
Molecular Interactions of Arterial Hypertension in Its Target Organs



Abstract
1. Introduction
2. Matrix Metalloproteinases and Hypertension-Mediated Organ Damage
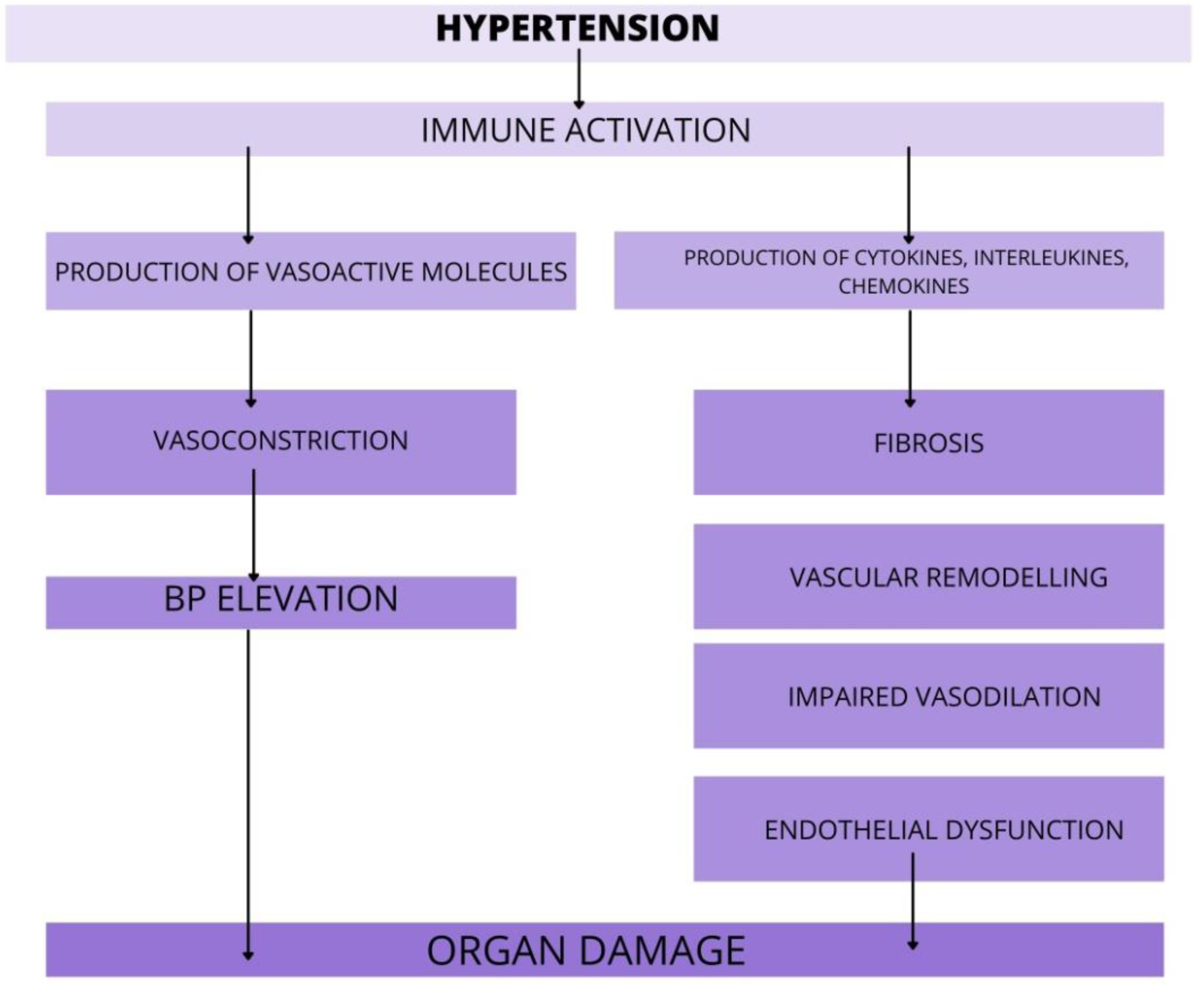
3. Role of Immune System in Hypertension and Target Organ Damage
4. Role of Renin-Angiotensin-Aldosterone-System (RAAS) in Hypertension and Target Organ Damage
5. Role of Oxidative Stress in Hypertension and Hypertensive Organ Damage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of ROS | Chemical Formula |
|---|---|
| Superoxide | O2▪− |
| Hydrogen peroxide | H2O2 |
| Hydroxyl radical | OH |
| Singlet oxygen | 1O2 |
| Peroxyl radical | LOO |
| Alkoxyl radical | LO |
| Lipid hydroperoxide | LOOH |
| Peroxynitrite | ONOO− |
| Hypochlorous acid | HOCl |
| Ozone | O3 |
6. Role of Endoplasmic Reticulum Stress in Hypertension and Its Target Organ Damage
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wermelt, J.A.; Schunkert, H. Management of arterial hypertension. Herz 2017, 42, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Pardell, H.; Armario, P.; Hernández, R. Pathogenesis and epidemiology of arterial hypertension. Drugs 1998, 56 (Suppl. 2), 1–10. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Kardiol. Pol. 2019, 77, 71–159. [Google Scholar] [CrossRef]
- Jordan, J.; Kurschat, C.; Reuter, H. Arterial Hypertension. Dtsch. Arztebl. Int. 2018, 115, 557–558. [Google Scholar] [CrossRef] [PubMed]
- Flack, J.M.; Adekola, B. Blood pressure and the new ACC/AHA hypertension guidelines. Trends Cardiovasc. Med. 2020, 30, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Durango, N.; Fuentes, C.A.; Castillo, A.E.; González-Gómez, L.M.; Vecchiola, A.; Fardella, C.E.; Kalergis, A.M. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: Molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int. J. Mol. Sci. 2016, 17, 797. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, S.F.; Scherrer, U.; Messerli, F.H. Secondary arterial hypertension: When, who, and how to screen? Eur. Heart J. 2014, 35, 1245–1254. [Google Scholar] [CrossRef]
- Forman, J.P.; Stampfer, M.J.; Curhan, G.C. Diet and lifestyle risk factors associated with incident hypertension in women. J. Am. Med. Assoc. 2009, 302, 401–411. [Google Scholar] [CrossRef]
- Virdis, A.; Giannarelli, C.; Neves, M.F.; Taddei, S.; Ghiadoni, L. Cigarette Smoking and Hypertension. Curr. Pharm. Des. 2010, 16, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Vilaplana, J.M. Blood pressure measurement. J. Ren. Care 2006, 32, 210–213. [Google Scholar] [CrossRef]
- Manrique, C.; Lastra, G.; Gardner, M.; Sowers, J.R. The Renin Angiotensin Aldosterone System in Hypertension: Roles of Insulin Resistance and Oxidative Stress. Med. Clin. N. Am. 2009, 93, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Mensah, G.A.; Croft, J.B.; Giles, W.H. The heart, kidney, and brain as target organs in hypertension. Cardiol. Clin. 2002, 20, 225–247. [Google Scholar] [CrossRef]
- Mensah, G.A. Hypertension and target organ damage: Don’t believe everything you think. Ethn. Dis. 2016, 26, 275–278. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [PubMed]
- Bisogni, V.; Cerasari, A.; Pucci, G.; Vaudo, G. Matrix metalloproteinases and hypertension-mediated organ damage: Current insights. Integr. Blood Press. Control 2020, 13, 157–169. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef]
- Chang, C.; Werb, Z. The many faces of metalloproteases: Cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 2001, 11, S37–S43. [Google Scholar] [CrossRef]
- DeLano, F.A.; Schmid-Schönbein, G.W. Proteinase activity and receptor cleavage: Mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension 2008, 52, 415–423. [Google Scholar] [CrossRef]
- Román-García, P.; Coto, E.; Reguero, J.R.; Cannata-Andía, J.B.; Lozano, I.; Avanzas, P.; Morís, C.; Rodríguez, I. Matrix metalloproteinase 1 promoter polymorphisms and risk of myocardial infarction: A case-control study in a Spanish population. Coron. Artery Dis. 2009, 20, 383–386. [Google Scholar] [CrossRef]
- Renkiewicz, R.; Qiu, L.; Lesch, C.; Sun, X.; Devalaraja, R.; Cody, T.; Kaldjian, E.; Welgus, H.; Baragi, V. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003, 48, 1742–1749. [Google Scholar] [CrossRef]
- Hu, J.; den Steen, P.E.V.; Sang, Q.X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. Emerging Role of the Inflammasome and Pyroptosis in Hypertension. Int. J. Mol. Sci. 2021, 22, 1064. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Crowley, S.D. Inflammation in Salt-Sensitive Hypertension and Renal Damage. Curr. Hypertens. Rep. 2018, 20, 103. [Google Scholar] [CrossRef]
- Wen, Y.; Crowley, S.D. Renal effects of cytokines in hypertension. Curr. Opin. Nephrol. Hypertens. 2018, 27, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-N.; Li, C.; Liu, Y.; Du, L.-J.; Zeng, M.-R.; Zheng, X.-J.; Zhang, W.-C.; Liu, Y.; Zhu, M.; Kong, D.; et al. T-Cell Mineralocorticoid Receptor Controls Blood Pressure by Regulating Interferon-Gamma. Circ. Res. 2017, 120, 1584–1597. [Google Scholar] [CrossRef]
- Rai, A.; Narisawa, M.; Li, P.; Piao, L.; Li, Y.; Yang, G.; Cheng, X.W. Adaptive immune disorders in hypertension and heart failure: Focusing on T-cell subset activation and clinical implications. J. Hypertens. 2020, 38, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.K.; Fehrenbach, D.J.; Madhur, M.S. Interleukin 17A: Key Player in the Pathogenesis of Hypertension and a Potential Therapeutic Target. Curr. Hypertens. Rep. 2021, 23, 13. [Google Scholar] [CrossRef]
- Amador, C.A.; Barrientos, V.; Peña, J.; Herrada, A.A.; González, M.; Valdés, S.; Carrasco, L.; Alzamora, R.; Figueroa, F.; Kalergis, A.M.; et al. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension 2014, 63, 797–803. [Google Scholar] [CrossRef]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, Immunity, and Hypertensive End-Organ Damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef]
- Guo, Y.; Lu, Y.; Lu, X.; He, S.; Li, S.; Shao, S.; Zhou, H.; Wang, R.; Li, X.; Gao, P. Krüppel-Like Factor 15/Interleukin 11 Axis-Mediated Adventitial Remodeling Depends on Extracellular Signal-Regulated Kinases 1 and 2 Activation in Angiotensin II–Induced Hypertension. J. Am. Heart Assoc. 2021, 10, e020554. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, S. Brain perivascular macrophages: Connecting inflammation to autonomic activity in hypertension. Hypertens. Res. 2020, 43, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Iyonaga, T.; Shinohara, K.; Mastuura, T.; Hirooka, Y.; Tsutsui, H. Brain perivascular macrophages contribute to the development of hypertension in stroke-prone spontaneously hypertensive rats via sympathetic activation. Hypertens. Res. 2020, 43, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Rudemiller, N.P.; Crowley, S.D. The role of chemokines in hypertension and consequent target organ damage. Pharmacol. Res. 2017, 119, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Brundel, B.J. Inflammasomes and Proteostasis Novel Molecular Mechanisms Associated with Atrial Fibrillation. Circ. Res. 2020, 127, 73–90. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef]
- Sun, H.J.; Ren, X.S.; Xiong, X.Q.; Chen, Y.Z.; Zhao, M.X.; Wang, J.J.; Zhou, Y.B.; Han, Y.; Chen, Q.; Li, Y.H.; et al. Nlrp3 inflammasome activation contributes to vsmc phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017, 8, e3074. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Lang, Y.; Shao, J.; Deng, Y.; Refuhati, R.; Cui, L. The Role of NLRP3 Inflammasome in Cerebrovascular Diseases Pathology and Possible Therapeutic Targets. ASN Neuro 2021, 13. [Google Scholar] [CrossRef]
- Müller, D.N.; Luft, F.C. Direct renin inhibition with aliskiren in hypertension and target organ damage. Clin. J. Am. Soc. Nephrol. 2006, 1, 221–228. [Google Scholar] [CrossRef]
- Luca, M.R.D.; Sorriento, D.; Massa, D.; Valente, V.; Luise, F.D.; Barbato, E.; Morisco, C. Effects of inhibition of the renin-angiotensin system on hypertension-induced target organ damage: Clinical and experimental evidence. Monaldi Arch. Chest Dis. 2021, 91. [Google Scholar] [CrossRef]
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, H.; Wang, J.; Venugopal, J.; Kleiman, K.; Guo, C.; Sun, Y.; Eitzman, D.T. Angiotensin II-induced Hypertension is Reduced by Deficiency of P-selectin Glycoprotein Ligand-1. Sci. Rep. 2018, 8, 3223. [Google Scholar] [CrossRef] [PubMed]
- Piqueras, L.; Sanz, M.J. Angiotensin II and leukocyte trafficking: New insights for an old vascular mediator. Role of redox-signaling pathways. Free Radic. Biol. Med. 2020, 157, 38–54. [Google Scholar] [CrossRef]
- Navar, L.G.; Harrison-Bernard, L.M.; Nishiyama, A.; Kobori, H. Regulation of intrarenal angiotensin II in hypertension. Hypertension 2002, 39, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Young, C.N.; Davisson, R.L. Angiotensin-II, the brain, and hypertension: An update. Hypertension 2015, 66, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yamazaki, Y.; Tezuka, Y.; Omata, K.; Ono, Y.; Morimoto, R.; Nakamura, Y.; Suzuki, T.; Satoh, F.; Sasano, H. Pathology of aldosterone biosynthesis and its action. Tohoku J. Exp. Med. 2021, 254, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mirzahosseini, G.; Ismael, S.; Ahmed, H.A.; Ishrat, T. Manifestation of renin angiotensin system modulation in traumatic brain injury. Metab. Brain Dis. 2021, 36, 1079–1086. [Google Scholar] [CrossRef]
- Fouda, A.; Artham, S.; El-Remessy, A.; Fagan, S. Renin–angiotensin system as a potential therapeutic target in stroke and retinopathy: Experimental and clinical evidence. Clin. Sci. 2016, 130, 221–238. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Williams, J.S.; Williams, G.H.; Jeunemaitre, X.; Hopkins, P.N.; Conlin, P.R. Influence of dietary sodium on the renin-angiotensin-aldosterone system and prevalence of left ventricular hypertrophy by EKG criteria. J. Hum. Hypertens. 2005, 19, 133–138. [Google Scholar] [CrossRef][Green Version]
- Mecca, A.P.; Regenhardt, R.W.; O’Connor, T.E.; Joseph, J.P.; Raizada, M.K.; Katovich, M.J.; Sumners, C. Cerebroprotection by angiotensin-(1–7) in endothelin-1-induced ischaemic stroke. Exp. Physiol. 2011, 96, 1084. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Q.; Xing, X.; Yang, L.; Xu, M.; Cao, C.; Wang, R.; Li, W.; Niu, X.; Gao, D. The antagonistic effects and mechanisms of microRNA-26a action in hypertensive vascular remodeling. Br. J. Pharmacol. 2021, 178, 1037–1054. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.; Rhaleb, I.A.; Kassem, K.M.; Rhaleb, N.-E. Role of Kinins in Hypertension and Heart Failure. Pharmaceuticals 2020, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Zheng, X.; Chen, X.; Zhao, C.; Zhu, D.; Zhang, J.; Zhuo, J.L. Genetic and genomic evidence for an important role of the Na+/H exchanger 3 in blood pressure regulation and angiotensin II-induced hypertension. Physiol. Genom. 2019, 51, 97–108. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659–670. [Google Scholar] [CrossRef]
- Li, Y.R.; Trush, M. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Sarniak, A.; Lipińska, J.; Tytman, K.; Lipińska, S. Endogenous mechanisms of reactive oxygen species (ROS) generation. Postepy Hig. Med. Dosw. 2016, 70, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Chan, J.Y. Mitochondria and reactive oxygen species contribute to neurogenic hypertension. Physiolog 2017, 32, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Antioxidants in human health and disease. Annu. Rev. Nutr. 1996, 16, 33–50. [Google Scholar] [CrossRef]
- Crowley, S.D. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid. Redox Signal. 2014, 20, 102–120. [Google Scholar] [CrossRef]
- Harrison, D.G. The Mosaic Theory revisited: Common molecular mechanisms coordinating diverse organ and cellular events in hypertension. J. Am. Soc. Hypertens. 2013, 7, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.; Elmarakby, A.A.; El-Remessy, A.B.; Sullivan, J.C. Oxidative stress contributes to sex differences in angiotensin II-mediated hypertension in spontaneously hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R274–R282. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling–from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Reckelhoff, J.F.; Romero, D.G.; Cardozo, L.L.Y. Sex, Oxidative Stress, and Hypertension: Insights from Animal Models. Physiology 2019, 34, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.D.; Yang, X.P.; Liu, Y.H.; Shesely, E.G.; Cavasin, M.A.; Kuziel, W.A.; Pagano, P.J.; Carretero, O.A. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension 2008, 52, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Spitler, K.M.; Webb, R.C. Endoplasmic Reticulum Stress Contributes to Aortic Stiffening via Proapoptotic and Fibrotic Signaling Mechanisms. Hypertension 2014, 63, e40–e45. [Google Scholar] [CrossRef] [PubMed]
- Ayala, P.; Montenegro, J.; Vivar, R.; Letelier, A.; Urroz, P.A.; Copaja, M.; Pivet, D.; Humeres, C.; Troncoso, R.; Vicencio, J.M.; et al. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp. Mol. Pathol. 2011, 92, 97–104. [Google Scholar] [CrossRef]
- Okada, K.I.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef]
- Maekawa, H.; Inagi, R. Pathophysiological Role of Organelle Stress/Crosstalk in AKI-to-CKD Transition. Semin. Nephrol. 2019, 39, 581–588. [Google Scholar] [CrossRef] [PubMed]





| CATEGORY | BP [mmHg] |
|---|---|
| Normal | systolic: <130 diastolic: <85 |
| Elevated | systolic: <140 diastolic: <90 |
| Stage 1 hypertension | systolic: 140–159 diastolic: 90–99 |
| Stage 2 hypertension | systolic: 160–179 diastolic: 100–109 |
| Stage 3 hypertension | systolic: ≥180 diastolic: ≥110 |
| Cause of Secondary AH | Prevalence |
|---|---|
| Sleep apnea | >5–15% |
| Hyperaldosteronism | 1.4–10% |
| Renal parenchymal diseases | 1.6–8% |
| Renal artery stenosis | 1–8% |
| Thyroid diseases | 1–2% |
| Aortic isthmus stenosis | <1% |
| Cushing syndrome | 0.5% |
| Pheochromocytoma | 0.2–0.5% |
| Organ | Pre-Clinical/Clinical Damage |
|---|---|
| Heart |
|
| Heart valves |
|
| Central arteries |
|
| Peripheral arteries |
|
| Kidneys |
|
| Eyes |
|
| Brain |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kućmierz, J.; Frąk, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Molecular Interactions of Arterial Hypertension in Its Target Organs. Int. J. Mol. Sci. 2021, 22, 9669. https://doi.org/10.3390/ijms22189669
Kućmierz J, Frąk W, Młynarska E, Franczyk B, Rysz J. Molecular Interactions of Arterial Hypertension in Its Target Organs. International Journal of Molecular Sciences. 2021; 22(18):9669. https://doi.org/10.3390/ijms22189669
Chicago/Turabian StyleKućmierz, Joanna, Weronika Frąk, Ewelina Młynarska, Beata Franczyk, and Jacek Rysz. 2021. "Molecular Interactions of Arterial Hypertension in Its Target Organs" International Journal of Molecular Sciences 22, no. 18: 9669. https://doi.org/10.3390/ijms22189669
APA StyleKućmierz, J., Frąk, W., Młynarska, E., Franczyk, B., & Rysz, J. (2021). Molecular Interactions of Arterial Hypertension in Its Target Organs. International Journal of Molecular Sciences, 22(18), 9669. https://doi.org/10.3390/ijms22189669