
Small Molecules Targeting Programmed Cell Death in Breast Cancer Cells



Abstract
1. Introduction
2. Apoptosis
2.1. Targeting Apoptotic Pathway
2.2. Targeting Signalling Pathways Involved in Genomic Instability
2.3. Targeting Pathways Involved in Proliferative Signalling
2.4. Targeting Signalling Pathways Involved in Modulating Tumour Microenvironment
2.4.1. Endoplasmic Reticulum Stress
2.4.2. Oxidative Stress
2.5. Other SMALL Molecules
3. Autophagy
3.1. Synthetic Small Molecules as Autophagic Inducers
3.2. Small Molecules from Natural Products as Autophagic Inducer
3.3. Small Molecules as Autophagic Inhibitors
3.4. Enhance Delivery
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef]
- Kerr, J.F.R. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J. Pathol. Bacteriol. 1965, 90, 419–435. [Google Scholar] [CrossRef]
- Williams, J.R.; Little, J.B.; Shipley, W.U. Association of mammalian cell death with a specific endonucleolytic degradation of DNA. Nature 1974, 252, 754–755. [Google Scholar] [CrossRef]
- Rozeboom, B.; Dey, N.; De, P. ER+ metastatic breast cancer: Past, present, and a prescription for an apoptosis-targeted future. Am. J. Cancer Res. 2019, 9, 2821–2831. [Google Scholar]
- MacKenzie, S.H.; Clark, A.C. Death by caspase dimerization. Adv. Exp. Med. Biol. 2012, 747, 55–73. [Google Scholar] [CrossRef]
- McKenna, S.; García-Gutiérrez, L.; Matallanas, D.; Fey, D. BAX and SMAC regulate bistable properties of the apoptotic caspase system. Sci. Rep. 2021, 11, 3272. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Sun, H.; Nikolovska-Coleska, Z.; Lu, J.; Meagher, J.L.; Yang, C.Y.; Qiu, S.; Tomita, Y.; Ueda, Y.; Jiang, S.; Krajewski, K.; et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J. Am. Chem. Soc. 2007, 129, 15279–15294. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; McEachern, D.; Li, W.; Davis, M.A.; Li, H.; Morgan, M.A.; Bai, L.; Sebolt, J.T.; Sun, H.; Lawrence, T.S.; et al. Radiosensitization of head and neck squamous cell carcinoma by a SMAC-mimetic compound, SM-164, requires activation of caspases. Mol. Cancer Ther. 2011, 10, 658–669. [Google Scholar] [CrossRef]
- Yang, D.; Zhao, Y.; Li, A.Y.; Wang, S.; Wang, G.; Sun, Y. Smac-mimetic compound SM-164 induces radiosensitization in breast cancer cells through activation of caspases and induction of apoptosis. Breast Cancer Res. Treat. 2012, 133, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.A.; Perry, C.; Dickinson, P.; Ball, G.; Moseley, P.; Madhusudan, S.; Ellis, I.O.; Chan, S.Y.T. Bcl2 is an independent prognostic marker of triple negative breast cancer (TNBC) and predicts response to anthracycline combination (ATC) chemotherapy (CT) in adjuvant and neoadjuvant settings. Ann. Oncol. 2013, 24, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Tekedereli, I.; Alpay, S.N.; Akar, U.; Yuca, E.; Ayugo-Rodriguez, C.; Han, H.D.; Sood, A.K.; Lopez-Berestein, G.; Ozpolat, B. Therapeutic silencing of Bcl-2 by systemically administered siRNA nanotherapeutics inhibits tumor growth by autophagy and apoptosis and enhances the efficacy of chemotherapy in orthotopic xenograft models of ER (−) and ER (+) breast cancer. Mol. Ther. Nucleic Acids 2013, 2, e121. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Dal Molin, G.Z.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Chen, H.; Wu, J.; Zhang, Z.; Tang, Y.; Li, X.; Liu, S.; Cao, S.; Li, X. Association between BRCA status and triple-negative breast cancer: A meta-analysis. Front. Pharmacol. 2018, 9, 909. [Google Scholar] [CrossRef]
- Ye, F.; He, M.; Huang, L.; Lang, G.; Hu, X.; Shao, Z.; Di, G.; Cao, A. Insights Into the Impacts of BRCA Mutations on Clinicopathology and Management of Early-Onset Triple-Negative Breast Cancer. Front. Oncol. 2021, 10, 574813. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, R.; Simionato, F.; Ciccarese, C.; Grego, E.; Cingarlini, S.; Iacovelli, R.; Bria, E.; Tortora, G.; Melisi, D. The development of PARP as a successful target for cancer therapy. Expert Rev. Anticancer. Ther. 2018, 18, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Go, A.; Jang, J.W.; Lee, W.; Du Ha, J.; Kim, H.J.; Nam, H.J. Augmentation of the antitumor effects of PARP inhibitors in triple-negative breast cancer via degradation by hydrophobic tagging modulation. Eur. J. Med. Chem. 2020, 204, 112635. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wang, S.; Wang, X.; Wang, P.; Zheng, Y.; Yao, D.; Guo, M.; Zhang, L.; Ouyang, L. Crystal structure-based discovery of a novel synthesized PARP1 inhibitor (OL-1) with apoptosis-inducing mechanisms in triple-negative breast cancer. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef]
- Ofori, S.; Awuah, S.G. Small-molecule poly(ADP-ribose) polymerase and PD-L1 inhibitor conjugates as dual-action anticancer agents. ACS Omega 2019, 4, 12584–12597. [Google Scholar] [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the cell cycle: A complex landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef]
- Engström, W.; Darbre, P.; Eriksson, S.; Gulliver, L.; Hultman, T.; Karamouzis, M.V.; Klaunig, J.E.; Mehta, R.; Moorwood, K.; Sanderson, T.; et al. The potential for chemical mixtures from the environment to enable the cancer hallmark of sustained proliferative signalling. Carcinogenesis 2015, 36, S38–S60. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Peptide-chain elongation in eukaryotes. Mol. Biol. Rep. 1994, 19, 161–170. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, X.; Liu, J.; Zhou, L.; Zhang, C.; Xu, L.; Qin, Q.; Zhan, L.; Lu, J.; Cheng, H.; et al. Eukaryotic elongation factor 2 kinase confers tolerance to stress conditions in cancer cells. Cell Stress Chaperones 2015, 20, 217–220. [Google Scholar] [CrossRef]
- Wang, X.; Xie, J.; Proud, C.G. Eukaryotic elongation factor 2 kinase (eEF2K) in cancer. Cancers 2017, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Liu, R.; Proud, C.G.; Wang, M.W. A high-throughput screening assay for eukaryotic elongation factor 2 kinase inhibitors. Acta Pharm. Sin. B 2016, 6, 557–563. [Google Scholar] [CrossRef][Green Version]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.; Jones, R.A.; Datti, A.; Uehling, D.; Al-awar, R.; Egan, S.E.; Bader, G.D.; et al. Combined deletion of P ten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on e EF 2 K. EMBO Mol. Med. 2014, 6, 1542–1560. [Google Scholar] [CrossRef]
- Liu, R.; Proud, C.G. Eukaryotic elongation factor 2 kinase as a drug target in cancer, and in cardiovascular and neurodegenerative diseases. Acta Pharmacol. Sin. 2016, 37, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhao, Y.; Wang, G.; Chen, Y.; Jiang, Y.; Ouyang, L.; Liu, B. Design, synthesis and structure-activity relationship of a focused library of β-phenylalanine derivatives as novel eEF2K inhibitors with apoptosis-inducing mechanisms in breast cancer. Eur. J. Med. Chem. 2018, 143, 402–418. [Google Scholar] [CrossRef]
- Liu, Y.; Zhen, Y.; Wang, G.; Yang, G.; Fu, L.; Liu, B.; Ouyang, L. Designing an eEF2K-Targeting PROTAC small molecule that induces apoptosis in MDA-MB-231 cells. Eur. J. Med. Chem. 2020, 204, 112505. [Google Scholar] [CrossRef]
- Karginova, O.; Weekley, C.M.; Raoul, A.; Alsayed, A.; Wu, T.; Lee, S.S.Y.; He, C.; Olopade, O.I. Inhibition of copper transport induces apoptosis in triple-negative breast cancer cells and suppresses tumor angiogenesis. Mol. Cancer Ther. 2019, 18, 873–885. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 10, 3083. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Akman, M.; Belisario, D.C.; Salaroglio, I.C.; Kopecka, J.; Donadelli, M.; De Smaele, E.; Riganti, C. Hypoxia, endoplasmic reticulum stress and chemoresistance: Dangerous liaisons. J. Exp. Clin. Cancer Res. 2021, 40, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Kho, D.; Xu, J.; Gajan, A.; Wu, K.; Wu, G.S. ONC201 activates ER stress to inhibit the growth of triple-negative breast cancer cells. Oncotarget 2017, 8, 21626–21638. [Google Scholar] [CrossRef]
- Yamanaka, K.; Nakata, M.; Kaneko, N.; Fushiki, H.; Kita, A.; Nakahara, T.; Koutoku, H.; Sasamata, M. YM155, a selective survivin suppressant, inhibits tumor spread and prolongs survival in a spontaneous metastatic model of human triple negative breast cancer. Int. J. Oncol. 2011, 39, 569–575. [Google Scholar] [CrossRef]
- Pennati, M.; Sbarra, S.; De Cesare, M.; Lopergolo, A.; Locatelli, S.L.; Campi, E.; Daidone, M.G.; Carlo-Stella, C.; Gianni, A.M.; Zaffaroni, N. YM155 sensitizes triple-negative breast cancer to membrane-bound TRAIL through p38 MAPK- and CHOP-mediated DR5 upregulation. Int. J. Cancer 2015, 136, 299–309. [Google Scholar] [CrossRef]
- Jia, L.; Zhou, Z.; Liang, H.; Wu, J.; Shi, P.; Li, F.; Wang, Z.; Wang, C.; Chen, W.; Zhang, H.; et al. KLF5 promotes breast cancer proliferation, migration and invasion in part by upregulating the transcription of TNFAIP2. Oncogene 2016, 35, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, Q.; Ding, Y.; Zhou, W.; Liu, R.; Chen, H.; Zhou, J.; Feng, J.; Chen, C. YD277 suppresses triple-negative breast cancer partially through activating the endoplasmic reticulum stress pathway. Theranostics 2017, 7, 2339–2349. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef]
- Maurais, A.J.; Weerapana, E. Reactive-cysteine profiling for drug discovery. Curr. Opin. Chem. Biol. 2019, 50, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; To, M.; Olzmann, J.A.; Nomura, D.K. Chemoproteomics-Enabled Covalent Ligand Screening Reveals a Thioredoxin-Caspase 3 Interaction Disruptor That Impairs Breast Cancer Pathogenicity. ACS Chem. Biol. 2017, 12, 2522–2528. [Google Scholar] [CrossRef]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding—A review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 2011, 9, 13. [Google Scholar] [CrossRef]
- Preet, R.; Mohapatra, P.; Mohanty, S.; Sahu, S.K.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine has anticancer activity in breast cancer cells through inhibition of topoisomerase activity. Int. J. Cancer 2012, 130, 1660–1670. [Google Scholar] [CrossRef]
- Sharma, P.; Dwivedee, B.P.; Bisht, D.; Dash, A.K.; Kumar, D. The chemical constituents and diverse pharmacological importance of Tinospora cordifolia. Heliyon 2019, 5, e02437. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, K.C.; Raj, M.H.; Paul, M.; Girish, K.S.; Salimath, B.P.; Aparna, H.S. A new pyrrole based small molecule from Tinospora cordifolia induces apoptosis in MDA-MB-231 breast cancer cells via ROS mediated mitochondrial damage and restoration of p53 activity. Chem. Biol. Interact. 2019, 299, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.C.; Gierke, S.; Pitt, C.; Sagolla, M.; Cheng, C.K.; Zhou, W.; Jubb, A.M.; Strickland, L.; Schmidt, M.; Duron, S.G.; et al. Small molecule inhibition of group I p21-activated kinases in breast cancer induces apoptosis and potentiates the activity of microtubule stabilizing agents. Breast Cancer Res. 2015, 17, 1–12. [Google Scholar] [CrossRef]
- Wang, J.; Kim, T.H.; Ahn, M.Y.; Lee, J.; Jung, J.H.; Choi, W.S.; Lee, B.M.; Yoon, K.S.; Yoon, S.; Kim, H.S. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 1101–1109. [Google Scholar] [CrossRef]
- Kalle, A.M.; Mallika, A.; Badiger, J.; Alinakhi; Talukdar, P.; Sachchidanand. Inhibition of SIRT1 by a small molecule induces apoptosis in breast cancer cells. Biochem. Biophys. Res. Commun. 2010, 401, 13–19. [Google Scholar] [CrossRef]
- Kondo, Y. Targeting histone methyltransferase EZH2 as cancer treatment. J. Biochem. 2014, 156, 249–257. [Google Scholar] [CrossRef]
- Song, X.; Gao, T.; Wang, N.; Feng, Q.; You, X.; Ye, T.; Lei, Q.; Zhu, Y.; Xiong, M.; Xia, Y.; et al. Selective inhibition of EZH2 by ZLD1039 blocks H3K27methylation and leads to potent anti-tumor activity in breast cancer. Sci. Rep. 2016, 6, 20864. [Google Scholar] [CrossRef]
- Chestukhin, A.; Pfeffer, C.; Milligan, S.; DeCaprio, J.A.; Pellman, D. Processing, localization, and requirement of human separase for normal anaphase progression. Proc. Natl. Acad. Sci. USA 2003, 100, 4574–4579. [Google Scholar] [CrossRef]
- Zhang, N.; Scorsone, K.; Ge, G.; Kaffes, C.C.; Dobrolecki, L.E.; Mukherjee, M.; Lewis, M.T.; Berg, S.; Stephan, C.C.; Pati, D. Identification and characterization of Separase inhibitors (Sepins) for cancer therapy. J. Biomol. Screen. 2014, 19, 878–889. [Google Scholar] [CrossRef]
- Wang, W.; Zafar, A.; Rajaei, M.; Zhang, R. Two Birds with One Stone: NFAT1-MDM2 Dual Inhibitors for Cancer Therapy. Cells 2020, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Wang, W.; Sarkar, S.; Voruganti, S.; Agarwal, R.; Zhang, R. Inulanolide A as a new dual inhibitor of NFAT1-MDM2 pathway for breast cancer therapy. Oncotarget 2016, 7, 32566–32578. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Liu, B.; Wen, X.; Cheng, Y. Survival or death: Disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013, 4, e892–e892. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef]
- Okamoto, K.; Kondo-Okamoto, N. Mitochondria and autophagy: Critical interplay between the two homeostats. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 595–600. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Nyfeler, B.; Eng, C.H. Revisiting autophagy addiction of tumor cells. Autophagy 2016, 12, 1206–1207. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef]
- Ren, X.S.; Sato, Y.; Harada, K.; Sasaki, M.; Furubo, S.; Song, J.Y.; Nakanuma, Y. Activation of the PI3K/mTOR pathway is involved in cystic proliferation of cholangiocytes of the PCK rat. PLoS ONE 2014, 9, e87660. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Liu, T.; Kamp, D.W.; Wang, Y.; He, H.; Zhou, X.; Li, D.; Yang, L.; Zhao, B.; Liu, G. AKT/mTOR and c-Jun N-terminal kinase signaling pathways are required for chrysotile asbestos-induced autophagy. Free Radic. Biol. Med. 2014, 72, 296–307. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Nakano, I.; Masterman-Smith, M.; Saigusa, K.; Paucar, A.A.; Horvath, S.; Shoemaker, L.; Watanabe, M.; Negro, A.; Bajpai, R.; Howes, A.; et al. Maternal embryonic leucine zipper kinase is a key regulator of the proliferation of malignant brain tumors, including brain tumor stem cells. J. Neurosci. Res. 2008, 86, 48–60. [Google Scholar] [CrossRef]
- Marie, S.K.; Okamoto, O.K.; Uno, M.; Hasegawa, A.P.; Oba-Shinjo, S.M.; Cohen, T.; Camargo, A.A.; Kosoy, A.; Carlotti, C.G., Jr.; Toledo, S.; et al. Maternal embryonic leucine zipper kinase transcript abundance correlates with malignancy grade in human astrocytomas. Int. J. Cancer 2008, 122, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Pickard, M.R.; Green, A.R.; Ellis, I.O.; Caldas, C.; Hedge, V.L.; Mourtada-Maarabouni, M.; Williams, G.T. Dysregulated expression of Fau and MELK is associated with poor prognosis in breast cancer. Breast Cancer Res. 2009, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Springer, M.Z.; Xu, D.; Liu, D.; Hudmon, A.; Macleod, K.F.; Meroueh, S.O. Small molecules inhibit STAT3 activation, autophagy, and cancer cell anchorage-independent growth. Bioorg. Med. Chem. 2017, 25, 2995–3005. [Google Scholar] [CrossRef]
- Vogel, R.I.; Coughlin, K.; Scotti, A.; Iizuka, Y.; Anchoori, R.; Roden, R.B.S.; Marastoni, M.; Bazzaro, M. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget 2015, 6, 4159–4170. [Google Scholar] [CrossRef]
- Chang, C.H.; Bijian, K.; Wernic, D.; Su, J.; da Silva, S.D.; Yu, H.; Qiu, D.; Asslan, M.; Alaoui-Jamali, M.A. A novel orally available seleno-purine molecule suppresses triple-negative breast cancer cell proliferation and progression to metastasis by inducing cytostatic autophagy. Autophagy 2019, 15, 1376–1390. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, J.; Yang, C.; Pluta, R.; Wang, G.; Ye, T.; Ouyang, L. Identification and optimization of 3-bromo-N’-(4-hydroxybenzylidene)-4-methylbenzohydrazide derivatives as mTOR inhibitors that induce autophagic cell death and apoptosis in triple-negative breast cancer. Eur. J. Med. Chem. 2021, 219, 113424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fu, L.; Zhang, S.; Zhang, J.; Zhao, Y.; Zheng, Y.; He, G.; Yang, S.; Ouyang, L.; Liu, B. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem. Sci. 2017, 8, 2687–2701. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Zhang, L.; Liu, J.; Fu, L.; Yao, D.; Zhao, Y.; Zhang, S.; Wang, G.; He, G.; Liu, B. Discovery of a Small-Molecule Bromodomain-Containing Protein 4 (BRD4) Inhibitor That Induces AMP-Activated Protein Kinase-Modulated Autophagy-Associated Cell Death in Breast Cancer. J. Med. Chem. 2017, 60, 9990–10012. [Google Scholar] [CrossRef] [PubMed]
- Kozyreva, V.K.; Kiseleva, A.A.; Ice, R.J.; Jones, B.; Loskutov, Y.V.; Matalkah, F.; Smolkin, M.B.; Marinak, K.; Livengood, R.H.; Salkeni, M.; et al. Combination of eribulin and aurora a inhibitor MLN8237 prevents metastatic colonization and induces cytotoxic autophagy in breast cancer. Mol. Cancer Ther. 2016, 15, 1809–1822. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, M.; Li, J.; Zheng, Y.; Zhang, S.; Xie, T.; Liu, B. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol. Biosyst. 2015, 11, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Chollat-Namy, M.; Ben Safta-Saadoun, T.; Haferssas, D.; Meurice, G.; Chouaib, S.; Thiery, J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell Death Dis. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Arihara, Y.; Takada, K.; Kamihara, Y.; Hayasaka, N.; Nakamura, H.; Murase, K.; Ikeda, H.; Iyama, S.; Sato, T.; Miyanishi, K.; et al. Small molecule CP-31398 induces reactive oxygen species-dependent apoptosis in human multiple myeloma. Oncotarget 2017, 8, 65889–65899. [Google Scholar] [CrossRef]
- Xu, J.; Timares, L.; Heilpern, C.; Weng, Z.; Li, C.; Xu, H.; Pressey, J.G.; Elmets, C.A.; Kopelovich, L.; Athar, M. Targeting wild-type and mutant p53 with small molecule CP-31398 blocks the growth of rhabdomyosarcoma by inducing reactive oxygen species-dependent apoptosis. Cancer Res. 2010, 70, 6566–6576. [Google Scholar] [CrossRef]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef]
- Rauch, A.; Hennig, D.; Schäfer, C.; Wirth, M.; Marx, C.; Heinzel, T.; Schneider, G.; Krämer, O.H. Survivin and YM155: How faithful is the liaison? Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 202–220. [Google Scholar] [CrossRef]
- Nakahara, T.; Takeuchi, M.; Kinoyama, I.; Minematsu, T.; Shirasuna, K.; Matsuhisa, A.; Kita, A.; Tominaga, F.; Yamanaka, K.; Kudoh, M.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef]
- Véquaud, E.; Séveno, C.; Loussouarn, D.; Engelhart, L.; Campone, M.; Juin, P.; Barillé-Nion, S. YM155 potently triggers cell death in breast cancer cells through an autophagy-NF-kB network. Oncotarget 2015, 6, 13476–13486. [Google Scholar] [CrossRef]
- Cheng, S.M.; Chang, Y.C.; Liu, C.Y.; Lee, J.Y.C.; Chan, H.H.; Kuo, C.W.; Lin, K.Y.; Tsai, S.L.; Chen, S.H.; Li, C.F.; et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br. J. Pharmacol. 2015, 172, 214–234. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Castellino, S.M.; Friedman, H.S.; Elion, G.B.; Ong, E.T.; Marcelli, S.L.; Page, R.; Bigner, D.D.; Dewhirst, M.W. Flunarizine enhancement of melphalan activity against drug-sensitive/resistant rhabdomyosarcoma. Br. J. Cancer 1995, 71, 1181–1187. [Google Scholar] [CrossRef][Green Version]
- Gornati, D.; Zaffaroni, N.; Villa, R.; De Marco, C.; Silvestrini, R. Modulation of melphalan and cisplatin cytotoxicity in human ovarian cancer cells resistant to alkylating drugs. Anti-Cancer Drugs 1997, 8, 509–516. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Li, J.; Li, F.; Zhu, Y.; Cui, K.; Wong, S.T.; Chang, E.C.; Liao, Y.H. Induction of N-Ras degradation by flunarizine-mediated autophagy. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moulder, S.L.; Yakes, F.M.; Muthuswamy, S.K.; Bianco, R.; Simpson, J.F.; Arteaga, C.L. Epidermal Growth Factor Receptor (HER1) Tyrosine Kinase Inhibitor ZD1839 (Iressa) Inhibits HER2/neu (erbB2)-overexpressing Breast Cancer Cells in Vitro and in Vivo. Cancer Res. 2001, 61, 8887–8895. [Google Scholar]
- Anido, J.; Matar, P.; Albanell, J.; Guzmán, M.; Rojo, F.; Arribas, J.; Averbuch, S.; Baselga, J. ZD1839, a Specific Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor, Induces the Formation of Inactive EGFR/HER2 and EGFR/HER3 Heterodimers and Prevents Heregulin Signaling in HER2-overexpressing Breast Cancer Cells. Clin. Cancer Res. 2003, 9, 1274–1283. [Google Scholar] [PubMed]
- Moasser, M.M.; Basso, A.; Averbuch, S.D.; Rosen, N. The Tyrosine Kinase Inhibitor ZD1839 (“Iressa”) Inhibits HER2-driven Signaling and Suppresses the Growth of HER2-overexpressing Tumor Cells. Cancer Res. 2001, 61, 7184–7188. [Google Scholar]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of Autophagy Is an Early Response to Gefitinib and a Potential Therapeutic Target in Breast Cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F.; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef]
- Young, C.D.; Arteaga, C.L.; Cook, R.S. Dual inhibition of Type I and Type III PI3 kinases increases tumor cell apoptosis in HER2+ breast cancers. Breast Cancer Res. 2015, 17, 1–11. [Google Scholar] [CrossRef]
- Farina, A.; Hattori, M.; Qin, J.; Nakatani, Y.; Minato, N.; Ozato, K. Bromodomain Protein Brd4 Binds to GTPase-Activating SPA-1, Modulating Its Activity and Subcellular Localization. Mol. Cell. Biol. 2004, 24, 9059–9069. [Google Scholar] [CrossRef]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmäki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain Protein BRD4 Is Required for Estrogen Receptor-Dependent Enhancer Activation and Gene Transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Cannizzaro, E.; Dawson, M.A. Bromodomains as therapeutic targets in cancer. Brief. Funct. Genom. 2013, 12, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.J.; Qi, M.; Li, N.; Lei, Y.H.; Zhang, D.M.; Chen, J.X. Natural products and their derivatives: Promising modulators of tumor immunotherapy. J. Leukoc. Biol. 2020, 108, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.E.F.; Moustafa, M.S.; El-Wahed, A.A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Kallifatidis, G.; Labsch, S.; Rausch, V.; Mattern, J.; Gladkich, J.; Moldenhauer, G.; Büchler, M.W.; Salnikov, A.V.; Herr, I. Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Mol. Ther. 2011, 19, 188–195. [Google Scholar] [CrossRef]
- Singh, S.V.; Herman-Antosiewicz, A.; Singh, A.V.; Lew, K.L.; Srivastava, S.K.; Kamath, R.; Brown, K.D.; Zhang, L.; Baskaran, R. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J. Biol. Chem. 2004, 279, 25813–25822. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Noh, E.M.; Han, J.H.; Kim, J.M.; Hwang, B.M.; Kim, B.S.; Lee, S.H.; Jung, S.H.; Youn, H.J.; Chung, E.Y.; et al. Sulforaphane controls TPA-induced MMP-9 expression through the NF-κB signaling pathway, but not AP-1, in MCF-7 breast cancer cells. BMB Rep. 2013, 46, 201–206. [Google Scholar] [CrossRef]
- Yang, F.; Wang, F.; Liu, Y.; Wang, S.; Li, X.; Huang, Y.; Xia, Y.; Cao, C. Sulforaphane induces autophagy by inhibition of HDAC6-mediated PTEN activation in triple negative breast cancer cells. Life Sci. 2018, 213, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Vasilatos, S.N.; Bhargava, R.; Fine, J.L.; Oesterreich, S.; Davidson, N.E.; Huang, Y. Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 2017, 36, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Wu, H.; Vasilatos, S.N.; Chandran, U.; Qin, Y.; Wan, Y.; Oesterreich, S.; Davidson, N.E.; Huang, Y. HDAC5–LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 2018, 143, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhou, Y.; Duan, D.; Yang, C.; Zhou, Z.; Li, F.; Kong, Y.; Hsieh, Y.C.; Zhang, R.; Ding, W.; et al. Targeting ubiquitin conjugating enzyme UbcH5b by a triterpenoid PC3-15 from Schisandra plants sensitizes triple-negative breast cancer cells to lapatinib. Cancer Lett. 2021, 504, 125–136. [Google Scholar] [CrossRef]
- Hoeller, D.; Dikic, I. Targeting the ubiquitin system in cancer therapy. Nature 2009, 458, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. Ubiquitin- and ubiquitin-like proteins-conjugating enzymes (E2s) in breast cancer. Mol. Biol. Rep. 2013, 40, 2019–2034. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Mok, S.W.F.; Chen, J.; Michelangeli, F.; Jiang, Z.H.; Han, Y.; Qu, Y.Q.; Qiu, A.C.L.; Xu, S.W.; Xue, W.W.; et al. N-desmethyldauricine induces autophagic cell death in apoptosis-defective cells via Ca2+ mobilization. Front. Pharmacol. 2017, 8, 388. [Google Scholar] [CrossRef]
- York, B.; O’Malley, B.W. Steroid Receptor Coactivator (SRC) family: Masters of systems biology. J. Biol. Chem. 2010, 285, 38743–38750. [Google Scholar] [CrossRef]
- Zhao, C.; Yasui, K.; Lee, C.J.; Kurioka, H.; Hosokawa, Y.; Oka, T.; Inazawa, J. Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer 2003, 98, 18–23. [Google Scholar] [CrossRef]
- Wang, L.; Yu, Y.; Chow, D.C.; Yan, F.; Hsu, C.C.; Stossi, F.; Mancini, M.A.; Palzkill, T.; Liao, L.; Zhou, S.; et al. Characterization of a Steroid Receptor Coactivator Small Molecule Stimulator that Overstimulates Cancer Cells and Leads to Cell Stress and Death. Cancer Cell 2015, 28, 240–252. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Dichloroacetate (DCA) sensitizes both wild-type and over expressing bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Shi, W. Autophagy regulates resistance of non-small cell lung cancer cells to paclitaxel. Tumor Biol. 2016, 37, 10539–10544. [Google Scholar] [CrossRef]
- Wang, M.; Liao, C.; Hu, Y.; Pan, Q.; Jiang, J. Sensitization of breast cancer cells to paclitaxel by dichloroacetate through inhibiting autophagy. Biochem. Biophys. Res. Commun. 2017, 489, 103–108. [Google Scholar] [CrossRef]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Schlütermann, D.; Skowron, M.A.; Berleth, N.; Böhler, P.; Deitersen, J.; Stuhldreier, F.; Wallot-Hieke, N.; Wu, W.; Peter, C.; Hoffmann, M.J.; et al. Targeting urothelial carcinoma cells by combining cisplatin with a specific inhibitor of the autophagy-inducing class III PtdIns3K complex. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 160.e1–160.e13. [Google Scholar] [CrossRef]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef]
- Chen, C.H.; Changou, C.A.; Hsieh, T.H.; Lee, Y.C.; Chu, C.Y.; Hsu, K.C.; Wang, H.C.; Lin, Y.C.; Lo, Y.N.; Liu, Y.R.; et al. Dual inhibition of PIK3C3 and FGFR as a new therapeutic approach to treat bladder cancer. Clin. Cancer Res. 2018, 24, 1176–1189. [Google Scholar] [CrossRef]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Dassonneville, L.; Bailly, C. Stimulation of topoisomerase II-mediated DNA cleavage by an indazole analogue of lucanthone. Biochem. Pharmacol. 1999, 58, 1307–1312. [Google Scholar] [CrossRef]
- LUO, M.; KELLEY, M.R. Inhibition of the Human Apurinic/Apyrimidinic Endonuclease (Ape1) Repair Activity and Sensitization of Breast Cancer Cells to DNA Alkylating Agents with Lucanthone. Anticancer Res. 2004, 24, 2127–2134. [Google Scholar]
- Carew, J.S.; Espitia, C.M.; Esquivel, J.A.; Mahalingam, D.; Kelly, K.R.; Reddy, G.; Giles, F.J.; Nawrocki, S.T. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J. Biol. Chem. 2011, 286, 6602–6613. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Bakas, N.A.; Vamos, M.; Chaikuad, A.; Limpert, A.S.; Wimer, C.D.; Brun, S.N.; Lambert, L.J.; Tautz, L.; Celeridad, M.; et al. Design, Synthesis, and Characterization of an Orally Active Dual-Specific ULK1/2 Autophagy Inhibitor that Synergizes with the PARP Inhibitor Olaparib for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2020, 63, 14609–14625. [Google Scholar] [CrossRef] [PubMed]
- Robke, L.; Futamura, Y.; Konstantinidis, G.; Wilke, J.; Aono, H.; Mahmoud, Z.; Watanabe, N.; Wu, Y.W.; Osada, H.; Laraia, L.; et al. Discovery of the novel autophagy inhibitor aumitin that targets mitochondrial complex i. Chem. Sci. 2018, 9, 3014–3022. [Google Scholar] [CrossRef]
- Eck, W.; Craig, G.; Sigdel, A.; Ritter, G.; Old, L.J.; Tang, L.; Brennan, M.F.; Allen, P.J.; Mason, M.D. PEGylated gold nanoparticles conjugated to monoclonal F19 antibodies as targeted labeling agents for human pancreatic carcinoma tissue. ACS Nano 2008, 2, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E.; Mwamuka, J.; Gole, A. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small 2005, 1, 325–327. [Google Scholar] [CrossRef]
- Sanders, M.A.; Brahemi, G.; Nangia-Makker, P.; Balan, V.; Morelli, M.; Kothayer, H.; Westwell, A.D.; Shekhar, M.P. Novel inhibitors of Rad6 ubiquitin conjugating enzyme: Design, synthesis, identification, and functional characterization. Mol. Cancer Ther. 2013, 12, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, S.; McGrath, J.P.; Varshavsky, A. The yeast DNA repair gene RAD6 encodes a ubiquitin-conjugating enzyme. Nature 1987, 329, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Koken, M.H.; Reynolds, P.; Jaspers-Dekker, I.; Prakash, L.O.; Prakash, S.; Bootsma, D.; Hoeijmakers, J.H. Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc. Natl. Acad. Sci. USA 1991, 88, 8865–8869. [Google Scholar] [CrossRef]
- Koken, M.H.; Smit, E.M.; Jaspers-Dekker, I.; Oostra, B.A.; Hagemeuer, A.; Bootsma, D.; Hoeumakers, J.H. Localization of two human homologs, HHR6A and HHR6B, of the yeast DNA repair gene RAD6 to chromosomes Xq24-q25 and 5q23-q31. Genomics 1992, 12, 447–453. [Google Scholar] [CrossRef]
- Haynes, B.; Zhang, Y.; Liu, F.; Li, J.; Petit, S.; Kothayer, H.; Bao, X.; Westwell, A.D.; Mao, G.; Shekhar, M.P. Gold nanoparticle conjugated Rad6 inhibitor induces cell death in triple negative breast cancer cells by inducing mitochondrial dysfunction and PARP-1 hyperactivation: Synthesis and characterization. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 745–757. [Google Scholar] [CrossRef]
- Werfel, T.A.; Wang, S.; Jackson, M.A.; Kavanaugh, T.E.; Joly, M.M.; Lee, L.H.; Hicks, D.J.; Sanchez, V.; Ericsson, P.G.; Kilchrist, K.V.; et al. Selective mTORC2 inhibitor therapeutically blocks breast cancer cell growth and survival. Cancer Res. 2018, 78, 1845–1858. [Google Scholar] [CrossRef]
- Ross, J.S.; Fletcher, J.A. The HER-2/neu oncogene in breast cancer: Prognostic factor, predictive factor, and target for therapy. Stem Cells 1998, 16, 413–428. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Rozengurt, E.; Soares, H.P.; Sinnet-Smith, J. Suppression of feedback loops mediated by pi3k/mtor induces multiple overactivation of compensatory pathways: An unintended consequence leading to drug resistance. Mol. Cancer Ther. 2014, 13, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Jain, K.; Paranandi, K.S.; Sridharan, S.; Basu, A. Autophagy in breast cancer and its implications for therapy. Am. J. Cancer Res. 2013, 3, 251–265. [Google Scholar] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, Y.X.; Qiao, Z.Y.; An, H.W.; Qiao, S.L.; Wang, L.; Rajapaksha, R.Y.; Wang, H. Self-assembled autophagy-inducing polymeric nanoparticles for breast cancer interference in-vivo. Adv. Mater. 2015, 27, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Röth, S.; Fulcher, L.J.; Sapkota, G.P. Advances in targeted degradation of endogenous proteins. Cell. Mol. Life Sci. 2019, 76, 2761–2777. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apoptosis | |||
|---|---|---|---|
| Structure | Code/Name | Pathway Affected | Refs |
 | SM-164 | mediate caspase activation via inhibition of XIAP and cIAP-1 | [18] |
| nanodelivery | nanoliposomal (NL)-Bcl-2 siRNA | reduce the expression of Bcl-2 | [20] |
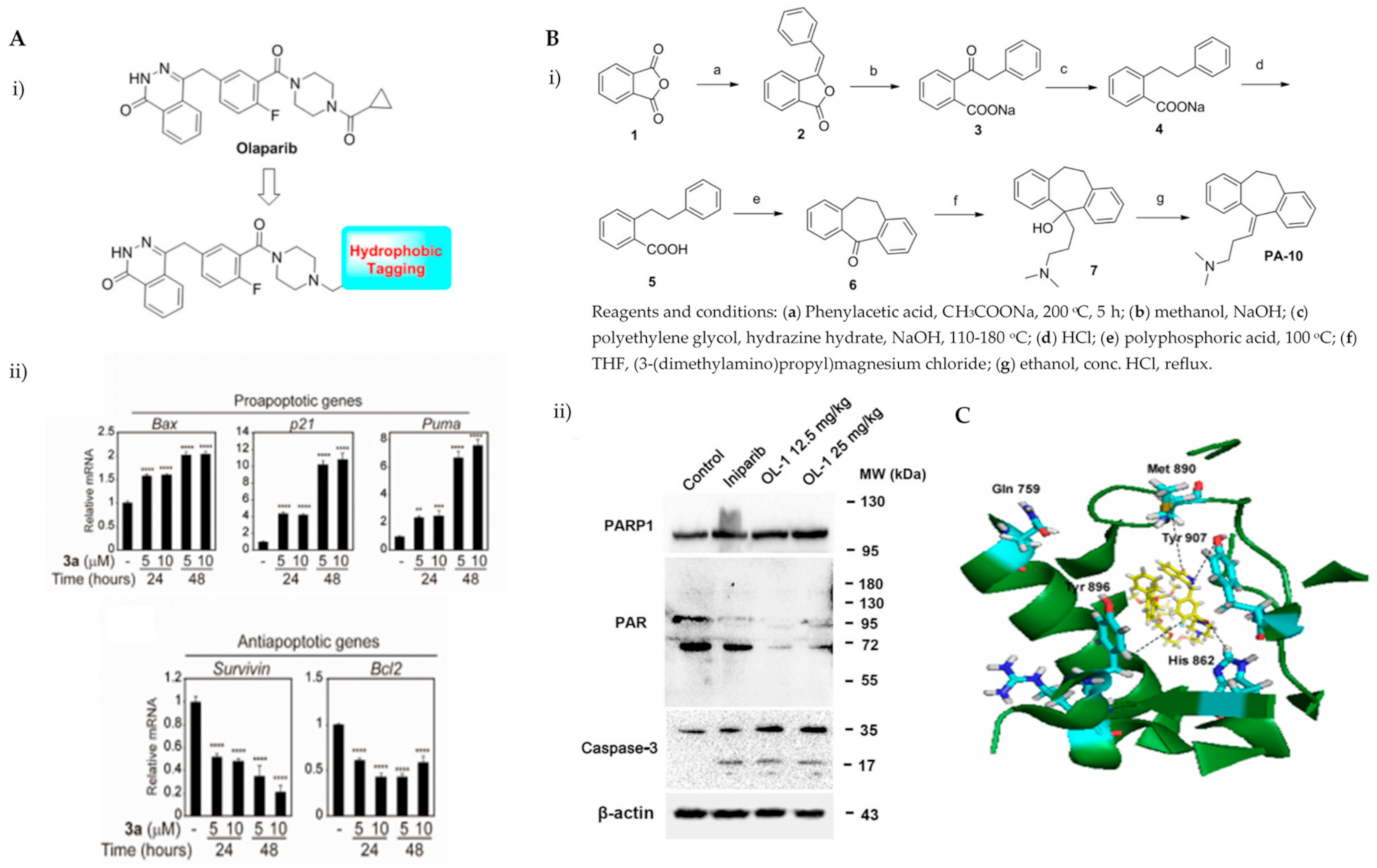
 | Compound 3a | induce PARP1 degradation | [26] |
 | OL-1 | PARP1 inhibition | [27] |
 | Compound 3 | induce proteasome-dependent PARP1 cleavage | [28] |
 | Conjugate 3 | target PARP1 and PD-L1 | [29] |
 | 21l | eEF2K inhibitor | [38] |
 | 11l | degradation via ubiquitin-proteasome-Mediated eEF2K and p-eEF2K | [39] |
 | ONC201 | activate ATF4/CHOP-mediated ER stress response | [46] |
 | YM155 | upregulates DR5 expression | [48] |
 | YD277 | induce IRE1α transcription which leads to JNK activation | [50] |
 | DCAC50 | ATOX1 and CCS inhibitor | [40] |
 | KEAI-97 | interaction disruption between thioredoxin and caspase-3 | [54] |
 | Quinacrine | S-phase arrest and upregulates p53 | [56] |
 | bis(2-ethylhexyl)-1H-pyrrole-3,4-dicarboxylate (TCCP) | elevates ROS and intracellular Ca2+ ion concentration | [58] |
 | FRAX1036 | increases PARP cleavage and reduces cyclin D1 expression | [59] |
 | JGB1741 | SIRT1 inhibitor | [61] |
 | ZLD1039 | EZH2 inhibitor | [63] |
 | Sepin-1 | separase inhibitor | [65] |
 | Inulanolide A (InuA) | cell cycle arrest at G2/M phase | [67] |
| Autophagy | |||
| Structure | Code/name | Target | Refs |
 | KIN-281 | multiple kinases (maternal leucine zipper kinase (MELK) and the non-receptor tyrosine kinase bone marrow X-linked (BMX)) | [86] |
 | b-AP15 | deubiquitinating enzymes | [87] |
 | RA-9 | ||
 | SLLN-15 | AKT-mTOR | [88] |
 | (E)-N-(1H-benzo[d]imidazol-2-yl)-4-(1-(2-(3-bromobenzoyl)hydrazineylidene)ethyl)benzamide | mTOR | [89] |
 | MLN8237 | Aurora A (AURKA) kinase | [92] |
 | Eribulin | ||
 | Flubendazole | Protein 4B(ATG4B) | [93] |
 | LYN-1604 | UNC-51-like kinase 1 (ULK1) | [90] |
 | CP-31398 | p53 | [94,95,96] |
 | Fluoxetine | eEF2K | [97] |
 | YM155 | Survivin | [102] |
 | FLN | N-Ras | [106] |
 | Gefitinib (Iressa®, ZD1839) | epidermal growth factor receptor (EGFR) tyrosine kinase | [110] |
 | SAR405 | Vps34 p110α and/or HER2 | [112] |
 | FL-411 | Bromodomain-containing protein 4 | [91] |
 | Sulforaphane | Histone deacetylase (HDAC) | [124,125] |
 | PC3-15 | Ubiquitin enzymes | [127,128] |
 | PC3-16 | ||
 | PC3-17 | ||
 | N-Desmethyldauricine (LP-4) | SERCA | [129] |
 | MCB-613 | steroid receptor coactivators | [132] |
 | Dichloroacetate | enzyme pyruvate dehydrogenase kinase | [137] |
| structure not known | SB02024 | Vps34 | [142] |
 | Lucanthone (Miracil D) | Cathepsin-D | [145] |
 | SBI-0206965 | ULK1 | [146] |
 | SBP-7455 | ULK1/2 and inhibits their enzymatic activity | [147] |
 | Aumitin | mitochondrial respiration complex 1 | [148] |
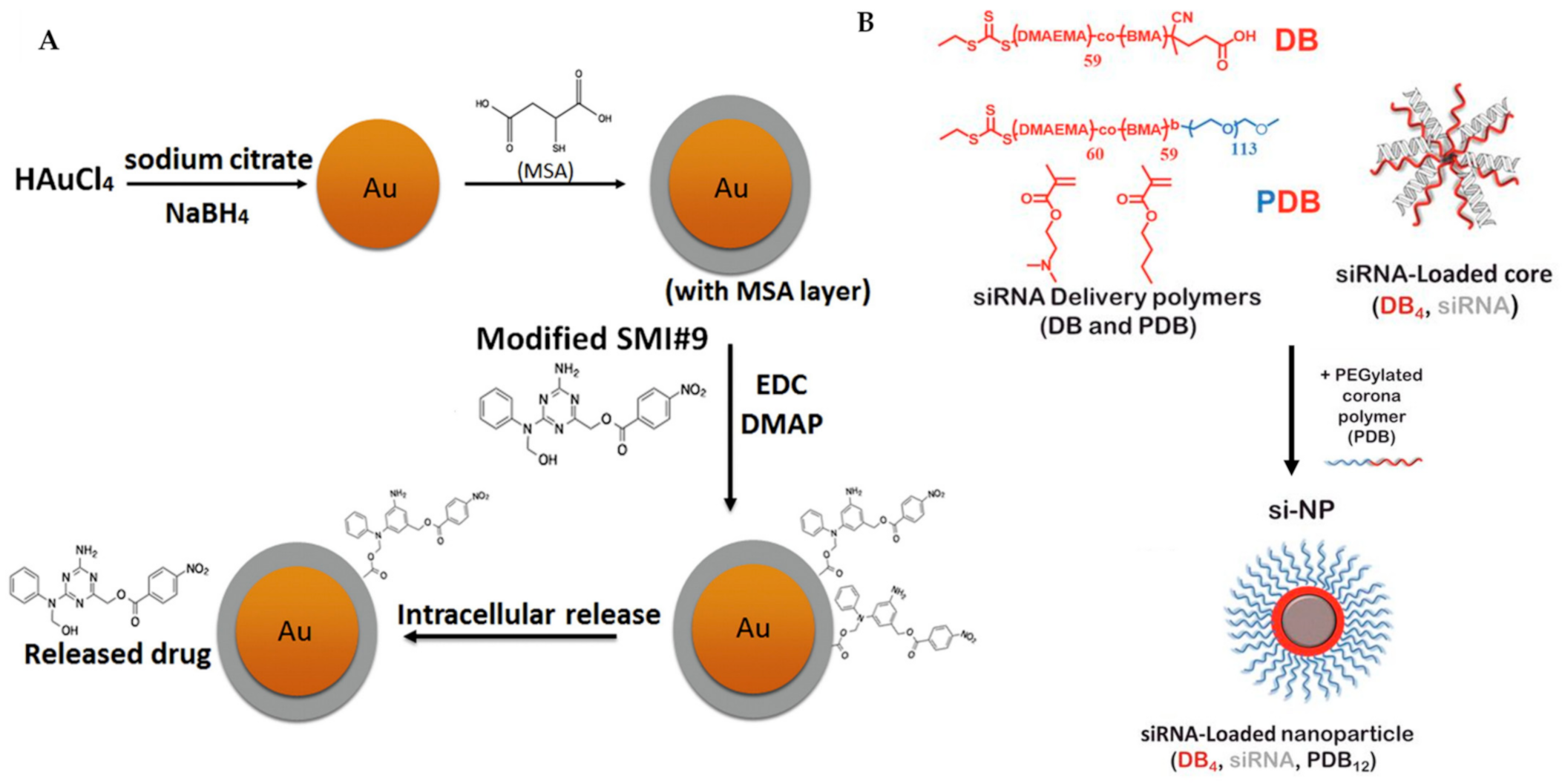
| Nanoparticle | SMI#9-gold nanoparticles | Rad6 | [155] |
| nanoparticle | Composed of siRNA, (poly(DMAEMA-co-BMA), DB4), and PEGylated, (PEG-b-poly(DMAEMA-co-BMA), | kinase mTORC2 | [156] |
| nanoparticle | Poly (β-amino ester) and poly (ethylene glycol) (PEG) | Beclin-1 | [167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maniam, S.; Maniam, S. Small Molecules Targeting Programmed Cell Death in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 9722. https://doi.org/10.3390/ijms22189722
Maniam S, Maniam S. Small Molecules Targeting Programmed Cell Death in Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(18):9722. https://doi.org/10.3390/ijms22189722
Chicago/Turabian StyleManiam, Subashani, and Sandra Maniam. 2021. "Small Molecules Targeting Programmed Cell Death in Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 18: 9722. https://doi.org/10.3390/ijms22189722
APA StyleManiam, S., & Maniam, S. (2021). Small Molecules Targeting Programmed Cell Death in Breast Cancer Cells. International Journal of Molecular Sciences, 22(18), 9722. https://doi.org/10.3390/ijms22189722