
Loss of Von Hippel–Lindau (VHL) Tumor Suppressor Gene Function: VHL–HIF Pathway and Advances in Treatments for Metastatic Renal Cell Carcinoma (RCC)


Abstract
:1. Introduction
2. VHL and the Loss of Chromosome 3p in ccRCC Tumorigenesis
3. VHL Inactivation and Targeted Therapy in ccRCC
3.1. The VHL–HIF–VEGF Pathway
3.2. Anti-VEGFR and VEGF Inhibitors in ccRCC
3.3. Other Targets: The Protein Kinase B–Mechanistic Target of Rapamycin (AKT–mTOR) and Epidermal Growth Factor Receptor (EGFR) Pathways
3.4. Resistance to VEGFR Inhibitors: Alternative Anti-Angiogenic Pathways
4. The TME and Anti-Cancer Immunity in ccRCC
4.1. Cellular Metabolism
4.2. Chronic Inflammation and TME
4.3. HIF, VEGF, and Immunosuppressive TME
4.4. Immunomodulatory Effects of Anti-VEGF and Anti-VEGFR Therapies
4.5. Immune Check Point Inhibitor-Combination Strategies
5. Outlook for the VHL–HIF Pathway in ccRCC Treatment
5.1. Return to HIFa
5.2. HIF2a Inhibitors
5.3. Other Targets: VHL Substrate Targets and Multi-Omics Approaches
6. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Cheng, S.C.; Signoretti, S.; Margolin, K.A.; Clark, J.I.; Sosman, J.A.; Dutcher, J.P.; Logan, T.F.; Curti, B.D.; Ernstoff, M.S.; et al. The high-dose aldesleukin “select” trial: A trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bex, A.; Mulders, P.; Jewett, M.; Wagstaff, J.; van Thienen, J.V.; Blank, C.U.; van Velthoven, R.; Del Pilar Laguna, M.; Wood, L.; van Melick, H.H.E.; et al. Comparison of immediate vs deferred cytoreductive nephrectomy in patients with synchronous metastatic renal cell carcinoma receiving sunitinib: The SURTIME randomized clinical trial. JAMA Oncol. 2019, 5, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Mejean, A.; Ravaud, A.; Thezenas, S.; Colas, S.; Beauval, J.B.; Bensalah, K.; Geoffrois, L.; Thiery-Vuillemin, A.; Cormier, L.; Lang, H.; et al. Sunitinib alone or after nephrectomy in metastatic renal-cell carcinoma. N. Engl. J. Med. 2018, 379, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Lynn, K.D.; Toombs, J.E.; Dineen, S.P.; Udugamasooriya, D.G.; Brekken, R.A. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS ONE 2009, 4, e7669. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [CrossRef] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 3698. [Google Scholar] [CrossRef]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell 2018, 173, 611–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Cubas, A.A.; Rathmell, W.K. Epigenetic modifiers: Activities in renal cell carcinoma. Nat. Rev. Urol. 2018, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Sasaki, H.; Kim, S.J.; Tobisu, K.; Kakizoe, T.; Tsukamoto, T.; Kumamoto, Y.; Sugimura, T.; Terada, M. Markedly increased amounts of messenger RNAs for vascular endothelial growth factor and placenta growth factor in renal cell carcinoma associated with angiogenesis. Cancer Res. 1994, 54, 4233–4237. [Google Scholar] [PubMed]
- Wizigmann-Voos, S.; Breier, G.; Risau, W.; Plate, K.H. Up-regulation of vascular endothelial growth factor and its receptors in von Hippel-Lindau disease-associated and sporadic hemangioblastomas. Cancer Res. 1995, 55, 1358–1364. [Google Scholar]
- Siemeister, G.; Weindel, K.; Mohrs, K.; Barleon, B.; Martiny-Baron, G.; Marmé, D. Reversion of deregulated expression of vascular endothelial growth factor in human renal carcinoma cells by von Hippel-Lindau tumor suppressor protein. Cancer Res. 1996, 56, 2299–2301. [Google Scholar] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993, 268, 21513–21518. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Koepp, D.M.; Conrad, M.N.; Skowyra, D.; Moreland, R.J.; Iliopoulos, O.; Lane, W.S.; Kaelin, W.G., Jr.; Elledge, S.J.; Conaway, R.C.; et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999, 284, 657–661. [Google Scholar] [CrossRef]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, K.M.; Iliopoulos, O.; Ohh, M.; Kamura, T.; Conaway, R.C.; Conaway, J.W.; Kaelin, W.G., Jr. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell. Biol. 1998, 18, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corn, P.G.; McDonald, E.R., 3rd; Herman, J.G.; El-Deiry, W.S. Tat-binding protein-1, a component of the 26S proteasome, contributes to the E3 ubiquitin ligase function of the von Hippel-Lindau protein. Nat. Genet. 2003, 35, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, Y.; Takata, K.; Moriyama, N.; Aso, Y.; Murakami, T.; Hirano, H. Immunohistochemical localization of glucose transporters in human renal cell carcinoma. J. Urol. 1995, 153, 798–801. [Google Scholar] [CrossRef]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Escudier, B.; Bellmunt, J.; Négrier, S.; Bajetta, E.; Melichar, B.; Bracarda, S.; Ravaud, A.; Golding, S.; Jethwa, S.; Sneller, V. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): Final analysis of overall survival. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2144–2150. [Google Scholar] [CrossRef] [Green Version]
- Kuenen, B.C.; Tabernero, J.; Baselga, J.; Cavalli, F.; Pfanner, E.; Conte, P.F.; Seeber, S.; Madhusudan, S.; Deplanque, G.; Huisman, H.; et al. Efficacy and toxicity of the angiogenesis inhibitor SU5416 as a single agent in patients with advanced renal cell carcinoma, melanoma, and soft tissue sarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1648–1655. [Google Scholar]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Hutson, T.E.; Lesovoy, V.; Al-Shukri, S.; Stus, V.P.; Lipatov, O.N.; Bair, A.H.; Rosbrook, B.; Chen, C.; Kim, S.; Vogelzang, N.J. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: A randomised open-label phase 3 trial. Lancet Oncol. 2013, 14, 1287–1294. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Nosov, D.; Eisen, T.; Bondarenko, I.; Lesovoy, V.; Lipatov, O.; Tomczak, P.; Lyulko, O.; Alyasova, A.; Harza, M.; et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: Results from a phase III trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3791–3799. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Flaifel, A.; Xie, W.; Braun, D.A.; Ficial, M.; Bakouny, Z.; Nassar, A.H.; Jennings, R.B.; Escudier, B.; George, D.J.; Motzer, R.J.; et al. PD-L1 Expression and Clinical outcomes to cabozantinib, everolimus, and sunitinib in patients with metastatic renal cell carcinoma: Analysis of the randomized clinical trials METEOR and CABOSUN. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6080–6088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Hessel, C.; Halabi, S.; Sanford, B.; Michaelson, M.D.; Hahn, O.; Walsh, M.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur. J. Cancer 2018, 94, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganner, A.; Gehrke, C.; Klein, M.; Thegtmeier, L.; Matulenski, T.; Wingendorf, L.; Wang, L.; Pilz, F.; Greidl, L.; Meid, L.; et al. VHL suppresses RAPTOR and inhibits mTORC1 signaling in clear cell renal cell carcinoma. Sci. Rep. 2021, 11, 14827. [Google Scholar] [CrossRef] [PubMed]
- Tumkur Sitaram, R.; Landström, M.; Roos, G.; Ljungberg, B. Significance of PI3K signalling pathway in clear cell renal cell carcinoma in relation to VHL and HIF status. J. Clin. Pathol. 2021, 74, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma : Final results and analysis of prognostic factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, L.; Wang, K.; Li, X.F.; Ge, L.Y.; Ma, R.Z.; Fan, Y.D.; Li, L.C.; Liu, Z.F.; Qiu, M.; et al. VHL-HIF-2α axis-induced SMYD3 upregulation drives renal cell carcinoma progression via direct trans-activation of EGFR. Oncogene 2020, 39, 4286–4298. [Google Scholar] [CrossRef]
- Gunaratnam, L.; Morley, M.; Franovic, A.; de Paulsen, N.; Mekhail, K.; Parolin, D.A.; Nakamura, E.; Lorimer, I.A.; Lee, S. Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(-/-) renal cell carcinoma cells. J. Biol. Chem. 2003, 278, 44966–44974. [Google Scholar] [CrossRef] [Green Version]
- Riazalhosseini, Y.; Lathrop, M. Precision medicine from the renal cancer genome. Nat. Rev. Nephrol. 2016, 12, 655–666. [Google Scholar] [CrossRef]
- Ding, C.; Li, L.; Yang, T.; Fan, X.; Wu, G. Combined application of anti-VEGF and anti-EGFR attenuates the growth and angiogenesis of colorectal cancer mainly through suppressing AKT and ERK signaling in mice model. BMC Cancer 2016, 16, 791. [Google Scholar] [CrossRef] [Green Version]
- Grépin, R.; Guyot, M.; Dumond, A.; Durivault, J.; Ambrosetti, D.; Roussel, J.F.; Dupré, F.; Quintens, H.; Pagès, G. The combination of bevacizumab/Avastin and erlotinib/Tarceva is relevant for the treatment of metastatic renal cell carcinoma: The role of a synonymous mutation of the EGFR receptor. Theranostics 2020, 10, 1107–1121. [Google Scholar] [CrossRef]
- Nassif, E.; Thibault, C.; Vano, Y.; Fournier, L.; Mauge, L.; Verkarre, V.; Timsit, M.O.; Mejean, A.; Tartour, E.; Oudard, S. Sunitinib in kidney cancer: 10 years of experience and development. Expert Rev. Anticancer Ther. 2017, 17, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamli, H.; Li, L.; Gobe, G.C. Limitations to the Therapeutic Potential of Tyrosine Kinase Inhibitors and Alternative Therapies for Kidney Cancer. Ochsner J. 2019, 19, 138–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef] [Green Version]
- Fei, M.; Guan, J.; Xue, T.; Qin, L.; Tang, C.; Cui, G.; Wang, Y.; Gong, H.; Feng, W. Hypoxia promotes the migration and invasion of human hepatocarcinoma cells through the HIF-1α-IL-8-Akt axis. Cell. Mol. Biol. Lett. 2018, 23, 46. [Google Scholar] [CrossRef]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Shields, L.B.E.; Rezazadeh Kalebasty, A. spontaneous regression of delayed pulmonary and mediastinal metastases from clear cell renal cell carcinoma. Case Rep. Oncol. 2020, 13, 1285–1294. [Google Scholar] [CrossRef]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Aghamir, S.M.K.; Heshmat, R.; Ebrahimi, M.; Ketabchi, S.E.; Parichehreh Dizaji, S.; Khatami, F. The impact of succinate dehydrogenase gene (SDH) mutations in renal cell carcinoma (RCC): A systematic review. OncoTargets Ther. 2019, 12, 7929–7940. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Kim, J.H.; Sun, B.K.; Song, S.U.; Suh, W.; Sung, J.H. Hypoxia induces glucose uptake and metabolism of adipose-derived stem cells. Mol. Med. Rep. 2016, 14, 4706–4714. [Google Scholar] [CrossRef] [Green Version]
- Shan, T.; Chen, S.; Chen, X.; Wu, T.; Yang, Y.; Li, S.; Ma, J.; Zhao, J.; Lin, W.; Li, W.; et al. M2-TAM subsets altered by lactic acid promote T-cell apoptosis through the PD-L1/PD-1 pathway. Oncol. Rep. 2020, 44, 1885–1894. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL Deficiency drives enhancer activation of oncogenes in clear cell renal cell carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Kai, J.; Wang, S.; Yu, Y.; Xie, S.; Zheng, H.; Wang, Y.; Liu, Y.; Zhu, K.; Guan, X.; et al. VHL regulates the sensitivity of clear cell renal cell carcinoma to SIRT4-mediated metabolic stress via HIF-1α/HO-1 pathway. Cell Death Dis. 2021, 12, 621. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Lin, C.H.; Hsu, T. VHL Inactivation in precancerous kidney cells induces an inflammatory response via er stress-activated ire1α signaling. Cancer Res. 2017, 77, 3406–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchett, T.L.; Bader, H.L.; Henderson, J.; Hsu, T. Conditional inactivation of the mouse von Hippel-Lindau tumor suppressor gene results in wide-spread hyperplastic, inflammatory and fibrotic lesions in the kidney. Oncogene 2015, 34, 2631–2639. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Chiu, V.; Hsieh, P.C.; Hsu, T.; Lin, T.Y. Loss of Function of von Hippel-Lindau Trigger Lipocalin 2-dependent inflammatory responses in cultured and primary renal tubular cells. Oxidative Med. Cell. Longev. 2021, 2021, 5571638. [Google Scholar] [CrossRef]
- Nguyen-Tran, H.H.; Nguyen, T.N.; Chen, C.Y.; Hsu, T. Endothelial reprograming stimulated by oncostatin M promotes inflammation and tumorigenesis in VHL-deficient kidney tissue. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Thomas, L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982, 55, 329–333. [Google Scholar]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kopecký, O.; Lukesová, S.; Vroblová, V.; Vokurková, D.; Morávek, P.; Safránek, H.; Hlávková, D.; Soucek, P. Phenotype analysis of tumour-infiltrating lymphocytes and lymphocytes in peripheral blood in patients with renal carcinoma. Acta Med. 2007, 50, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Kawashima, A.; Kanazawa, T.; Kidani, Y.; Yoshida, T.; Hirata, M.; Yamamoto, K.; Yamamoto, Y.; Sawada, M.; Kato, R.; et al. Clinical importance of the expression of CD4+CD8+ T cells in renal cell carcinoma. Int. Immunol. 2020, 32, 347–357. [Google Scholar] [CrossRef]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A axis in cytotoxic t cells regulates tumor progression. Cancer Cell 2017, 32, 669–683.e665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera-Domingo, C.; Audige, A.; Granja, S.; Cheng, W.C.; Ho, P.C.; Baltazar, F.; Stockmann, C.; Mazzone, M. Immunity, hypoxia, and metabolism-the menage a trois of cancer: Implications for immunotherapy. Physiol. Rev. 2020, 100, 1–102. [Google Scholar] [CrossRef] [PubMed]
- Sethumadhavan, S.; Silva, M.; Philbrook, P.; Nguyen, T.; Hatfield, S.M.; Ohta, A.; Sitkovsky, M.V. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS ONE 2017, 12, e0187314. [Google Scholar] [CrossRef] [Green Version]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Yoon, S.H.; Jang, H.; Jeong, J.H.; Lee, Y.M. Decursin promotes HIF-1α proteasomal degradation and immune responses in hypoxic tumour microenvironment. Phytomedicine Int. J. Phytother. Phytopharm. 2020, 78, 153318. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Onishi, H.; Wada, J.; Yamasaki, A.; Tanaka, H.; Nakano, K.; Morisaki, T.; Katano, M. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur. J. Immunol. 2010, 40, 197–203. [Google Scholar] [CrossRef]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhang, E.; Long, J.; Hu, Z.; Peng, J.; Liu, L.; Tang, F.; Li, L.; Ouyang, Y.; Zeng, Z. Immune infiltration in renal cell carcinoma. Cancer Sci. 2019, 110, 1564–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, K.; Miyake, M.; Onishi, K.; Hori, S.; Morizawa, Y.; Gotoh, D.; Itami, Y.; Onishi, S.; Nakai, Y.; Anai, S.; et al. Prognostic impact of tumor-infiltrating CD276/Foxp3-positive lymphocytes and associated circulating cytokines in patients undergoing radical nephrectomy for localized renal cell carcinoma. Oncol. Lett. 2019, 17, 4004–4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-γ drives t(reg) fragility to promote anti-tumor immunity. Cell 2017, 169, 1130–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Deng, J.; Li, J.; Sarde, A.; Lines, J.L.; Lee, Y.C.; Qian, D.C.; Pechenick, D.A.; Manivanh, R.; Le Mercier, I.; Lowrey, C.H.; et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol. Res. 2019, 7, 1079–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Xu, W.; Puligandla, M.; Manola, J.; Bullock, A.J.; Tamasauskas, D.; McDermott, D.F.; Atkins, M.B.; Haas, N.B.; Flaherty, K.; Uzzo, R.G.; et al. Angiogenic factor and cytokine analysis among patients treated with adjuvant VEGFR TKIs in resected renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6098–6106. [Google Scholar] [CrossRef] [Green Version]
- Shigeta, K.; Matsui, A.; Kikuchi, H.; Klein, S.; Mamessier, E.; Chen, I.X.; Aoki, S.; Kitahara, S.; Inoue, K.; Shigeta, A.; et al. Regorafenib combined with PD1 blockade increases CD8 T-cell infiltration by inducing CXCL10 expression in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Hirsch, L.; Flippot, R.; Escudier, B.; Albiges, L. Immunomodulatory roles of VEGF pathway inhibitors in renal cell carcinoma. Drugs 2020, 80, 1169–1181. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Zizzari, I.G.; Napoletano, C.; Botticelli, A.; Caponnetto, S.; Calabrò, F.; Gelibter, A.; Rughetti, A.; Ruscito, I.; Rahimi, H.; Rossi, E.; et al. TK inhibitor pazopanib primes DCs by downregulation of the β-catenin pathway. Cancer Immunol. Res. 2018, 6, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Arén Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet. Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet. Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for advanced renal cell carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Maroto, P.; Esteban, E.; Parra, E.F.; Mendez-Vidal, M.J.; Domenech, M.; Pérez-Valderrama, B.; Calderero, V.; Pérez-Gracia, J.L.; Grande, E.; Algaba, F. HIF pathway and c-Myc as biomarkers for response to sunitinib in metastatic clear-cell renal cell carcinoma. OncoTargets Ther. 2017, 10, 4635–4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [Green Version]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 271–273. [Google Scholar]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Isono, T.; Chano, T.; Yoshida, T.; Kageyama, S.; Kawauchi, A.; Suzaki, M.; Yuasa, T. Hydroxyl-HIF2-alpha is potential therapeutic target for renal cell carcinomas. Am. J. Cancer Res. 2016, 6, 2263–2276. [Google Scholar] [PubMed]
- Elvert, G.; Kappel, A.; Heidenreich, R.; Englmeier, U.; Lanz, S.; Acker, T.; Rauter, M.; Plate, K.; Sieweke, M.; Breier, G.; et al. Cooperative interaction of hypoxia-inducible factor-2alpha (HIF-2alpha ) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1). J. Biol. Chem. 2003, 278, 7520–7530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yu, Q.; Zhang, X. Allosteric inhibition of HIF-2alpha as a novel therapy for clear cell renal cell carcinoma. Drug Discov. Today 2019, 24, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Gagliano, T.; Giamas, G. Direct effects of anti-angiogenic therapies on tumor cells: VEGF signaling. Trends Mol. Med. 2017, 23, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Grampp, S.; Platt, J.L.; Lauer, V.; Salama, R.; Kranz, F.; Neumann, V.K.; Wach, S.; Stöhr, C.; Hartmann, A.; Eckardt, K.U.; et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat. Commun. 2016, 7, 13183. [Google Scholar] [CrossRef]
- Rasti, A.; Abolhasani, M.; Zanjani, L.S.; Asgari, M.; Mehrazma, M.; Madjd, Z. Reduced expression of CXCR4, a novel renal cancer stem cell marker, is associated with high-grade renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2017, 143, 95–104. [Google Scholar] [CrossRef]
- Chen, Y.S.; Meng, F.; Li, H.L.; Liu, Q.H.; Hou, P.F.; Bai, J.; Zheng, J.N. Dicer suppresses MMP-2-mediated invasion and VEGFA-induced angiogenesis and serves as a promising prognostic biomarker in human clear cell renal cell carcinoma. Oncotarget 2016, 7, 84299–84313. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Xu, R.; Wang, K.; Rizzi, J.P.; Huang, H.; Grina, J.A.; Schlachter, S.T.; Wang, B.; Wehn, P.M.; Yang, H.; Dixon, D.D.; et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell renal cell carcinoma. J. Med. Chem. 2019, 62, 6876–6893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Bauer, T.M.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: A phase 1 trial and biomarker analysis. Nat. Med. 2021, 27, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Simon, J.M.; Xie, H.; Hu, L.; Wang, J.; Zurlo, G.; Fan, C.; Ptacek, T.S.; Herring, L.; Tan, X.; et al. Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Mol. Cell 2020, 77, 1294–1306.e1295. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, L.; Xue, S.; Shi, J.; He, C.; Zhang, Q. Novel VHL substrate targets SFMBT1 and ZHX2 may be important prognostic predictors in patients with ccRCC. Oncol. Lett. 2021, 21, 379. [Google Scholar] [CrossRef]
- Czyzyk-Krzeska, M.F.; Landero Figueroa, J.A.; Gulati, S.; Cunningham, J.T.; Meller, J.; Shamsae, I.B.; Vemuri, B.; Plas, D.R. Molecular and Metabolic Subtypes in Sporadic and Inherited Clear Cell Renal Cell Carcinoma. Genes 2021, 12, 388. [Google Scholar] [CrossRef]
- Lin, P.; Lin, Y.Q.; Gao, R.Z.; Wen, R.; Qin, H.; He, Y.; Yang, H. Radiomic profiling of clear cell renal cell carcinoma reveals subtypes with distinct prognoses and molecular pathways. Transl. Oncol. 2021, 14, 101078. [Google Scholar] [CrossRef]
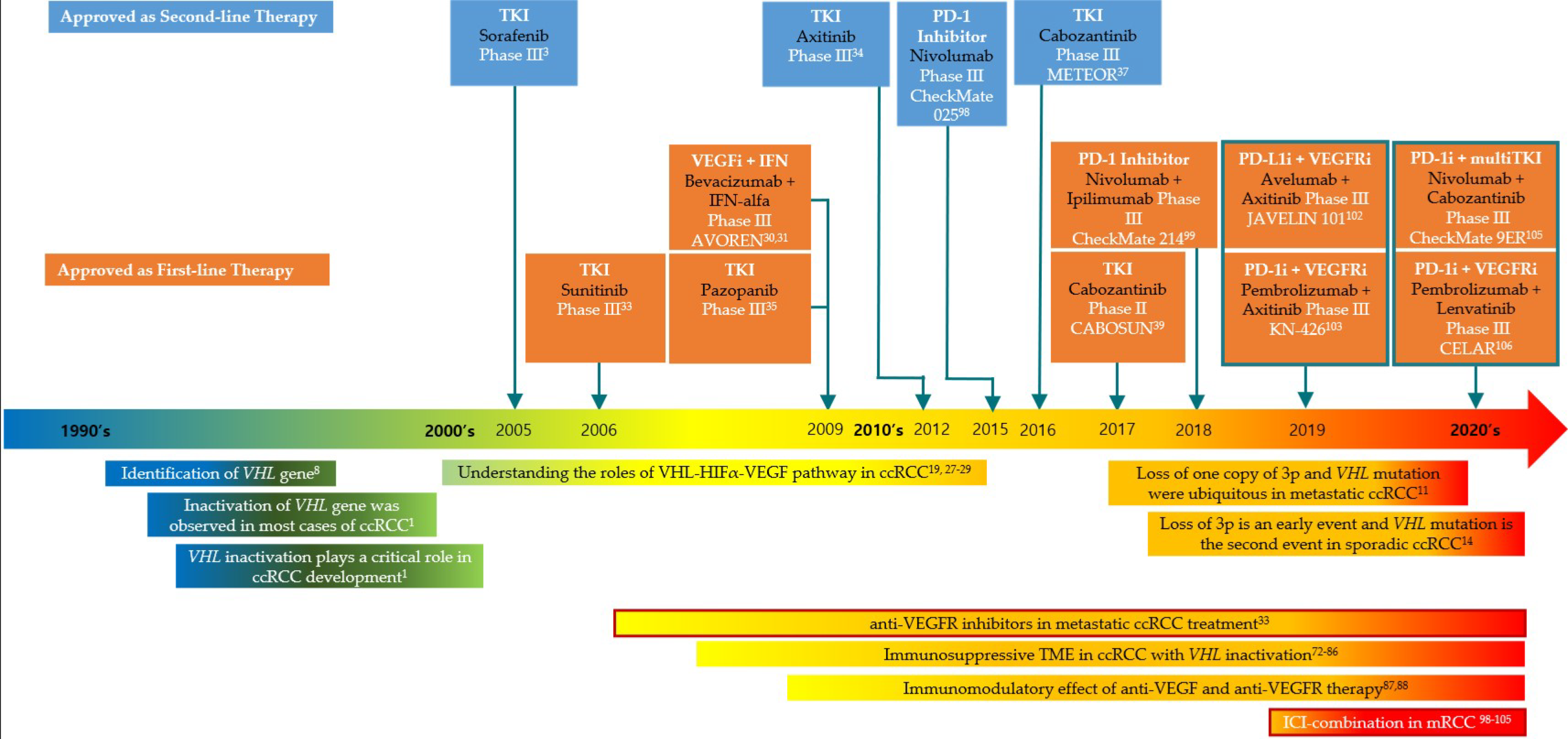

{kind=link}
| Study | Agents | N | Primary Endpoint | PFS (HR) | OS (HR) |
|---|---|---|---|---|---|
| CheckMate 214 99 (NCT02231749) | 1. Ipilimumab + Nivolumab | 1096 | PFS; OS; ORR | Met (0.85) | Met (0.71) |
| 2. Sunitinib | |||||
| JAVELIN Renal 101 102 (NCT02684006) | 1. Avelumab + Axitinib | 886 | PFS; OS | Met (0.69) | Unmet (0.78) |
| 2. Sunitinib | |||||
| IMmotion151 101 (NCT02420821) | 1. Atezolizumab + Bevacizumab | 915 | PFS; OS | Met (0.83) | Unmet (0.81) |
| 2. Sunitinib | |||||
| KEYNOTE-426 103 (NCT02853331) | 1. Pembrolizumab + Axitinib | 861 | PFS; OS | Met (0.69) | Met (0.53) |
| 2. Sunitinb | |||||
| CLEAR 106 (NCT02811861) | 1. Pembrolizumab + Lenvatinib | 1069 | PFS | Met (0.39) | Met (0.66) |
| 2. Sunitinib | |||||
| CheckMate 9ER 105 (NCT03141177) | 1. Nivolumab + Cabozantinib | 651 | PFS | Met (0.51) | Met (0.60) |
| 2. Sunitinib |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Shim, B.Y.; Lee, S.-J.; Lee, J.Y.; Lee, H.-J.; Kim, I.-H. Loss of Von Hippel–Lindau (VHL) Tumor Suppressor Gene Function: VHL–HIF Pathway and Advances in Treatments for Metastatic Renal Cell Carcinoma (RCC). Int. J. Mol. Sci. 2021, 22, 9795. https://doi.org/10.3390/ijms22189795
Kim H, Shim BY, Lee S-J, Lee JY, Lee H-J, Kim I-H. Loss of Von Hippel–Lindau (VHL) Tumor Suppressor Gene Function: VHL–HIF Pathway and Advances in Treatments for Metastatic Renal Cell Carcinoma (RCC). International Journal of Molecular Sciences. 2021; 22(18):9795. https://doi.org/10.3390/ijms22189795
Chicago/Turabian StyleKim, Hyunho, Byoung Yong Shim, Seung-Ju Lee, Ji Youl Lee, Hyo-Jin Lee, and In-Ho Kim. 2021. "Loss of Von Hippel–Lindau (VHL) Tumor Suppressor Gene Function: VHL–HIF Pathway and Advances in Treatments for Metastatic Renal Cell Carcinoma (RCC)" International Journal of Molecular Sciences 22, no. 18: 9795. https://doi.org/10.3390/ijms22189795
APA StyleKim, H., Shim, B. Y., Lee, S.-J., Lee, J. Y., Lee, H.-J., & Kim, I.-H. (2021). Loss of Von Hippel–Lindau (VHL) Tumor Suppressor Gene Function: VHL–HIF Pathway and Advances in Treatments for Metastatic Renal Cell Carcinoma (RCC). International Journal of Molecular Sciences, 22(18), 9795. https://doi.org/10.3390/ijms22189795