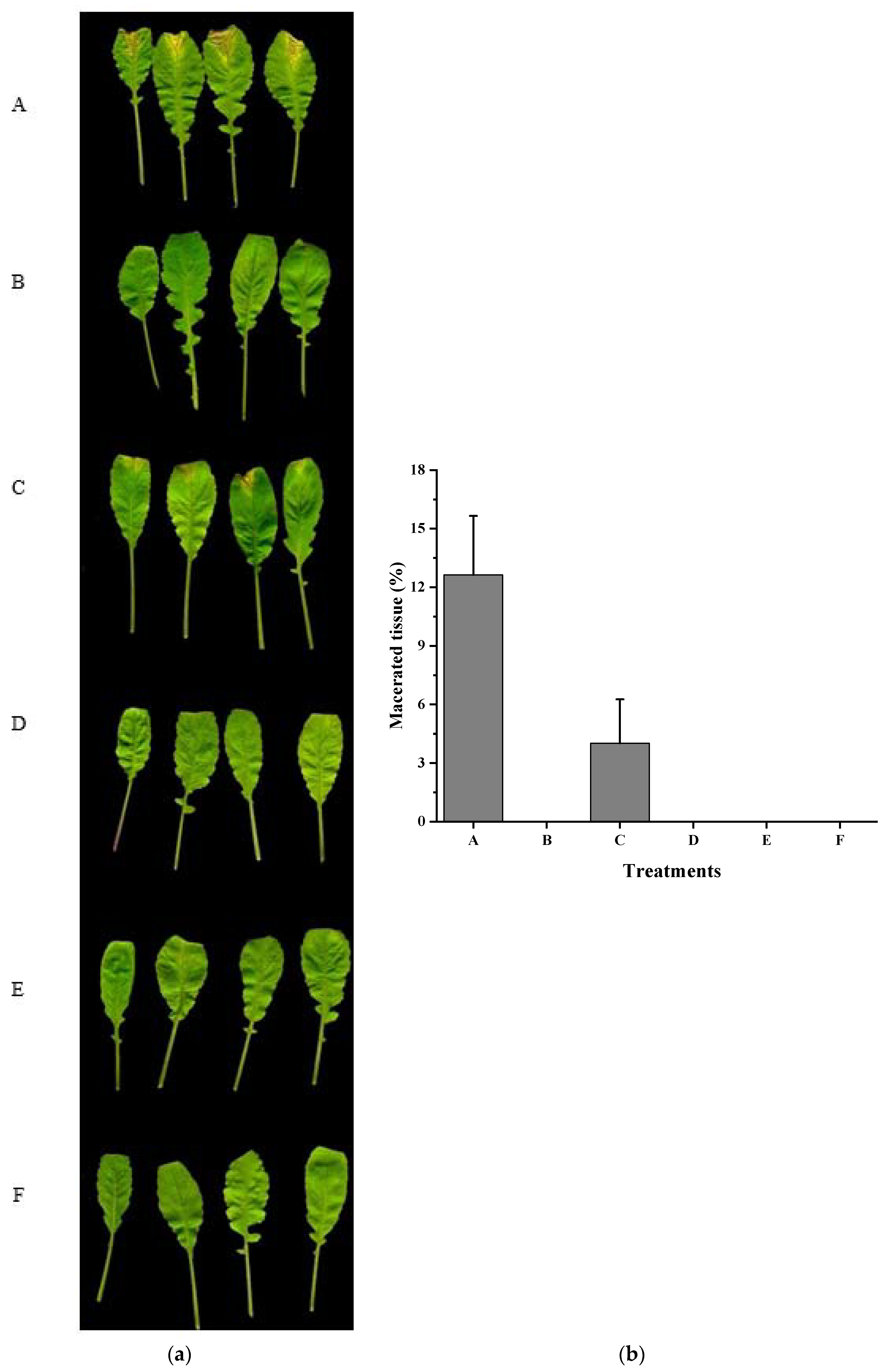
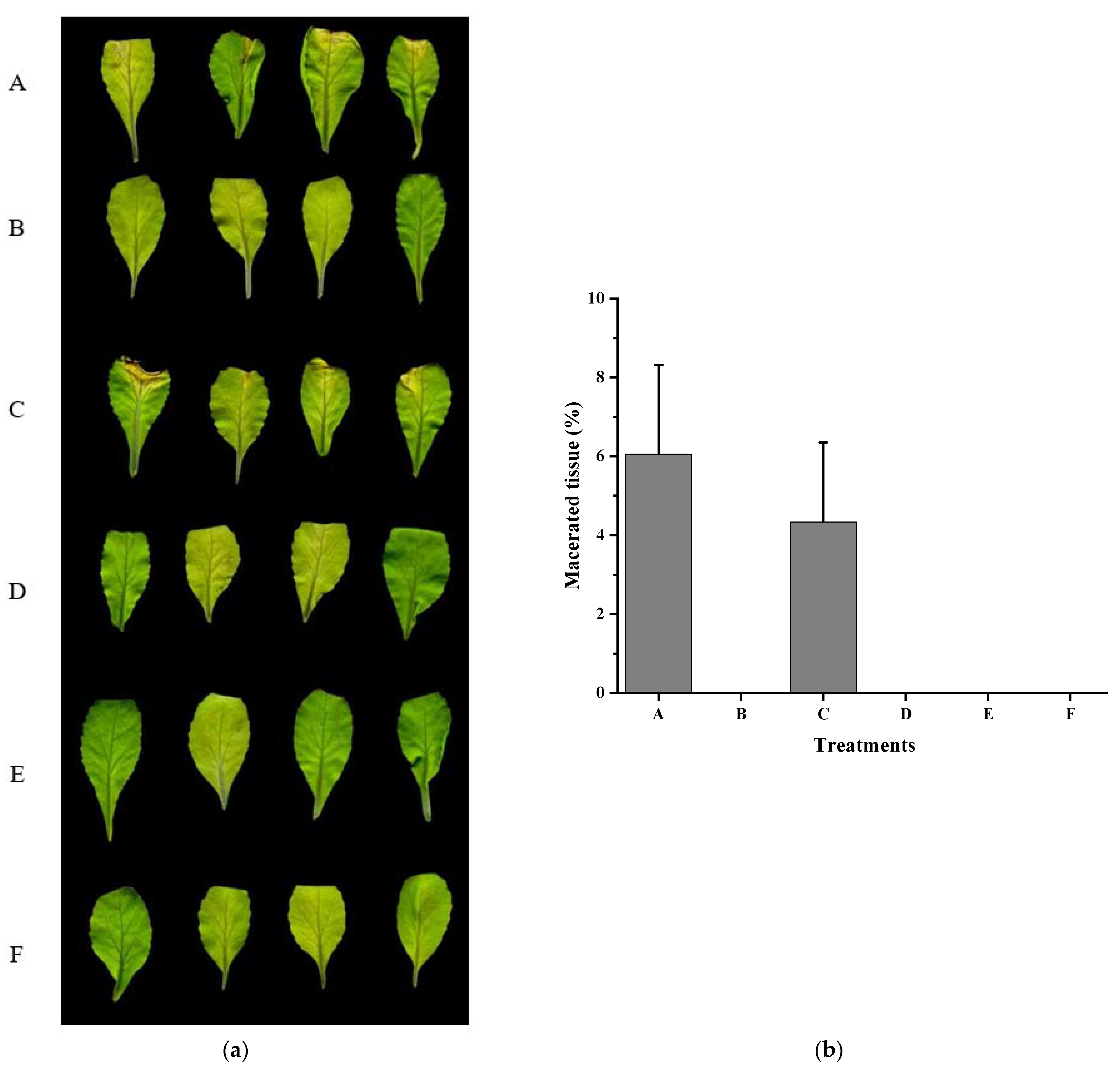
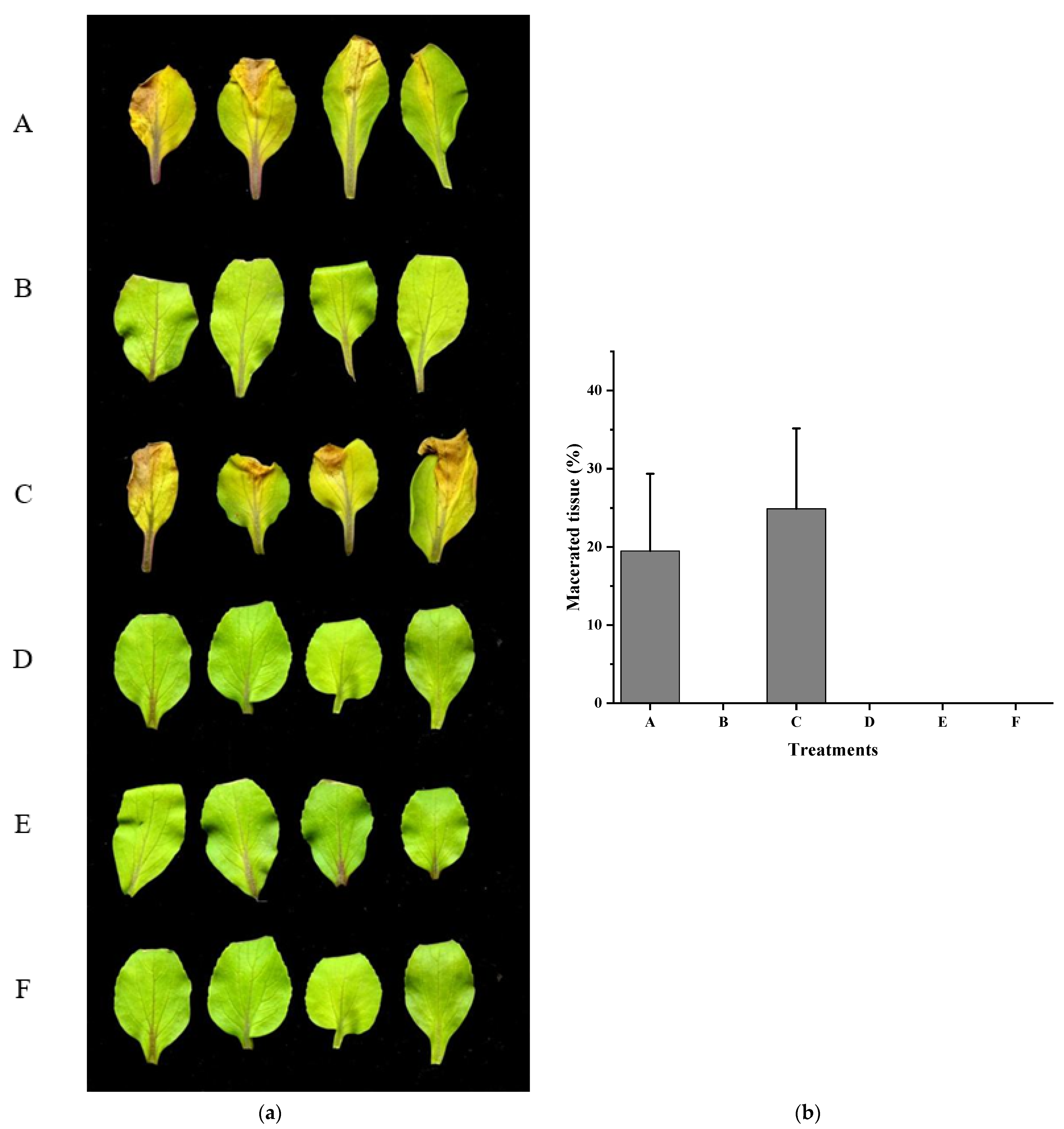
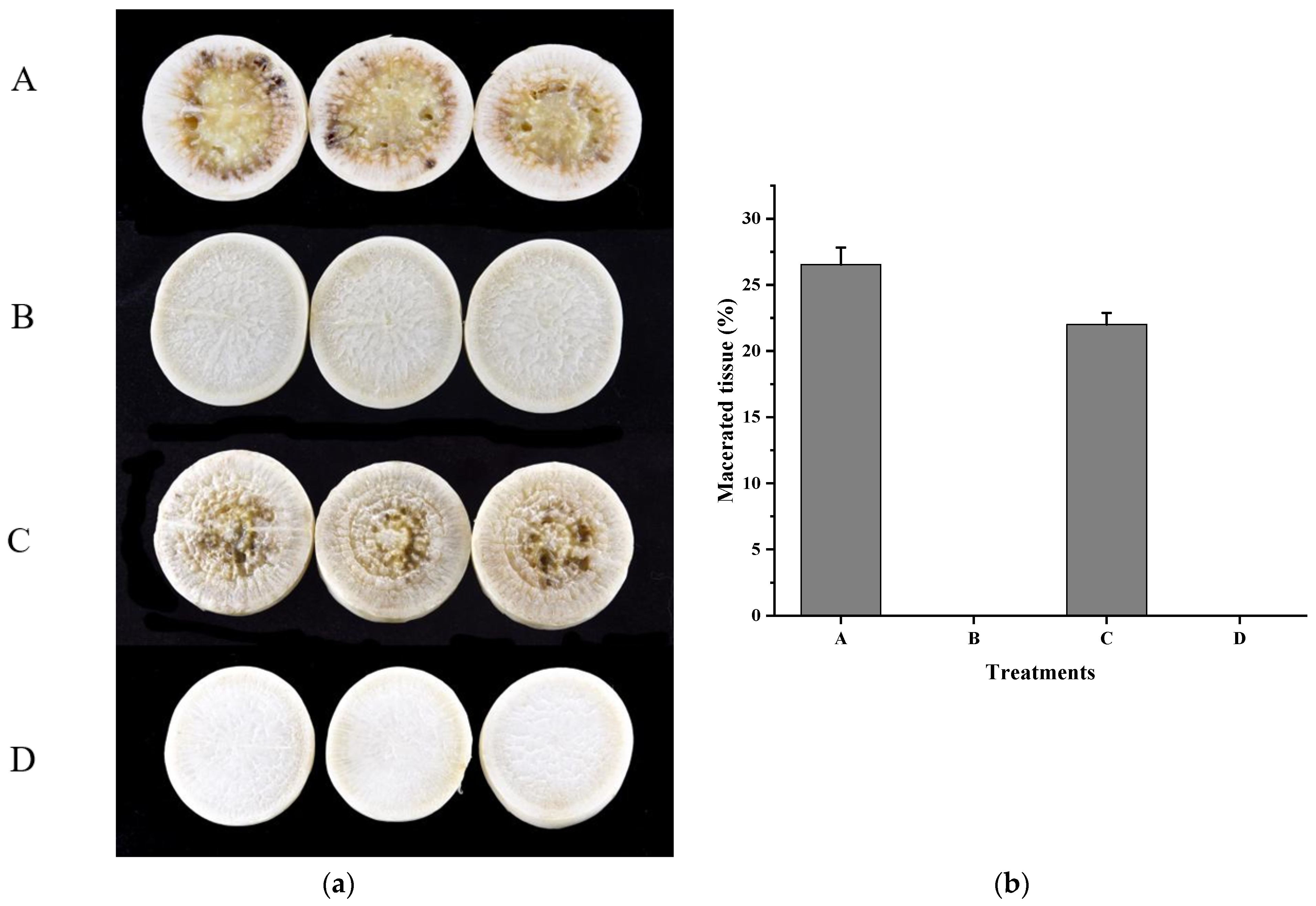
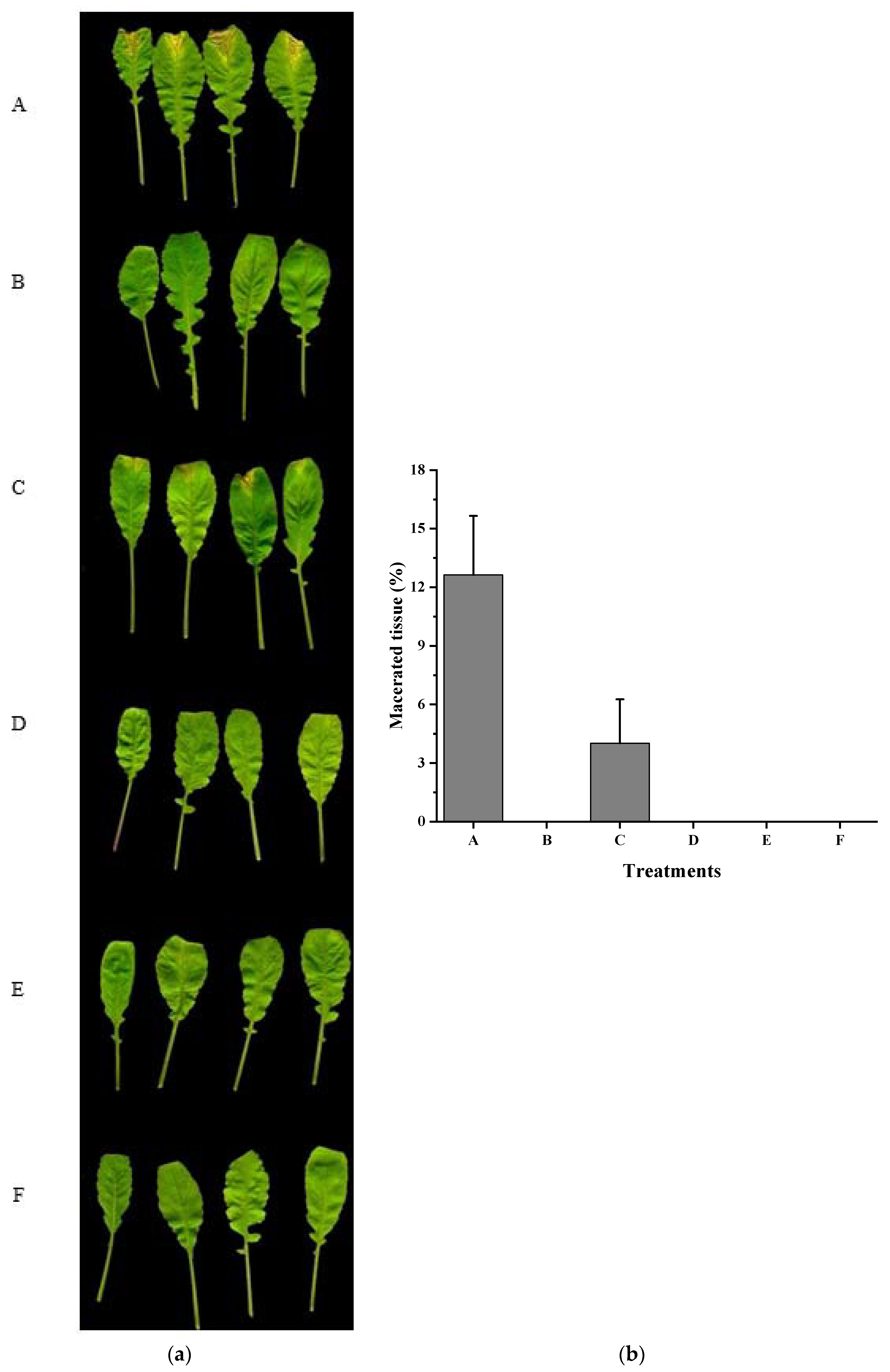
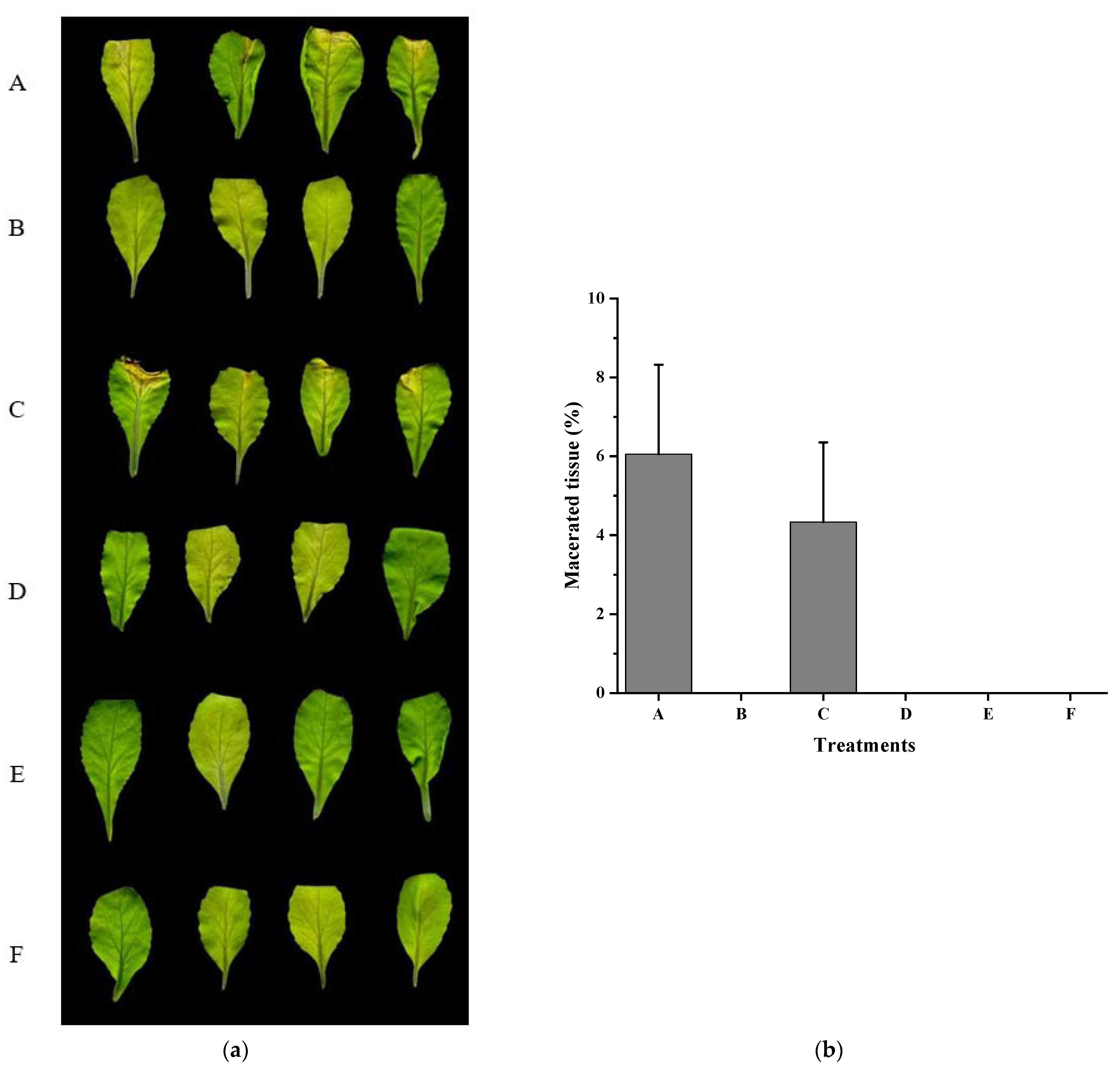
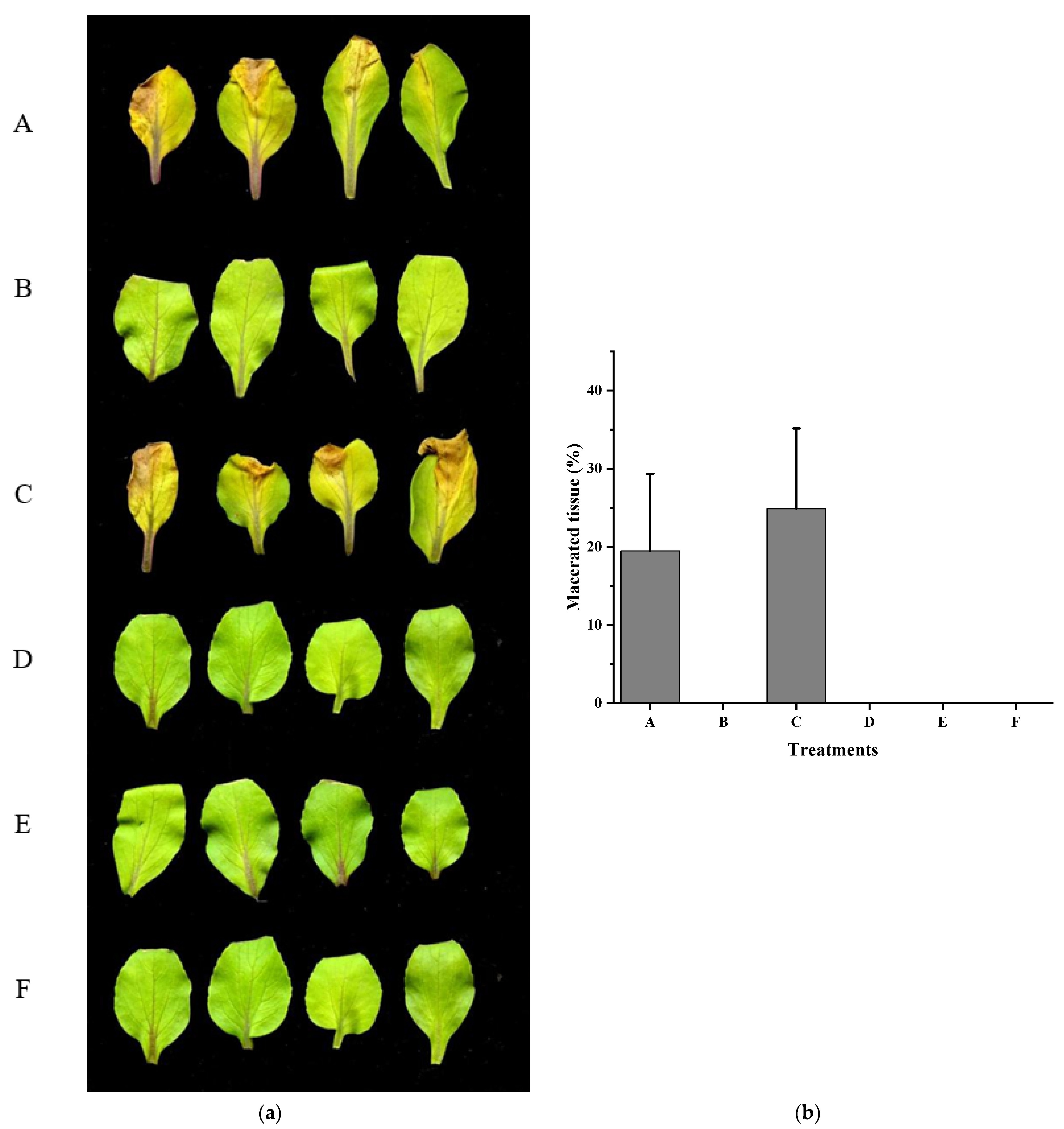
3.4. Xcc Expressing fadT Loses Virulence in Planta
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gupta, M.; Vikram, A.; Bharat, N. Black rot—A devastating disease of crucifers: A review. Agric. Rev. 2013, 34, 269. [Google Scholar] [CrossRef]
- Basavaraju, M.; Sisnity, V.S.; Palaparthy, R.; Addanki, P.K. Quorum quenching: Signal jamming in dental plaque biofilms. J. Dent. Sci. 2016, 11, 349–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turan, N.B.; Chormey, D.S.; Büyükpınar, Ç.; Engin, G.O.; Bakirdere, S. Quorum sensing: Little talks for an effective bacterial coordination. TrAC Trends Anal. Chem. 2017, 91, 1–11. [Google Scholar] [CrossRef]
- Wang, L.-H.; He, Y.; Gao, Y.; Wu, J.E.; Dong, Y.-H.; He, C.; Wang, S.X.; Weng, L.-X.; Xu, J.-L.; Tay, L.; et al. A bacterial cell-cell communication signal with cross-kingdom structural analogues. Mol. Microbiol. 2004, 51, 903–912. [Google Scholar] [CrossRef]
- Newman, K.L.; Chatterjee, S.; Ho, K.A.; Lindow, S.E. Virulence of Plant Pathogenic Bacteria Attenuated by Degradation of Fatty Acid Cell-to-Cell Signaling Factors. Mol. Plant-Microbe Interact. 2008, 21, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhang, L.-H.; Cámara, M.; He, Y.-W. The DSF Family of Quorum Sensing Signals: Diversity, Biosynthesis, and Turnover. Trends Microbiol. 2017, 25, 293–303. [Google Scholar] [CrossRef]
- Ryan, R.P.; An, S.-Q.; Allan, J.H.; McCarthy, Y.; Dow, J.M. The DSF Family of Cell-Cell Signals: An Expanding Class of Bacterial Virulence Regulators. PLoS Pathog. 2015, 11, e1004986. [Google Scholar] [CrossRef]
- Ma, F.; Xu, S.; Tang, Z.; Li, Z.; Zhang, L. Use of antimicrobials in food animals and impact of transmission of antimicrobial resistance on humans. Biosaf. Health 2021, 3, 32–38. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, T.; Wang, J.; Wang, Y.; Zhang, X.-H. The Mechanisms and Applications of Quorum Sensing (QS) and Quorum Quenching (QQ). J. Ocean Univ. China 2019, 18, 1427–1442. [Google Scholar] [CrossRef]
- Zhang, L.-H. Quorum quenching and proactive host defense. Trends Plant Sci. 2003, 8, 238–244. [Google Scholar] [CrossRef]
- Fetzner, S. Quorum quenching enzymes. J. Biotechnol. 2015, 201, 2–14. [Google Scholar] [CrossRef]
- Schuster, M.; Sexton, D.J.; Diggle, S.P.; Greenberg, E.P. Acyl-Homoserine Lactone Quorum Sensing: From Evolution to Application. Annu. Rev. Microbiol. 2013, 67, 43–63. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, X.-Y.; Sun, S.; Yang, L.-C.; Jiang, B.-L.; He, Y.-W. Identification and characterization of naturally occurring DSF-family quorum sensing signal turnover system in the phytopathogen Xanthomonas. Environ. Microbiol. 2015, 17, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Yu, Y.; Dong, H.; Wang, H.; Cronan, J.E. Xanthomonas campestris RpfB is a fatty Acyl-CoA ligase required to counteract the thioesterase activity of the RpfF diffusible signal factor (DSF) synthase. Mol. Microbiol. 2014, 93, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, T.; Zhou, T.; Li, Q.; Xu, X.; Fan, X.; Zhang, L.; Chen, S. Cupriavidus sp. HN-2, a Novel Quorum Quenching Bacterial Isolate, is a Potent Biocontrol Agent Against Xanthomonas campestris pv. campestris. Microorganisms 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Huang, Y.; Pang, S.; Zhou, T.; Lin, Z.; Yu, H.; Zhang, G.; Bhatt, P.; Chen, S. Novel Mechanism and Kinetics of Tetramethrin Degradation Using an Indigenous Gordonia cholesterolivorans A16. Int. J. Mol. Sci. 2021, 22, 9242. [Google Scholar] [CrossRef]
- Reiner, J.; Pisani, L.; Qiao, W.; Singh, R.; Yang, Y.; Shi, L.; Khan, W.A.; Sebra, R.; Cohen, N.; Babu, A.; et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet–Biedl Syndrome 9 (BBS9) deletion. npj Genom. Med. 2018, 3, 3. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichinger, V.; Nussbaumer, T.; Platzer, A.; Jehl, M.E.; Arnold, R.; Rattei, T. EffectiveDB—Updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 2016, 44, D669–D674. [Google Scholar] [CrossRef]
- Hsiao, W.; Wan, I.; Jones, S.; Brinkman, F.S.L. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Kanehisa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galperin, M.; Makarova, K.S.; Wolf, Y.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Sayer, M.H., Jr.; Reddy, V.S.; Tamang, D.G.; Västermark, Å. The Transporter Classification Database. Nucleic Acids Res. 2014, 42, D251–D258. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012, 40, D641–D645. [Google Scholar] [CrossRef]
- Chen, G.; Gan, J.; Yang, C.; Zuo, Y.; Peng, J.; Li, M.; Huo, W.; Xie, Y.; Zhang, Y.; Wang, T.; et al. The SiaA/B/C/D signaling network regulates biofilm formation in Pseudomonas aeruginosa. EMBO J. 2020, 39, e105997. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 43, D645–D655. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Goeker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Kreft, L.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinformatics 2017, 33, 2946–2947. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, J.; Birch, R.G. High affinity binding of albicidin phytotoxins by the AlbA protein from Klebsiella oxytoca. Microbiology 1998, 144, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Ye, T.; Zhang, W.; Feng, Z.; Fan, X.; Xu, X.; Mishra, S.; Zhang, L.; Chen, S. Characterization of a Novel Quorum-Quenching Bacterial Strain, Burkholderia anthina HN-8, and Its Biocontrol Potential against Black Rot Disease Caused by Xanthomonas campestris pv. campestris. Microorganisms 2020, 8, 1485. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, S.; Hu, M.; Hu, Q.; Luo, J.; Li, Y. Purification and Characterization of a Novel Chlorpyrifos Hydrolase from Cladosporium cladosporioides Hu-01. PLoS ONE 2012, 7, e38137. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lin, Q.; Xiao, Y.; Deng, Y.; Chang, C.; Zhong, G.; Hu, M.; Zhang, L.-H. Monooxygenase, a Novel Beta-Cypermethrin Degrading Enzyme from Streptomyces sp. PLoS ONE 2013, 8, e75450. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, P.; Rene, E.R.; Kumar, A.J.; Zhang, W.; Chen, S. Binding interaction of allethrin with esterase: Bioremediation potential and mechanism. Bioresour. Technol. 2020, 315, 123845. [Google Scholar] [CrossRef]
- Bhatt, P.; Zhou, X.; Huang, Y.; Zhang, W.; Chen, S. Characterization of the role of esterases in the biodegradation of organophosphate, carbamate, and pyrethroid pesticides. J. Hazard. Mater. 2021, 411, 125026. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.; Fouhy, Y.; Lucey, J.F.; Jiang, B.-L.; He, Y.-Q.; Feng, J.-X.; Tang, J.-L.; Dow, J.M. Cyclic di-GMP signalling in the virulence and environmental adaptation of Xanthomonas campestris. Mol. Microbiol. 2007, 63, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fan, X.; Li, J.; Ye, T.; Mishra, S.; Zhang, L.; Chen, S. Exploration of the Quorum-Quenching Mechanism in Pseudomonas nitroreducens W-7 and Its Potential to Attenuate the Virulence of Dickeya zeae EC1. Front. Microbiol. 2021, 12, 694161. [Google Scholar] [CrossRef]
- Li, J.; Hu, M.; Xue, Y.; Chen, X.; Lu, G.; Zhang, L.; Zhou, J. Screening, Identification and Efficacy Evaluation of Antagonistic Bacteria for Biocontrol of Soft Rot Disease Caused by Dickeya zeae. Microorganisms 2020, 8, 697. [Google Scholar] [CrossRef]
- Lv, M.; Hu, M.; Li, P.; Jiang, Z.; Zhang, L.; Zhou, J. A two-component regulatory system VfmIH modulates multiple virulence traits in Dickeya zeae. Mol. Microbiol. 2019, 111, 1493–1509. [Google Scholar] [CrossRef]
- Li, S.; Tang, Y.; Fang, X.; Qiao, T.; Han, S.; Zhu, T. Whole-genome sequence of Arthrinium phaeospermum, a globally distributed pathogenic fungus. Genomics 2020, 112, 919–929. [Google Scholar] [CrossRef]
- Ye, T.; Zhou, T.; Xu, X.; Zhang, W.; Fan, X.; Mishra, S.; Zhang, L.; Zhou, X.; Chen, S. Whole-Genome Sequencing Analysis of Quorum Quenching Bacterial Strain Acinetobacter lactucae QL-1 Identifies the FadY Enzyme for Degradation of the Diffusible Signal Factor. Int. J. Mol. Sci. 2020, 21, 6729. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gao, Y.; Chen, X.; Yu, Z.; Li, X. Quorum Quenching Enzymes and Their Application in Degrading Signal Molecules to Block Quorum Sensing-Dependent Infection. Int. J. Mol. Sci. 2013, 14, 17477–17500. [Google Scholar] [CrossRef]
- Zhang, W.; Luo, Q.; Zhang, Y.; Fan, X.; Ye, T.; Mishra, S.; Bhatt, P.; Zhang, L.; Chen, S. Quorum Quenching in a Novel Acinetobacter sp. XN-10 Bacterial Strain against Pectobacterium carotovorum subsp. carotovorum. Microorganisms 2020, 8, 1100. [Google Scholar] [CrossRef]
- Caicedo, J.C.; Villamizar, S.; Ferro, M.I.T.; Kupper, K.C.; Ferro, J.A. Bacteria from the citrus phylloplane can disrupt cell–cell signalling in Xanthomonas citri and reduce citrus canker disease severity. Plant Pathol. 2016, 65, 5. [Google Scholar] [CrossRef]
- Ye, T.; Zhou, T.; Fan, X.; Bhatt, P.; Zhang, L.; Chen, S. Acinetobacter lactucae Strain QL-1, a Novel Quorum Quenching Candidate Against Bacterial Pathogen Xanthomonas campestris pv. campestris. Front. Microbiol. 2019, 10, 2867. [Google Scholar] [CrossRef]
- Dong, Y.H.; Xu, J.L.; Li, X.Z.; Zhang, L.H. AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora. Proc. Natl. Acad. Sci. USA 2000, 97, 3526–3531. [Google Scholar] [CrossRef]
- An, S.-Q.; Potnis, N.; Dow, M.; Vorhölter, F.-J.; He, Y.-Q.; Becker, A.; Teper, D.; Li, Y.; Wang, N.; Bleris, L.; et al. Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol. Rev. 2020, 44, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Hu, M.; Xue, Y.; Li, Z.; Zhang, Y.; Zheng, D.; Lu, G.; Wang, J.; Zhou, J. Five Fungal Pathogens Are Responsible for Bayberry Twig Blight and Fungicides Were Screened for Disease Control. Microorganisms 2020, 8, 689. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, L.; Chen, S.; Zhang, L.-H. A Quorum Quenching Bacterial Isolate Contains Multiple Substrate-Inducible Genes Conferring Degradation of Diffusible Signal Factor. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef] [PubMed]
- Azimi, S.; Klementiev, A.D.; Whiteley, M.; Diggle, S.P. Bacterial Quorum Sensing During Infection. Annu. Rev. Microbiol. 2020, 74. [Google Scholar] [CrossRef]
- Ban, H.; Chai, X.; Lin, Y.; Zhou, Y.; Peng, D.; Zhou, Y.; Zou, Y.; Yu, Z.; Sun, M. Transgenic Amorphophallus konjac expressing synthesized acyl-homoserine lactonase (aiiA) gene exhibit enhanced resistance to soft rot disease. Plant Cell Rep. 2009, 28, 1847–1855. [Google Scholar] [CrossRef]
- Cho, H.S.; Park, S.Y.; Ryu, C.M.; Kim, J.F.; Kim, J.G.; Park, S.H. Interference of quorum sensing and virulence of the rice pathogen Burkholderia glumae by an engineered endophytic bacterium. FEMS Microbiol. Ecol. 2007, 60, 14–23. [Google Scholar] [CrossRef]
- Defoirdt, T.; Thanh, L.D.; Van Delsen, B.; De Schryver, P.; Sorgeloos, P.; Boon, N.; Bossier, P. N-acylhomoserine lactone-degrading Bacillus strains isolated from aquaculture animals. Aquaculture 2011, 311, 258–260. [Google Scholar] [CrossRef]
- Fan, X.; Ye, T.; Li, Q.; Bhatt, P.; Zhang, L.; Chen, S. Potential of a Quorum Quenching Bacteria Isolate Ochrobactrum intermedium D-2 Against Soft Rot Pathogen Pectobacterium carotovorum subsp. carotovorum. Front. Microbiol. 2020, 11, 898. [Google Scholar] [CrossRef] [PubMed]
- Dwidar, M.; Jang, H.; Sangwan, N.; Mun, W.; Im, H.; Yoon, S.; Choi, S.; Nam, D.; Mitchell, R.J. Diffusible signaling factor, a quorum-sensing molecule, interferes with and is toxic towards Bdellovibrio bacteriovorus 109J. Microb. Ecol. 2021, 81, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cha, E.; Ham, S.; Park, J.; Nam, S.; Kwon, H.; Byun, Y.; Park, H. Linoleic acid inhibits Pseudomonas aeruginosa biofilm formation by activating diffusible signal factor-mediated quorum sensing. Biotechnol. Bioeng. 2021, 118, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wu, J.; Tao, F.; Zhang, L.-H. Listening to a New Language: DSF-Based Quorum Sensing in Gram-Negative Bacteria. Chem. Rev. 2011, 111, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, X.; Wu, J.; Lee, J.; Chen, S.; Cheng, Y.; Zhang, C.; Zhang, L.-H. The Host Plant Metabolite Glucose is the Precursor of Diffusible Signal Factor (DSF) Family Signals in Xanthomonas campestris. Appl. Environ. Microbiol. 2015, 81, 2861–2868. [Google Scholar] [CrossRef] [Green Version]
- Whiteley, M.; Diggle, S.P.; Greenberg, E. Progress in and promise of bacterial quorum sensing research. Nature 2017, 551, 313–320. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains or Plasmids | Relevant Genotype or Phenotype | Sources |
|---|---|---|
| HN-2 strains | Laboratory storage | This study |
| ∆fadT | fadT deletion mutant of strain HN-2 with 4323-nt internal coding region deleted | This study |
| Xcc XC1 | Pathogenic bacteria responsible for black rot | Lab collection |
| Escherichia coli strains | ||
| DH5α | spuE44 ∆lacU169(ϕ80lacZ∆M15) hsdR17λpir recA1 endA1 gyrA96 thi-1 relA1 | Lab collection |
| BL21 | F−ompThsdS (rB−mB−) dcm+ Tet r gal (DE3) endA | Lab collection |
| pRK2013 | Tra+, Mob−, ColE1-replicon, Kan r, Spe r | Lab collection |
| Plasmids | ||
| pBBR1-MCS5 | Broad-host-range expression vector; Gm r | Lab collection |
| pK18mobsacB | Broad-host-range gene knockout vector, sacB, Gmr | Lab collection |
| pBBR1-MCS5-fadT | pBBR1-MCS5 containing fadT under control of Plac | This study |
| pK18-fadT | pK18mobsacB containing flanking sequences of fadT | This study |
| pGEX-6p-1 | GST fusion protein expression vector, Amp r | Invitrogen |
| pGEX-fadT | pGEX-6p-1 containing fadT | This study |
| Primers | Sequence (5′–3′) | Purposes |
|---|---|---|
| DefadTupF | GAGCTCGGTACCCGGGGATCCGGCCCGGTGTTCGCGGTG | For amplification of the 5′-region of fadT |
| DefadTupR | ATAGCGGCGCGGGGGTGTCTCCATGCGGGGC | |
| DefadTdnF | AGACACCCCCGCGCCGCTATCCGTG | For amplification of the 3′-region of fadT |
| DefadTdnR | ACGACGGCCAGTGCCAAGCTTTCGATGCCACGTTGATGATG | |
| TsfadT-F | CTTATGGATGTCCGGGGTCG | For identification of ∆fadT |
| TsfadT-R | TTCATGACCTTGTCCCAGGC | |
| cfadT-F | GTCGACGGTATCGATAAGCTTAGTACGTCGTCGTGACGATGTG | For construction of pBBR1-MCS5-fadT |
| cfadT-R | CGCTCTAGAACTAGTGGATCCGGTTCCCTGATGTTCCTCGAT | |
| pfadT-F | TTCCAGGGGCCCCTGGGATCC ATGTCGCTGATCCTTTCCCG | For construction of pGEX-6P-1-fadT |
| pfadT-R | CTCGAGTCGACCCGGGAATTC TCAGAACCAGGCGTCCTGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Ye, T.; Zhang, W.; Zhou, T.; Zhou, X.; Dai, W.; Chen, S. Identification of FadT as a Novel Quorum Quenching Enzyme for the Degradation of Diffusible Signal Factor in Cupriavidus pinatubonensis Strain HN-2. Int. J. Mol. Sci. 2021, 22, 9862. https://doi.org/10.3390/ijms22189862
Xu X, Ye T, Zhang W, Zhou T, Zhou X, Dai W, Chen S. Identification of FadT as a Novel Quorum Quenching Enzyme for the Degradation of Diffusible Signal Factor in Cupriavidus pinatubonensis Strain HN-2. International Journal of Molecular Sciences. 2021; 22(18):9862. https://doi.org/10.3390/ijms22189862
Chicago/Turabian StyleXu, Xudan, Tian Ye, Wenping Zhang, Tian Zhou, Xiaofan Zhou, Weijun Dai, and Shaohua Chen. 2021. "Identification of FadT as a Novel Quorum Quenching Enzyme for the Degradation of Diffusible Signal Factor in Cupriavidus pinatubonensis Strain HN-2" International Journal of Molecular Sciences 22, no. 18: 9862. https://doi.org/10.3390/ijms22189862
APA StyleXu, X., Ye, T., Zhang, W., Zhou, T., Zhou, X., Dai, W., & Chen, S. (2021). Identification of FadT as a Novel Quorum Quenching Enzyme for the Degradation of Diffusible Signal Factor in Cupriavidus pinatubonensis Strain HN-2. International Journal of Molecular Sciences, 22(18), 9862. https://doi.org/10.3390/ijms22189862