
The Impact of Hypoxia in Early Pregnancy on Placental Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
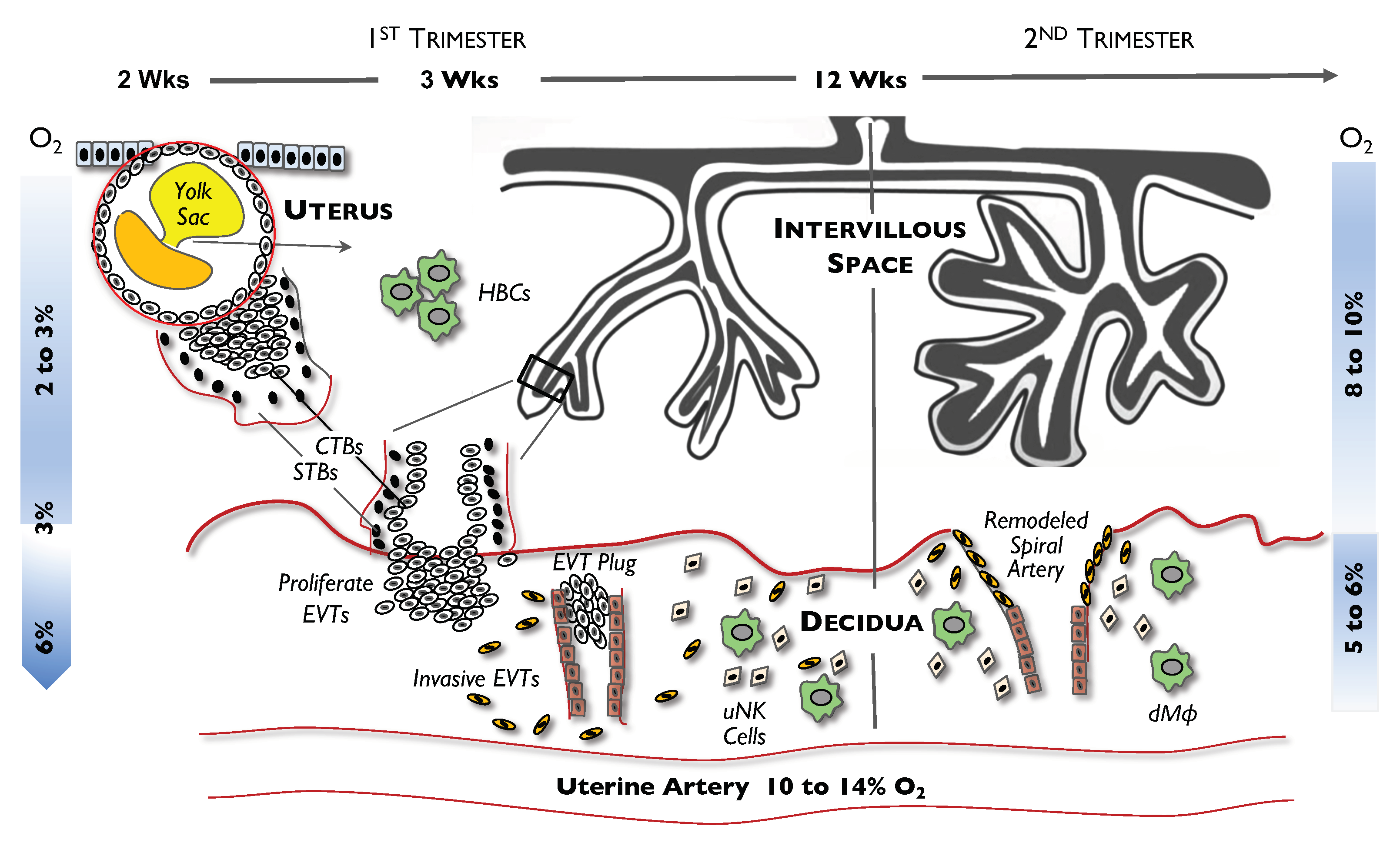
2. Cells in Early Placental Development
3. Hypoxic Conditions in Early Placental Development
4. General Cellular Response Mechanisms to Hypoxia
4.1. Hypoxia-Inducible Factor (HIF)
4.2. mTORs
4.3. Metabolic Changes
4.4. Autophagy
4.5. Epigenetic Alterations and miRNA Function
5. Effect of Hypoxia on Placental Cells
5.1. Hypoxia on CTB Expansion
5.2. Effects of Hypoxia on Trophoblast Differentiation
5.2.1. EVT Differentiation
5.2.2. STB Differentiation
5.3. Hypoxia and uNK Cells
5.4. Effect of Hypoxia on Macrophages
5.4.1. HBCs
5.4.2. dMϕs
6. A Mouse Model Representing a Failure to Response to Hypoxia
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | Adenosine 5′ monophosphate-activated protein kinase |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| Bcl-1 | Beclin-1 |
| BM | Bone marrow |
| CAT | Catalase |
| cNK | Circulating NK |
| CO | Carbon monoxide |
| CTBs | Cytotrophoblasts |
| DC-SIGN | Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin |
| dMϕ | Decidual macrophages |
| EGFR | Epidermal growth factor receptor |
| Elf5 | E74-like factor 5 |
| EPAS | Endothelial PAS domain protein |
| ER | Endoplasmic reticulum |
| ESCs | Embryonic stem cells |
| EVTs | Extravillous trophoblasts |
| Fe2+ | Ferrous iron |
| FGR | Fetal growth restriction |
| GPx | Glutathione peroxidase |
| HBCs | Hofbauer cells |
| HCG | Human chorionic gonadotropin |
| Het | Heterozygous |
| HIF | Hypoxia-inducible factor |
| HLA-G | Human leukocyte antigen-G |
| HO-1 | Heme oxygenase-1 |
| HPL | Human placental lactogen |
| HRES | Hypoxic response elements |
| HSCs | Hematopoietic stem cells |
| IVF | In vitro fertilization |
| KO | Knockout |
| M1 | Classically activated macrophages |
| M2 | Alternatively activated macrophages |
| MDSCs | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| miRNAs | MicroRNAs |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| O2 | Oxygen |
| PaO2 | Partial pressure of arterial O2 (in mm Hg) |
| Plet1 | Placenta expressed transcript 1 |
| RAPTOR | Rapamycin-associated TOR protein |
| RICTOR | Rapamycin-insensitive companion of mTOR |
| ROS | Reactive oxygen species |
| sFlt-1 | Soluble fms-like tyrosine kinase-1 |
| SOD | Superoxide dismutase |
| STBs | Syncytiotrophoblasts |
| sVEGR1 | Soluble VEGR receptor-1 |
| TAMs | Tumor-associated macrophages |
| TLR | Toll-like receptor |
| trNK | Tissue resident NK |
| uNK | Uterine natural killer |
| VEGR | Vascular endothelial growth factor |
| VHL | von Hippel–Lindau protein |
| WT | Wild-type |
References
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Morin, S.J. Oxygen tension in embryo culture: Does a shift to 2% O2 in extended culture represent the most physiologic system? J. Assist. Reprod. Genet. 2017, 34, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Fathollahipour, S.; Patil, P.S.; Leipzig, N.D. Oxygen regulation in development: Lessons from embryogenesis towards tissue engineering. Cells Tissues Organs 2018, 205, 350–371. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Wakeland, A.K.; Parast, M.M. Trophoblast lineage specification, differentiation and their regulation by oxygen tension. J. Endocrinol. 2018, 236, R43–R56. [Google Scholar] [CrossRef]
- Cervar, M.; Blaschitz, A.; Dohr, G.; Desoye, G. Paracrine regulation of distinct trophoblast functions in vitro by placental macrophages. Cell. Tissue Res. 1999, 295, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Seval, Y.; Korgun, E.T.; Demir, R. Hofbauer cells in early human placenta: Possible implications in vasculogenesis and angiogenesis. Placenta 2007, 28, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Ottosen, L.D.; Hindkaer, J.; Husth, M.; Petersen, D.E.; Kirk, J.; Ingerslev, H.J. Observations on intrauterine oxygen tension measured by fibre-optic microsensors. Reprod. Biomed. Online 2006, 13, 380–385. [Google Scholar] [CrossRef]
- Yedwab, G.A.; Paz, G.; Homonnai, T.Z.; David, M.P.; Kraicer, P.F. The temperature, pH, and partial pressure of oxygen in the cervix and uterus of women and uterus of rats during the cycle. Fertil. Steril. 1976, 27, 304–309. [Google Scholar] [CrossRef]
- Burton, G.J.; Cindrova-Davies, T.; Yung, H.W.; Jauniaux, E. Hypoxia and reproductive health: Oxygen and development of the human placenta. Reproduction 2021, 161, F53–F65. [Google Scholar] [CrossRef] [PubMed]
- Rodesch, F.; Simon, P.; Donner, C.; Jauniaux, E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet. Gynecol. 1992, 80, 283–285. [Google Scholar]
- Fischer, B.; Bavister, B.D. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J. Reprod. Fertil. 1993, 99, 673–679. [Google Scholar] [CrossRef]
- Soares, M.J.; Iqbal, K.; Kozai, K. Hypoxia and placental development. Birth Defects Res. 2017, 109, 1309–1329. [Google Scholar] [CrossRef]
- Burton, G.J. Oxygen, the Janus gas; its effects on human placental development and function. J. Anat. 2009, 215, 27–35. [Google Scholar] [CrossRef]
- Van Patot, M.C.; Valdez, M.; Becky, V.; Cindrova-Davies, T.; Johns, J.; Zwerdling, L.; Jauniaux, E.; Burton, G.J. Impact of pregnancy at high altitude on placental morphology in non-native women with and without preeclampsia. Placenta 2009, 30, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.Z.; Burton, G.J.; Morad, N.; Ali, M.E. Does hypercapillarization influence the branching pattern of terminal villi in the human placenta at high altitude? Placenta 1996, 17, 677–682. [Google Scholar] [CrossRef]
- Jackson, M.R.; Mayhew, T.M.; Haas, J.D. On the factors which contribute to thinning of the villous membrane in human placentae at high altitude. II. An increase in the degree of peripheralization of fetal capillaries. Placenta 1988, 9, 9–18. [Google Scholar] [CrossRef]
- Reshetnikova, O.S.; Burton, G.J.; Milovanov, A.P. Effects of hypobaric hypoxia on the fetoplacental unit: The morphometric diffusing capacity of the villous membrane at high altitude. Am. J. Obstet. Gynecol. 1994, 171, 1560–1565. [Google Scholar] [CrossRef]
- Burton, G.J.; Reshetnikova, O.S.; Milovanov, A.P.; Teleshova, O.V. Stereological evaluation of vascular adaptations in human placental villi to differing forms of hypoxic stress. Placenta 1996, 17, 49–55. [Google Scholar] [CrossRef]
- Thiel, M.; Chouker, A.; Ohta, A.; Jackson, E.; Caldwell, C.; Smith, P.; Lukashev, D.; Bittmann, I.; Sitkovsky, M.V. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005, 3, e174. [Google Scholar] [CrossRef]
- Haase, V.H. The VHL tumor suppressor: Master regulator of HIF. Curr. Pharm. Des. 2009, 15, 3895–3903. [Google Scholar] [CrossRef]
- Aplin, J.D. Hypoxia and human placental development. J. Clin. Investig. 2000, 105, 559–560. [Google Scholar] [CrossRef][Green Version]
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J. 2001, 15, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Caniggia, I.; Winter, J.; Lye, S.J.; Post, M. Oxygen and placental development during the first trimester: Implications for the pathophysiology of pre-eclampsia. Placenta 2000, 21, S25–S30. [Google Scholar] [CrossRef]
- Ietta, F.; Wu, Y.; Romagnoli, R.; Soleymanlou, N.; Orsini, B.; Zamudio, S.; Paulesu, L.; Caniggia, I. Oxygen regulation of macrophage migration inhibitory factor in human placenta. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E272–E280. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Watson, A.; Burton, G. Evaluation of respiratory gases and acid-base gradients in human fetal fluids and uteroplacental tissue between 7 and 16 weeks’ gestation. Am. J. Obstet. Gynecol. 2001, 184, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Narasimhan, P.; Kalish, F.; Wong, R.J.; Stevenson, D.K. Dysregulation of hypoxia-inducible factor-1alpha (Hif1alpha) expression in the Hmox1-deficient placenta. Placenta 2020, 99, 108–116. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- Kozak, K.R.; Abbott, B.; Hankinson, O. ARNT-deficient mice and placental differentiation. Dev. Biol. 1997, 191, 297–305. [Google Scholar] [CrossRef]
- Maltepe, E.; Schmidt, J.V.; Baunoch, D.; Bradfield, C.A.; Simon, M.C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature 1997, 386, 403–407. [Google Scholar] [CrossRef]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Highet, A.R.; Khoda, S.M.; Buckberry, S.; Leemaqz, S.; Bianco-Miotto, T.; Harrington, E.; Ricciardelli, C.; Roberts, C.T. Hypoxia induced HIF-1/HIF-2 activity alters trophoblast transcriptional regulation and promotes invasion. Eur. J. Cell. Biol. 2015, 94, 589–602. [Google Scholar] [CrossRef]
- Colson, A.; Depoix, C.L.; Baldin, P.; Hubinont, C.; Sonveaux, P.; Debieve, F. Hypoxia-inducible factor 2 alpha impairs human cytotrophoblast syncytialization: New insights into placental dysfunction and fetal growth restriction. FASEB J. 2020, 34, 15222–15235. [Google Scholar] [CrossRef]
- Beevers, C.S.; Li, F.; Liu, L.; Huang, S. Curcumin inhibits the mammalian target of rapamycin-mediated signaling pathways in cancer cells. Int. J. Cancer 2006, 119, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Lamming, D.W. The mechanistic target of rapamycin: The grand conductor of metabolism and aging. Cell. Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Gangloff, Y.G.; Mueller, M.; Dann, S.G.; Svoboda, P.; Sticker, M.; Spetz, J.F.; Um, S.H.; Brown, E.J.; Cereghini, S.; Thomas, G.; et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell. Biol. 2004, 24, 9508–9516. [Google Scholar] [CrossRef]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell. 2006, 11, 859–871. [Google Scholar] [CrossRef]
- Tuuli, M.G.; Longtine, M.S.; Nelson, D.M. Review: Oxygen and trophoblast biology—A source of controversy. Placenta 2011, 32 (Suppl. S2), S109–S118. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E.; Murray, A.J. Oxygen and placental development; parallels and differences with tumour biology. Placenta 2017, 56, 14–18. [Google Scholar] [CrossRef]
- Jauniaux, E.; Hempstock, J.; Greenwold, N.; Burton, G.J. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am. J. Pathol. 2003, 162, 115–125. [Google Scholar] [CrossRef]
- Leese, H.J. Quiet please, do not disturb: A hypothesis of embryo metabolism and viability. Bioessays 2002, 24, 845–849. [Google Scholar] [CrossRef]
- Sturmey, R.G.; Hawkhead, J.A.; Barker, E.A.; Leese, H.J. DNA damage and metabolic activity in the preimplantation embryo. Hum. Reprod. 2009, 24, 81–91. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef]
- Cieslar-Pobuda, A.; Yue, J.; Lee, H.C.; Skonieczna, M.; Wei, Y.H. ROS and oxidative stress in stem cells. Oxid. Med. Cell. Longev. 2017, 2017, 5047168. [Google Scholar] [CrossRef]
- Lewandowski, D.; Barroca, V.; Duconge, F.; Bayer, J.; Van Nhieu, J.T.; Pestourie, C.; Fouchet, P.; Tavitian, B.; Romeo, P.H. In vivo cellular imaging pinpoints the role of reactive oxygen species in the early steps of adult hematopoietic reconstitution. Blood 2010, 115, 443–452. [Google Scholar] [CrossRef]
- Spaans, F.; de Vos, P.; Bakker, W.W.; van Goor, H.; Faas, M.M. Danger signals from ATP and adenosine in pregnancy and preeclampsia. Hypertension 2014, 63, 1154–1160. [Google Scholar] [CrossRef]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef]
- Cui, J.; Mao, X.; Olman, V.; Hastings, P.J.; Xu, Y. Hypoxia and miscoupling between reduced energy efficiency and signaling to cell proliferation drive cancer to grow increasingly faster. J. Mol. Cell. Biol. 2012, 4, 174–176. [Google Scholar] [CrossRef]
- Louie, M.C.; Ton, J.; Brady, M.L.; Le, D.T.; Mar, J.N.; Lerner, C.A.; Gerencser, A.A.; Mookerjee, S.A. Total cellular ATP production changes with primary substrate in MCF7 breast cancer cells. Front. Oncol. 2020, 10, 1703. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Liu, F.; Li, S. Metabolic adaptations in pregnancy: A review. Ann. Nutr. Metab. 2017, 70, 59–65. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Oh, S.Y.; Roh, C.R. Autophagy in the placenta. Obstet. Gynecol. Sci. 2017, 60, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, L.; Terraneo, L.; Virgili, E.; Martinelli, C.; Doi, P.; Samaja, M.; Bulfamante, G.P.; Marconi, A.M. Autophagy in normal and abnormal early human pregnancies. Reprod. Sci. 2015, 22, 838–844. [Google Scholar] [CrossRef]
- Murray, A.; Sienerth, A.R.; Hemberger, M. Plet1 is an epigenetically regulated cell surface protein that provides essential cues to direct trophoblast stem cell differentiation. Sci. Rep. 2016, 6, 25112. [Google Scholar] [CrossRef] [PubMed]
- Logan, P.C.; Mitchell, M.D.; Lobie, P.E. DNA methyltransferases and TETs in the regulation of differentiation and invasiveness of extra-villous trophoblasts. Front. Genet. 2013, 4, 265. [Google Scholar] [CrossRef]
- Gamage, T.; Schierding, W.; Hurley, D.; Tsai, P.; Ludgate, J.L.; Bhoothpur, C.; Chamley, L.W.; Weeks, R.J.; Macaulay, E.C.; James, J.L. The role of DNA methylation in human trophoblast differentiation. Epigenetics 2018, 13, 1154–1173. [Google Scholar] [CrossRef]
- Oda, M.; Oxley, D.; Dean, W.; Reik, W. Regulation of lineage specific DNA hypomethylation in mouse trophectoderm. PLoS ONE 2013, 8, e68846. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Mayne, B.T.; Buckberry, S.; Breen, J.; Rodriguez Lopez, C.M.; Roberts, C.T. Recent progress towards understanding the role of DNA methylation in human placental development. Reproduction 2016, 152, R23–R30. [Google Scholar] [CrossRef]
- Yuen, R.K.; Chen, B.; Blair, J.D.; Robinson, W.P.; Nelson, D.M. Hypoxia alters the epigenetic profile in cultured human placental trophoblasts. Epigenetics 2013, 8, 192–202. [Google Scholar] [CrossRef]
- Bao, L.; Chen, Y.; Lai, H.T.; Wu, S.Y.; Wang, J.E.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.M.; et al. Methylation of hypoxia-inducible factor (HIF)-1alpha by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef]
- Chelbi, S.T.; Mondon, F.; Jammes, H.; Buffat, C.; Mignot, T.M.; Tost, J.; Busato, F.; Gut, I.; Rebourcet, R.; Laissue, P.; et al. Expressional and epigenetic alterations of placental serine protease inhibitors: SERPINA3 is a potential marker of preeclampsia. Hypertension 2007, 49, 76–83. [Google Scholar] [CrossRef]
- Branco, M.R.; King, M.; Perez-Garcia, V.; Bogutz, A.B.; Caley, M.; Fineberg, E.; Lefebvre, L.; Cook, S.J.; Dean, W.; Hemberger, M.; et al. Maternal DNA Methylation regulates early trophoblast development. Dev. Cell 2016, 36, 152–163. [Google Scholar] [CrossRef]
- Hayder, H.; O’Brien, J.; Nadeem, U.; Peng, C. MicroRNAs: Crucial regulators of placental development. Reproduction 2018, 155, R259–R271. [Google Scholar] [CrossRef] [PubMed]
- Macharia, L.W.; Wanjiru, C.M.; Mureithi, M.W.; Pereira, C.M.; Ferrer, V.P.; Moura-Neto, V. MicroRNAs, hypoxia and the stem-like state as contributors to cancer aggressiveness. Front. Genet. 2019, 10, 125. [Google Scholar] [CrossRef]
- Shahbazi, A.; Safa, M.; Alikarami, F.; Kargozar, S.; Asadi, M.H.; Joghataei, M.T.; Soleimani, M. Rapid Induction of neural differentiation in human umbilical cord matrix mesenchymal stem cells by camp-elevating agents. Int. J. Mol. Cell. Med. 2016, 5, 167–177. [Google Scholar]
- Yang, Y.; Arenas-Hernandez, M.; Gomez-Lopez, N.; Dai, J.; Parker, G.C.; Puscheck, E.E.; Rappolee, D.A. Hypoxic stress forces irreversible differentiation of a majority of mouse trophoblast stem cells despite FGF4. Biol. Reprod. 2016, 95, 110. [Google Scholar] [CrossRef]
- Red-Horse, K.; Zhou, Y.; Genbacev, O.; Prakobphol, A.; Foulk, R.; McMaster, M.; Fisher, S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J. Clin. Investig. 2004, 114, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Kenchegowda, D.; Natale, B.; Lemus, M.A.; Natale, D.R.; Fisher, S.A. Inactivation of maternal Hif-1alpha at mid-pregnancy causes placental defects and deficits in oxygen delivery to the fetal organs under hypoxic stress. Dev. Biol. 2017, 422, 171–185. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Hempstock, J.; Jauniaux, E.; Greenwold, N.; Burton, G.J. The contribution of placental oxidative stress to early pregnancy failure. Hum. Pathol. 2003, 34, 1265–1275. [Google Scholar] [CrossRef]
- Rosario, G.X.; Konno, T.; Soares, M.J. Maternal hypoxia activates endovascular trophoblast cell invasion. Dev. Biol. 2008, 314, 362–375. [Google Scholar] [CrossRef]
- Zhou, Y.; Chiu, K.; Brescia, R.J.; Combs, C.A.; Katz, M.A.; Kitzmiller, J.L.; Heilbron, D.C.; Fisher, S.J. Increased depth of trophoblast invasion after chronic constriction of the lower aorta in rhesus monkeys. Am. J. Obstet. Gynecol. 1993, 169, 224–229. [Google Scholar] [CrossRef]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef]
- Myatt, L.; Cui, X. Oxidative stress in the placenta. Histochem. Cell. Biol. 2004, 122, 369–382. [Google Scholar] [CrossRef]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef]
- Wakeland, A.K.; Soncin, F.; Moretto-Zita, M.; Chang, C.W.; Horii, M.; Pizzo, D.; Nelson, K.K.; Laurent, L.C.; Parast, M.M. Hypoxia directs human extravillous trophoblast differentiation in a hypoxia-inducible factor-dependent manner. Am. J. Pathol. 2017, 187, 767–780. [Google Scholar] [CrossRef]
- Adelman, D.M.; Gertsenstein, M.; Nagy, A.; Simon, M.C.; Maltepe, E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000, 14, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Cowden Dahl, K.D.; Fryer, B.H.; Mack, F.A.; Compernolle, V.; Maltepe, E.; Adelman, D.M.; Carmeliet, P.; Simon, M.C. Hypoxia-inducible factors 1alpha and 2alpha regulate trophoblast differentiation. Mol. Cell. Biol. 2005, 25, 10479–10491. [Google Scholar] [CrossRef]
- Maltepe, E.; Krampitz, G.W.; Okazaki, K.M.; Red-Horse, K.; Mak, W.; Simon, M.C.; Fisher, S.J. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development 2005, 132, 3393–3403. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Wang, Y.L.; Fan, H.C.; Lo, W.T.; Wang, C.C.; Sytwu, H.K. Current status of the immunomodulation and immunomediated therapeutic strategies for multiple sclerosis. Clin. Dev. Immunol. 2012, 2012, 970789. [Google Scholar] [CrossRef]
- PrabhuDas, M.; Bonney, E.; Caron, K.; Dey, S.; Erlebacher, A.; Fazleabas, A.; Fisher, S.; Golos, T.; Matzuk, M.; McCune, J.M.; et al. Immune mechanisms at the maternal-fetal interface: Perspectives and challenges. Nat. Immunol. 2015, 16, 328–334. [Google Scholar] [CrossRef]
- Alsat, E.; Wyplosz, P.; Malassine, A.; Guibourdenche, J.; Porquet, D.; Nessmann, C.; Evain-Brion, D. Hypoxia impairs cell fusion and differentiation process in human cytotrophoblast, in vitro. J. Cell. Physiol. 1996, 168, 346–353. [Google Scholar] [CrossRef]
- Horii, M.; Li, Y.; Wakeland, A.K.; Pizzo, D.P.; Nelson, K.K.; Sabatini, K.; Laurent, L.C.; Liu, Y.; Parast, M.M. Human pluripotent stem cells as a model of trophoblast differentiation in both normal development and disease. Proc. Natl. Acad. Sci. USA 2016, 113, E3882–E3891. [Google Scholar] [CrossRef]
- Albers, R.E.; Kaufman, M.R.; Natale, B.V.; Keoni, C.; Kulkarni-Datar, K.; Min, S.; Williams, C.R.; Natale, D.R.C.; Brown, T.L. Trophoblast-specific expression of hif-1alpha results in preeclampsia-like symptoms and fetal growth restriction. Sci. Rep. 2019, 9, 2742. [Google Scholar] [CrossRef]
- Sojka, D.K.; Yang, L.; Yokoyama, W.M. Uterine natural killer cells. Front. Immunol. 2019, 10, 960. [Google Scholar] [CrossRef]
- Croy, B.A.; van den Heuvel, M.J.; Borzychowski, A.M.; Tayade, C. Uterine natural killer cells: A specialized differentiation regulated by ovarian hormones. Immunol. Rev. 2006, 214, 161–185. [Google Scholar] [CrossRef]
- Chakraborty, D.; Rumi, M.A.; Soares, M.J. NK cells, hypoxia and trophoblast cell differentiation. Cell Cycle 2012, 11, 2427–2430. [Google Scholar] [CrossRef]
- Strunz, B.; Bister, J.; Jonsson, H.; Filipovic, I.; Crona-Guterstam, Y.; Kvedaraite, E.; Sleiers, N.; Dumitrescu, B.; Brannstrom, M.; Lentini, A.; et al. Continuous human uterine NK cell differentiation in response to endometrial regeneration and pregnancy. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef]
- Ohta, A.; Diwanji, R.; Kini, R.; Subramanian, M.; Ohta, A.; Sitkovsky, M. In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Front. Immunol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Sceneay, J.; Chow, M.T.; Chen, A.; Halse, H.M.; Wong, C.S.; Andrews, D.M.; Sloan, E.K.; Parker, B.S.; Bowtell, D.D.; Smyth, M.J.; et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Chambers, A.M.; Matosevic, S. Immunometabolic dysfunction of natural killer cells mediated by the hypoxia-CD73 axis in solid tumors. Front. Mol. Biosci. 2019, 6, 60. [Google Scholar] [CrossRef]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1597. [Google Scholar] [CrossRef]
- Chakraborty, D.; Rumi, M.A.; Konno, T.; Soares, M.J. Natural killer cells direct hemochorial placentation by regulating hypoxia-inducible factor dependent trophoblast lineage decisions. Proc. Natl. Acad. Sci. USA 2011, 108, 16295–16300. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.R.; Appios, A.; Zhao, X.; Dutkiewicz, R.; Donde, M.; Lee, C.Y.C.; Naidu, P.; Lee, C.; Cerveira, J.; Liu, B.; et al. Phenotypic and functional characterization of first-trimester human placental macrophages, Hofbauer cells. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.D.; Hamilton, W.J. The Human Placenta; Heffer: Cambrdige, MA, USA, 1970. [Google Scholar]
- Castellucci, M.; Celona, A.; Bartels, H.; Steininger, B.; Benedetto, V.; Kaufmann, P. Mitosis of the Hofbauer cell: Possible implications for a fetal macrophage. Placenta 1987, 8, 65–76. [Google Scholar] [CrossRef]
- Ingman, K.; Cookson, V.J.; Jones, C.J.; Aplin, J.D. Characterisation of Hofbauer cells in first and second trimester placenta: Incidence, phenotype, survival in vitro and motility. Placenta 2010, 31, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Van Handel, B.; Prashad, S.L.; Hassanzadeh-Kiabi, N.; Huang, A.; Magnusson, M.; Atanassova, B.; Chen, A.; Hamalainen, E.I.; Mikkola, H.K. The first trimester human placenta is a site for terminal maturation of primitive erythroid cells. Blood 2010, 116, 3321–3330. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, C.; Schuchert, R.; Wagner, F.; Thaler, R.; Weinberger, T.; Pick, R.; Mass, E.; Ishikawa-Ankerhold, H.C.; Margraf, A.; Hutter, S.; et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat. Commun. 2018, 9, 75. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Ontogeny of tissue-resident macrophages. Front. Immunol. 2015, 6, 486. [Google Scholar] [CrossRef]
- Cipolleschi, M.G.; D'Ippolito, G.; Bernabei, P.A.; Caporale, R.; Nannini, R.; Mariani, M.; Fabbiani, M.; Rossi-Ferrini, P.; Olivotto, M.; Dello Sbarba, P. Severe hypoxia enhances the formation of erythroid bursts from human cord blood cells and the maintenance of BFU-E in vitro. Exp. Hematol. 1997, 25, 1187–1194. [Google Scholar] [PubMed]
- Adelman, D.M.; Maltepe, E.; Simon, M.C. Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev. 1999, 13, 2478–2483. [Google Scholar] [CrossRef]
- Reyes, L.; Golos, T.G. Hofbauer cells: Their role in healthy and complicated pregnancy. Front. Immunol. 2018, 9, 2628. [Google Scholar] [CrossRef]
- Ning, F.; Liu, H.; Lash, G.E. The role of decidual macrophages during normal and pathological pregnancy. Am. J. Reprod. Immunol. 2016, 75, 298–309. [Google Scholar] [CrossRef]
- Jena, M.K.; Nayak, N.; Chen, K.; Nayak, N.R. Role of macrophages in pregnancy and related complications. Arch. Immunol. Ther. Exp. 2019, 67, 295–309. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.S. Tumor interstitial fluid pressure-a link between tumor hypoxia, microvascular density, and lymph node metastasis. Neoplasia 2014, 16, 586–594. [Google Scholar] [CrossRef]
- Lewis, D.M.; Park, K.M.; Tang, V.; Xu, Y.; Pak, K.; Eisinger-Mathason, T.S.; Simon, M.C.; Gerecht, S. Intratumoral oxygen gradients mediate sarcoma cell invasion. Proc. Natl. Acad. Sci. USA 2016, 113, 9292–9297. [Google Scholar] [CrossRef]
- Campillo, N.; Falcones, B.; Otero, J.; Colina, R.; Gozal, D.; Navajas, D.; Farre, R.; Almendros, I. Differential oxygenation in tumor microenvironment modulates macrophage and cancer cell crosstalk: Novel experimental setting and proof of concept. Front. Oncol. 2019, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Konrad, F.M.; Zwergel, C.; Ngamsri, K.C.; Reutershan, J. Anti-inflammatory Effects of Heme Oxygenase-1 Depend on Adenosine A2A- and A2B-Receptor Signaling in Acute Pulmonary Inflammation. Front. Immunol. 2017, 8, 1874. [Google Scholar] [CrossRef]
- Min, K.J.; Kim, J.H.; Jou, I.; Joe, E.H. Adenosine induces hemeoxygenase-1 expression in microglia through the activation of phosphatidylinositol 3-kinase and nuclear factor E2-related factor 2. Glia 2008, 56, 1028–1037. [Google Scholar] [CrossRef]
- Cao, Y.A.; Wagers, A.J.; Karsunky, H.; Zhao, H.; Reeves, R.; Wong, R.J.; Stevenson, D.K.; Weissman, I.L.; Contag, C.H. Heme oxygenase-1 deficiency leads to disrupted response to acute stress in stem cells and progenitors. Blood 2008, 112, 4494–4502. [Google Scholar] [CrossRef]
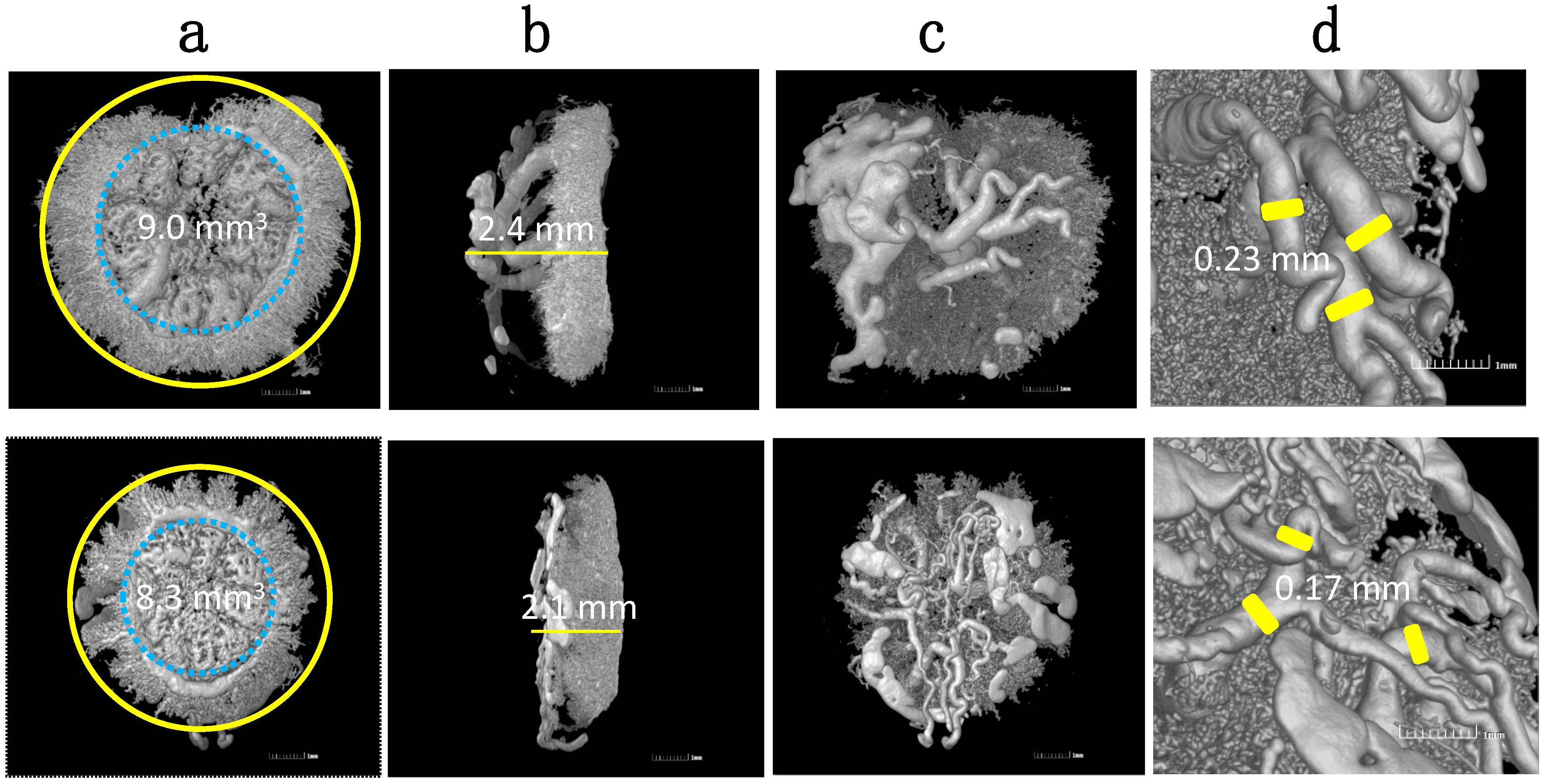
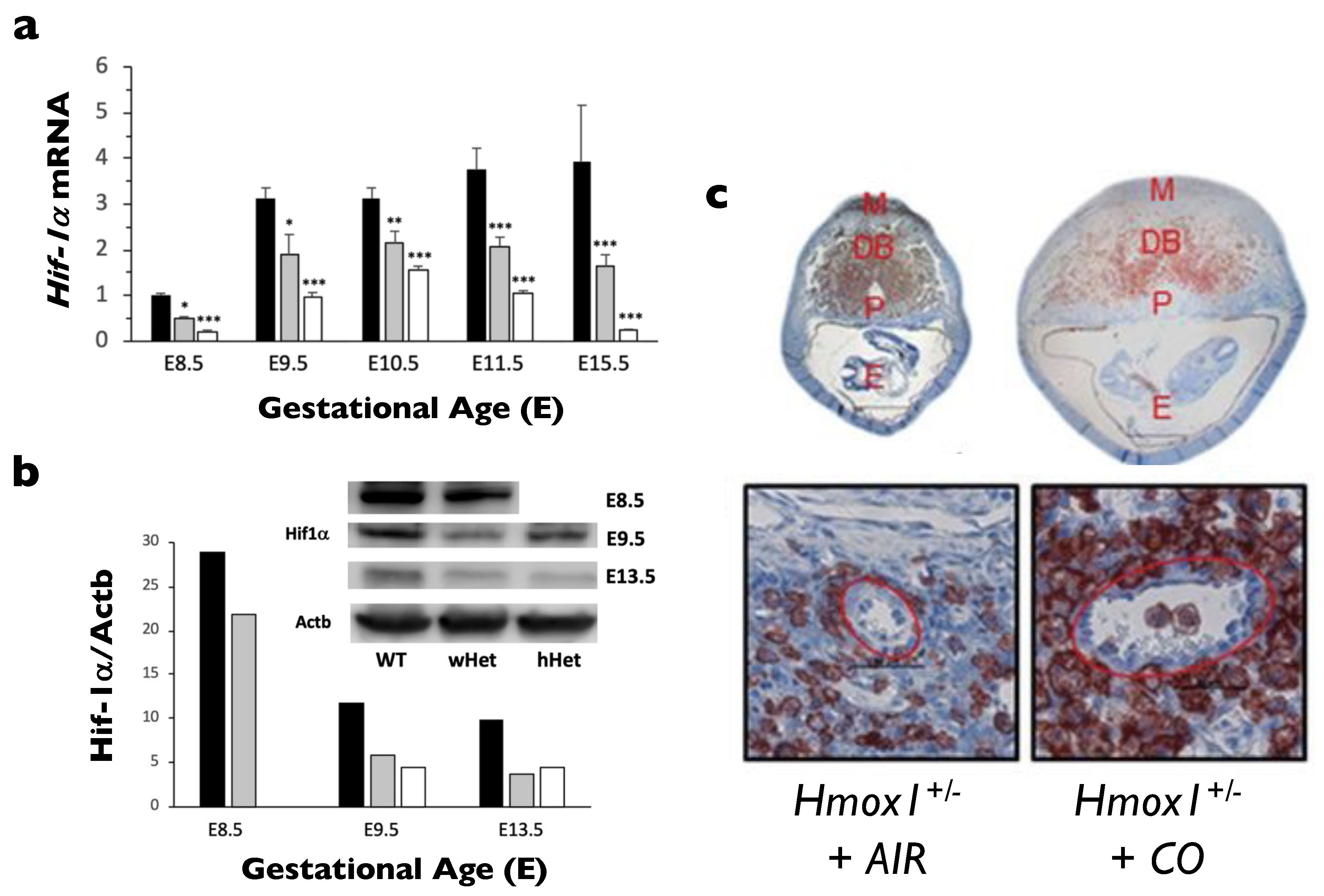
- Zhao, H.; Wong, R.J.; Kalish, F.S.; Nayak, N.R.; Stevenson, D.K. Effect of heme oxygenase-1 deficiency on placental development. Placenta 2009, 30, 861–868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wong, R.J.; Zhao, H.; Stevenson, D.K. A deficiency in haem oxygenase-1 induces foetal growth restriction by placental vasculature defects. Acta Paediatr. 2012, 101, 827–834. [Google Scholar] [CrossRef]
- Zhao, H.; Azuma, J.; Kalish, F.; Wong, R.J.; Stevenson, D.K. Maternal heme oxygenase 1 regulates placental vasculature development via angiogenic factors in mice. Biol. Reprod. 2011, 85, 1005–1012. [Google Scholar] [CrossRef]
- Linzke, N.; Schumacher, A.; Woidacki, K.; Croy, B.A.; Zenclussen, A.C. Carbon monoxide promotes proliferation of uterine natural killer cells and remodeling of spiral arteries in pregnant hypertensive heme oxygenase-1 mutant mice. Hypertension 2014, 63, 580–588. [Google Scholar] [CrossRef]
- Zhao, H.; Kalish, F.; Wong, R.J.; Stevenson, D.K. Infiltration of myeloid cells in the pregnant uterus is affected by heme oxygenase-1. J. Leukoc. Biol. 2017, 101, 217–226. [Google Scholar] [CrossRef]
- Lindqvist, P.G.; Marsal, K. Moderate smoking during pregnancy is associated with a reduced risk of preeclampsia. Acta Obstet. Gynecol. Scand. 1999, 78, 693–697. [Google Scholar]
- Wikstrom, A.K.; Stephansson, O.; Cnattingius, S. Tobacco use during pregnancy and preeclampsia risk: Effects of cigarette smoking and snuff. Hypertension 2010, 55, 1254–1259. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Wong, R.J.; Stevenson, D.K. The Impact of Hypoxia in Early Pregnancy on Placental Cells. Int. J. Mol. Sci. 2021, 22, 9675. https://doi.org/10.3390/ijms22189675
Zhao H, Wong RJ, Stevenson DK. The Impact of Hypoxia in Early Pregnancy on Placental Cells. International Journal of Molecular Sciences. 2021; 22(18):9675. https://doi.org/10.3390/ijms22189675
Chicago/Turabian StyleZhao, Hui, Ronald J. Wong, and David K. Stevenson. 2021. "The Impact of Hypoxia in Early Pregnancy on Placental Cells" International Journal of Molecular Sciences 22, no. 18: 9675. https://doi.org/10.3390/ijms22189675
APA StyleZhao, H., Wong, R. J., & Stevenson, D. K. (2021). The Impact of Hypoxia in Early Pregnancy on Placental Cells. International Journal of Molecular Sciences, 22(18), 9675. https://doi.org/10.3390/ijms22189675