
SREBP1-Induced Glutamine Synthetase Triggers a Feedforward Loop to Upregulate SREBP1 through Sp1 O-GlcNAcylation and Augments Lipid Droplet Formation in Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Insulin and Glutamine Deprivation Upregulate GS Expression through SREBP1
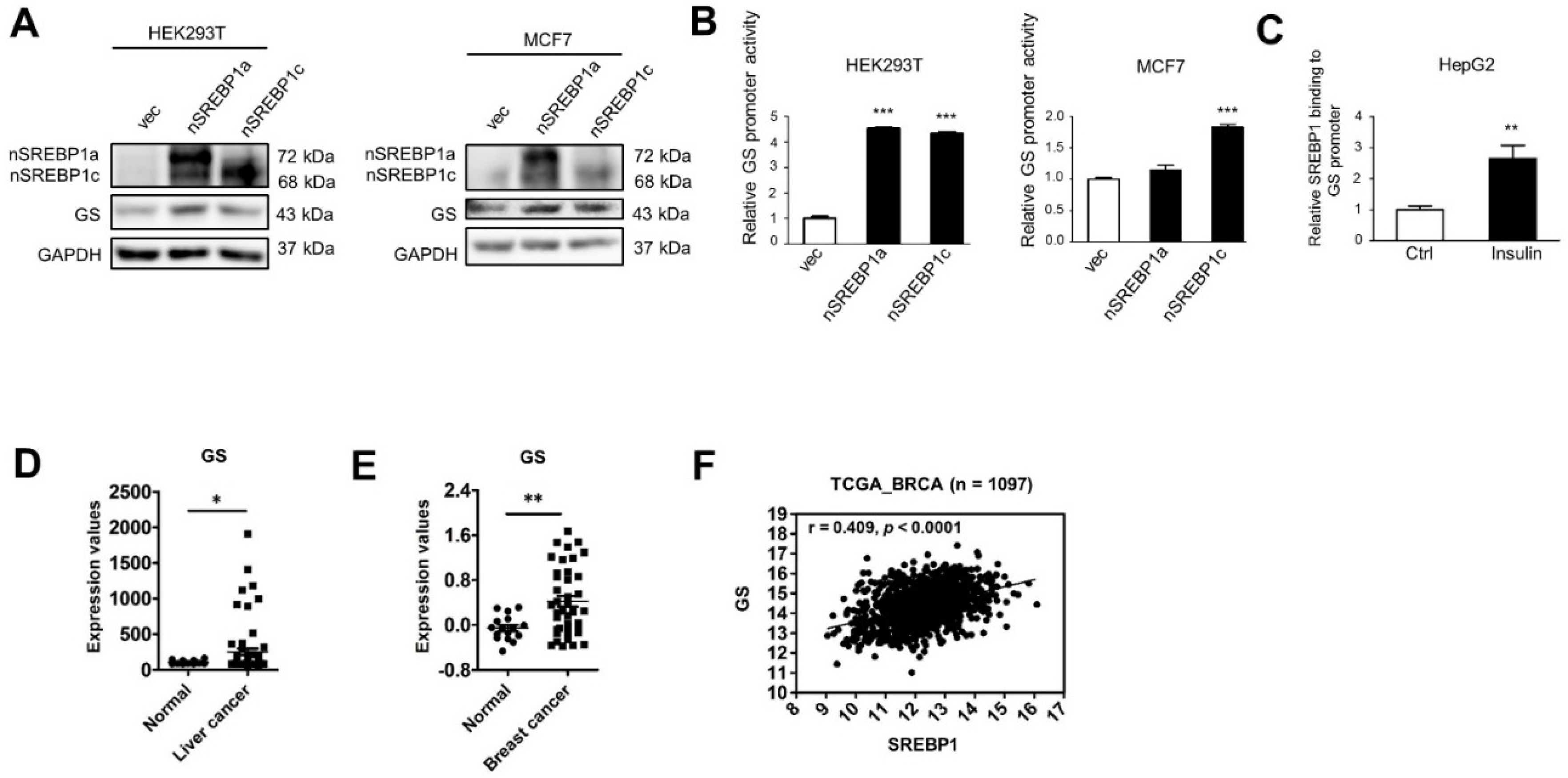
2.2. SREBP1 Binds to the GS Promoter Leading to GS Transactivation
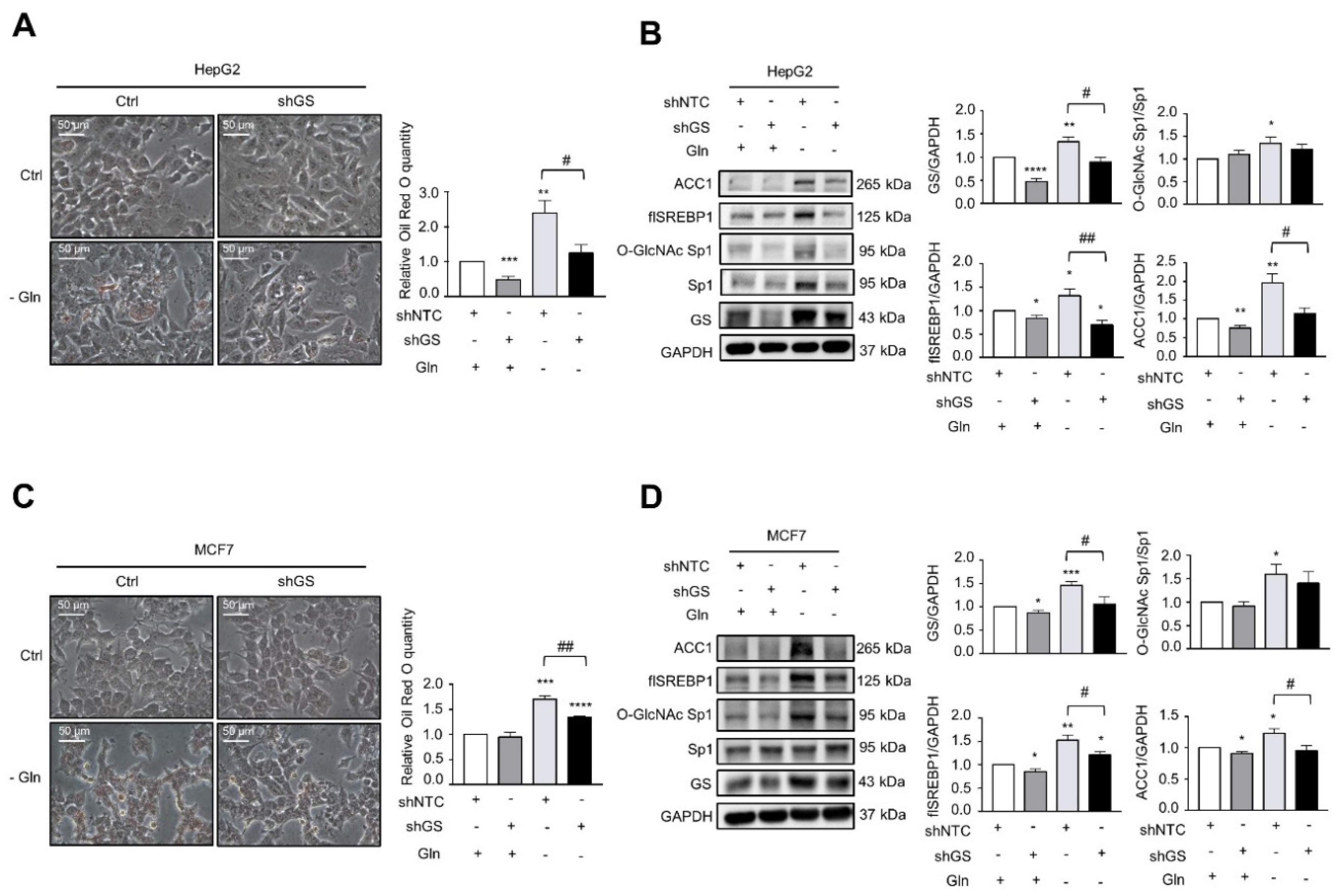
2.3. GS Triggers a Feedforward Loop of O-GlcNAc-Sp1/SREBP1/ACC1 Signaling That Induces LD Formation upon Insulin Treatment
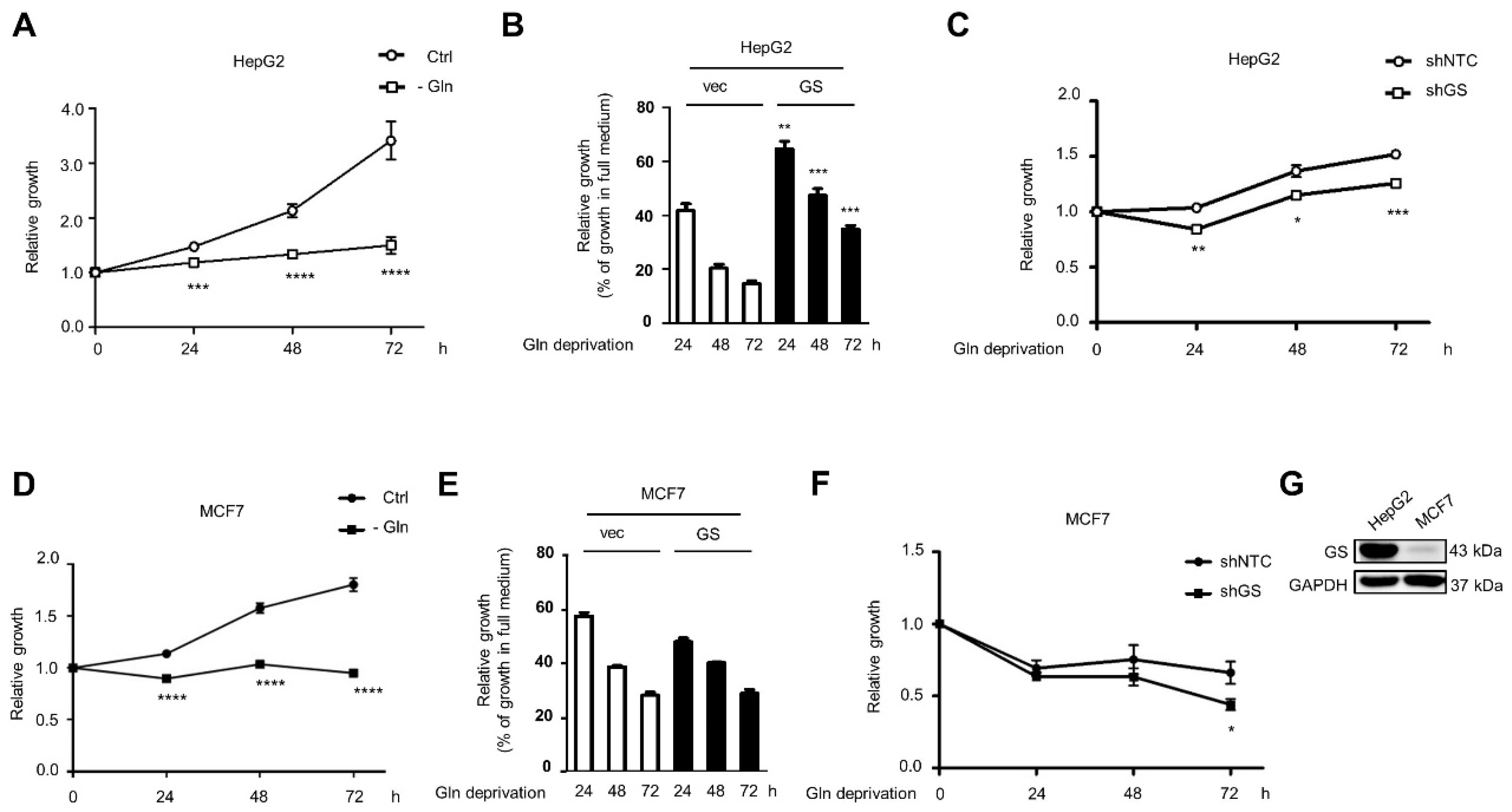
2.4. Glutamine Deprivation Induces LD Formation through GS-Mediated O-GlcNAc-Sp1/SREBP1/ACC1 Signaling, and GS Promotes Cell Survival under Glutamine Deprivation
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Lines and Culture
4.3. Western Blotting
4.4. Quantitative Reverse Transcription PCR (qRT-PCR)
4.5. Luciferase Assay
4.6. Intracellular Glutamine Measurement
4.7. ChIP Assay
4.8. Lipid Staining
4.9. Cell Growth Assay
4.10. Database Analyses
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC1 | acetyl-CoA carboxylase 1 |
| ChIP | chromatin immunoprecipitation |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | dimethyl sulfoxide |
| ECL | enhanced chemiluminescence |
| FBS | fetal bovine serum |
| GS | glutamine synthetase |
| HBP | hexosamine biosynthetic pathway |
| HCC | hepatocellular carcinoma |
| IDH1 | isocitrate dehydrogenase 1 |
| IDH2 | isocitrate dehydrogenase 2 |
| LD | lipid droplet |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| O-GlcNAcylation | O-linked N-acetylglucosaminylation |
| PMSF | phenylmethylsulfonyl fluoride |
| PVDF | polyvinylidene fluoride |
| PBS | phosphate buffer saline |
| qPCR | quantitative PCR |
| SREBP1 | sterol regulatory element-binding protein 1 |
| Sp1 | specificity protein 1 |
| SRE | sterol regulatory element |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEM | standard error of the mean |
| UDP-GlcNAc | uridine-diphosphate-N-acetylglucosamine |
References
- Osborne, T.F. Sterol regulatory element-binding proteins (SREBPs): Key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000, 275, 32379–32382. [Google Scholar] [CrossRef]
- Peng, I.-C.; Chen, Z.; Sun, W.; Li, Y.-S.; Marin, T.L.; Hsu, P.-H.; Su, M.-I.; Cui, X.; Pan, S.; Lytle, C.Y. Glucagon regulates ACC activity in adipocytes through the CAMKKβ/AMPK pathway. Am. J. Physiol.-Endocrinol. Metab. 2012, 302, E1560–E1568. [Google Scholar] [CrossRef][Green Version]
- Yan, J.B.; Lai, C.C.; Jhu, J.W.; Gongol, B.; Marin, T.L.; Lin, S.C.; Chiu, H.Y.; Yen, C.J.; Wang, L.Y.; Peng, I.C. Insulin and Metformin Control Cell Proliferation by Regulating TDG-Mediated DNA Demethylation in Liver and Breast Cancer Cells. Mol. Ther. Oncolytics 2020, 18, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.K.; Chong, H.K.; Infante, A.M.; Im, S.S.; Xie, X.; Osborne, T.F. Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc. Natl. Acad. Sci. USA 2009, 106, 13765–13769. [Google Scholar] [CrossRef] [PubMed]
- Dorotea, D.; Koya, D.; Ha, H. Recent Insights into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front. Pharmacol. 2020, 11, 265. [Google Scholar] [CrossRef]
- Uttarwar, L.; Gao, B.; Ingram, A.J.; Krepinsky, J.C. SREBP-1 activation by glucose mediates TGF-beta upregulation in mesangial cells. Am. J. Physiol. Ren. Physiol. 2012, 302, F329–F341. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alvarez, M.; Finger, F.; Arias-Garcia Mdel, M.; Bousgouni, V.; Pascual-Vargas, P.; Bakal, C. Signaling networks converge on TORC1-SREBP activity to promote endoplasmic reticulum homeostasis. PLoS ONE 2014, 9, e101164. [Google Scholar] [CrossRef] [PubMed]
- Gilardi, F.; Migliavacca, E.; Naldi, A.; Baruchet, M.; Canella, D.; Le Martelot, G.; Guex, N.; Desvergne, B.; Cycli, X.C. Genome-wide analysis of SREBP1 activity around the clock reveals its combined dependency on nutrient and circadian signals. PLoS Genet. 2014, 10, e1004155. [Google Scholar] [CrossRef]
- Bott, A.J.; Maimouni, S.; Zong, W.X. The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef]
- Kung, H.N.; Marks, J.R.; Chi, J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef]
- Bott, A.J.; Peng, I.-C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Issaq, S.H.; Mendoza, A.; Fox, S.D.; Helman, L.J. Glutamine synthetase is necessary for sarcoma adaptation to glutamine deprivation and tumor growth. Oncogenesis 2019, 8, 20. [Google Scholar] [CrossRef]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298 e1286. [Google Scholar] [CrossRef] [PubMed]
- Ryczko, M.C.; Pawling, J.; Chen, R.; Abdel Rahman, A.M.; Yau, K.; Copeland, J.K.; Zhang, C.; Surendra, A.; Guttman, D.S.; Figeys, D.; et al. Metabolic Reprogramming by Hexosamine Biosynthetic and Golgi N-Glycan Branching Pathways. Sci. Rep. 2016, 6, 23043. [Google Scholar] [CrossRef] [PubMed]
- De Vitto, H.; Perez-Valencia, J.; Radosevich, J.A. Glutamine at focus: Versatile roles in cancer. Tumor Biol. 2016, 37, 1541–1558. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Deng, X.; Yellaturu, C.; Cagen, L.; Wilcox, H.G.; Park, E.A.; Raghow, R.; Elam, M.B. Expression of the rat sterol regulatory element-binding protein-1c gene in response to insulin is mediated by increased transactivating capacity of specificity protein 1 (Sp1). J. Biol. Chem. 2007, 282, 17517–17529. [Google Scholar] [CrossRef]
- Lu, S.; Archer, M.C. Sp1 coordinately regulates de novo lipogenesis and proliferation in cancer cells. Int. J. Cancer 2010, 126, 416–425. [Google Scholar] [CrossRef]
- Penque, B.A.; Hoggatt, A.M.; Herring, B.P.; Elmendorf, J.S. Hexosamine Biosynthesis Impairs Insulin Action via a Cholesterolgenic Response. Mol. Endocrinol. 2013, 27, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Ito, Y.; Shimada, S.; Satoh, S.I.; Sasaki, T.; Hashidume, T.; Kamoshida, Y.; Shimizu, M.; Sato, R. Glutamine stimulates the gene expression and processing of sterol regulatory element binding proteins, thereby increasing the expression of their target genes. FEBS J. 2011, 278, 2739–2750. [Google Scholar] [CrossRef]
- Briggs, M.R.; Yokoyama, C.; Wang, X.; Brown, M.S.; Goldstein, J.L. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. I. Identification of the protein and delineation of its target nucleotide sequence. J. Biol. Chem. 1993, 268, 14490–14496. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, L.; Destro, A.; Seok, J.Y.; Balladore, E.; Terracciano, L.; Sangiovanni, A.; Iavarone, M.; Colombo, M.; Jang, J.J.; Yu, E.; et al. The application of markers (HSP70 GPC3 and GS) in liver biopsies is useful for detection of hepatocellular carcinoma. J. Hepatol. 2009, 50, 746–754. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2016. [Google Scholar]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef]
- Anastasiou, D.; Cantley, L.C. Breathless cancer cells get fat on glutamine. Cell Res. 2012, 22, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.; Dibble, C.C.; Asara, J.M.; Manning, B.D. Sterol Regulatory Element Binding Protein Regulates the Expression and Metabolic Functions of Wild-Type and Oncogenic IDH1. Mol. Cell. Biol. 2016, 36, 2384–2395. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.; Pathak, S.; Grzes, K.M.; Damerow, S.; Sinclair, L.V.; van Aalten, D.M.; Cantrell, D.A. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 2016, 17, 712–720. [Google Scholar] [CrossRef]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef]
- Lee, H.J.; Ryu, J.M.; Jung, Y.H.; Lee, K.H.; Kim, D.I.; Han, H.J. Glycerol-3-phosphate acyltransferase-1 upregulation by O-GlcNAcylation of Sp1 protects against hypoxia-induced mouse embryonic stem cell apoptosis via mTOR activation. Cell Death Dis. 2016, 7, e2158. [Google Scholar] [CrossRef] [PubMed]
- Baldini, S.F.; Wavelet, C.; Hainault, I.; Guinez, C.; Lefebvre, T. The Nutrient-Dependent O-GlcNAc Modification Controls the Expression of Liver Fatty Acid Synthase. J. Mol. Biol. 2016, 428, 3295–3304. [Google Scholar] [CrossRef]
- Dif, N.; Euthine, V.; Gonnet, E.; Laville, M.; Vidal, H.; Lefai, E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem. J. 2006, 400, 179–188. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jhu, J.-W.; Yan, J.-B.; Lin, Z.-H.; Lin, S.-C.; Peng, I.-C. SREBP1-Induced Glutamine Synthetase Triggers a Feedforward Loop to Upregulate SREBP1 through Sp1 O-GlcNAcylation and Augments Lipid Droplet Formation in Cancer Cells. Int. J. Mol. Sci. 2021, 22, 9814. https://doi.org/10.3390/ijms22189814
Jhu J-W, Yan J-B, Lin Z-H, Lin S-C, Peng I-C. SREBP1-Induced Glutamine Synthetase Triggers a Feedforward Loop to Upregulate SREBP1 through Sp1 O-GlcNAcylation and Augments Lipid Droplet Formation in Cancer Cells. International Journal of Molecular Sciences. 2021; 22(18):9814. https://doi.org/10.3390/ijms22189814
Chicago/Turabian StyleJhu, Jin-Wei, Jia-Bao Yan, Zou-Han Lin, Shih-Chieh Lin, and I-Chen Peng. 2021. "SREBP1-Induced Glutamine Synthetase Triggers a Feedforward Loop to Upregulate SREBP1 through Sp1 O-GlcNAcylation and Augments Lipid Droplet Formation in Cancer Cells" International Journal of Molecular Sciences 22, no. 18: 9814. https://doi.org/10.3390/ijms22189814
APA StyleJhu, J.-W., Yan, J.-B., Lin, Z.-H., Lin, S.-C., & Peng, I.-C. (2021). SREBP1-Induced Glutamine Synthetase Triggers a Feedforward Loop to Upregulate SREBP1 through Sp1 O-GlcNAcylation and Augments Lipid Droplet Formation in Cancer Cells. International Journal of Molecular Sciences, 22(18), 9814. https://doi.org/10.3390/ijms22189814