
HPTE-Induced Embryonic Thymocyte Death and Alteration of Differentiation Is Not Rescued by ERα or GPER Inhibition but Is Exacerbated by Concurrent TCR Signaling

 ,
,  and
and
Abstract
:1. Introduction
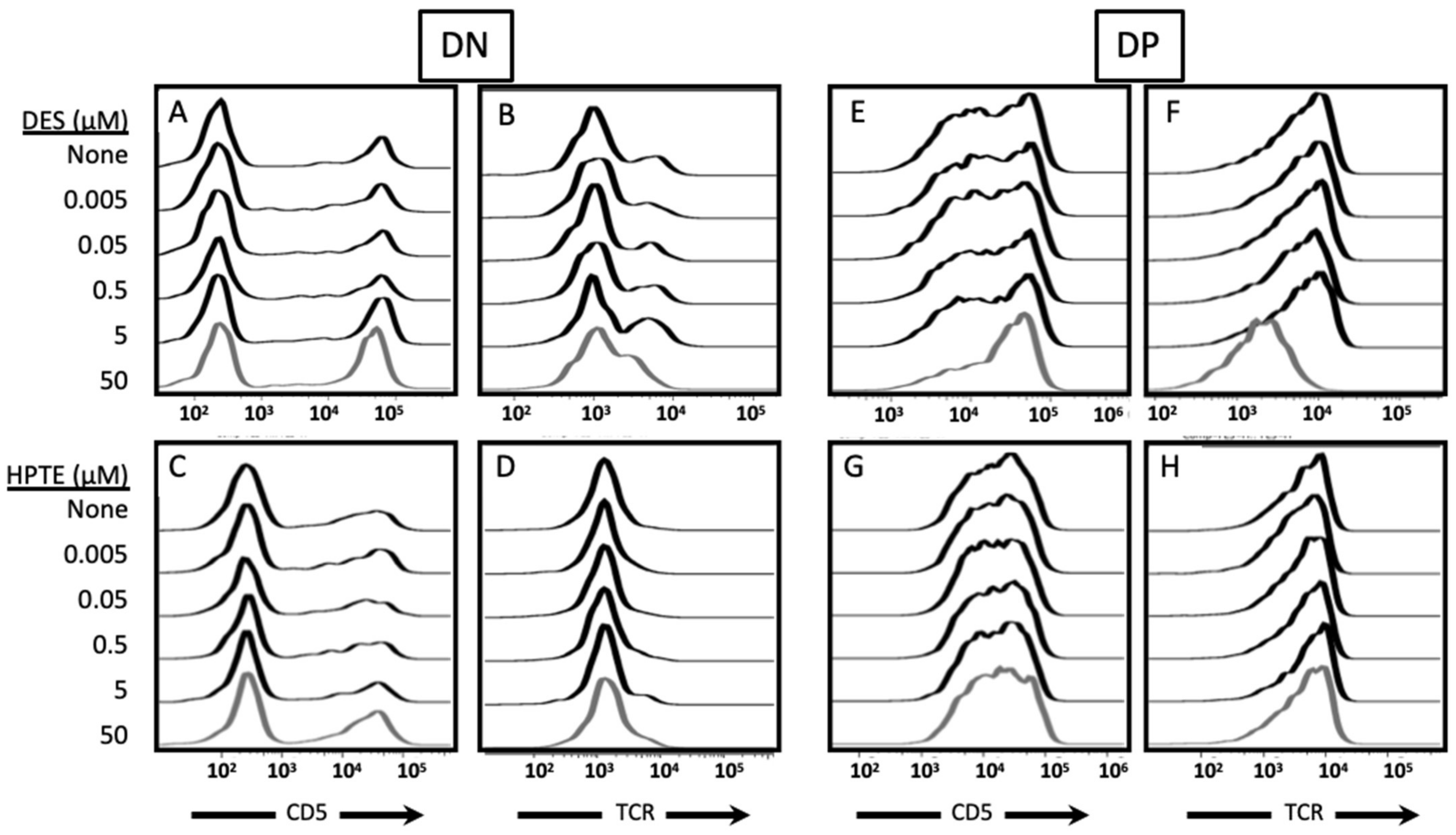
2. Results
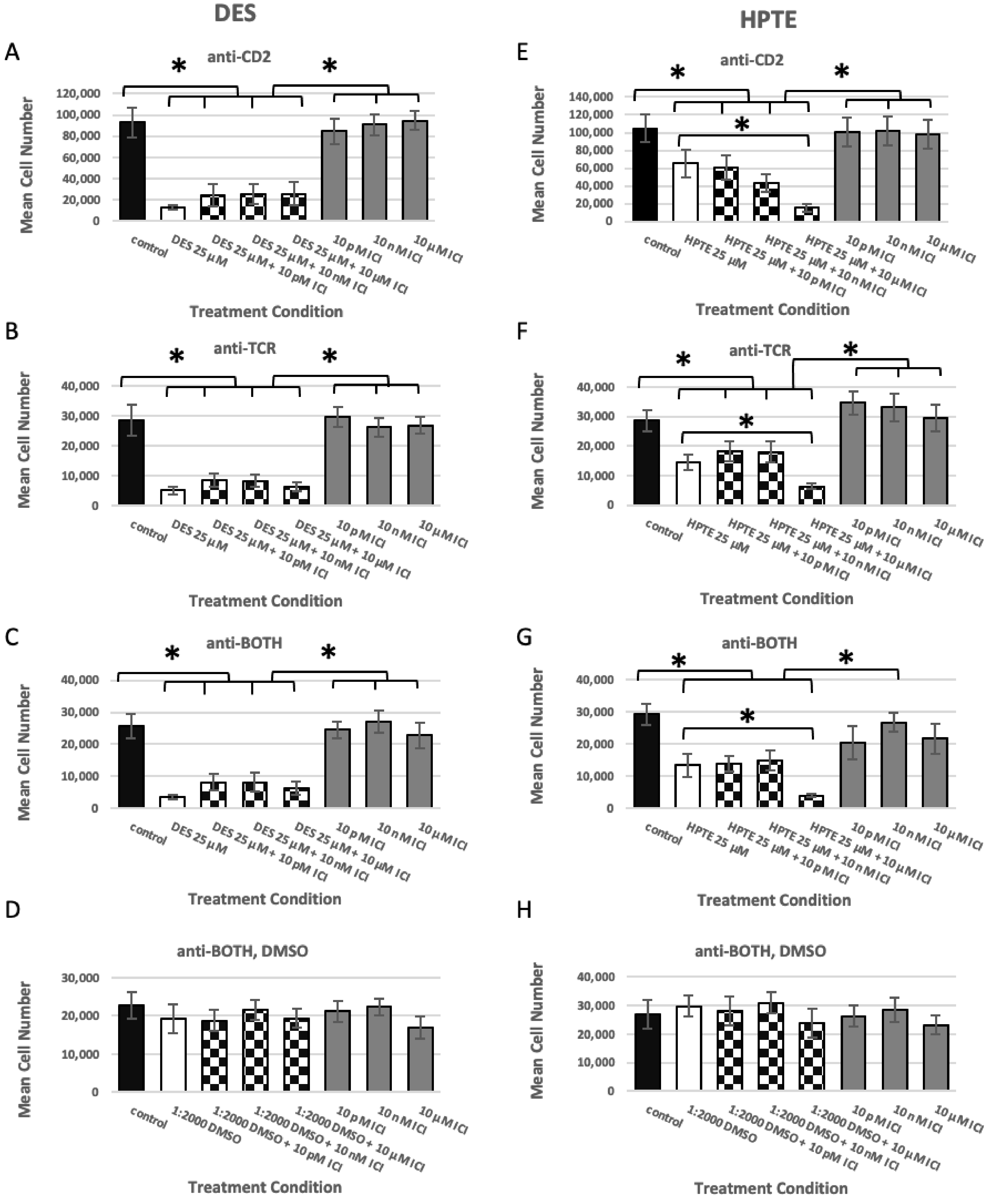
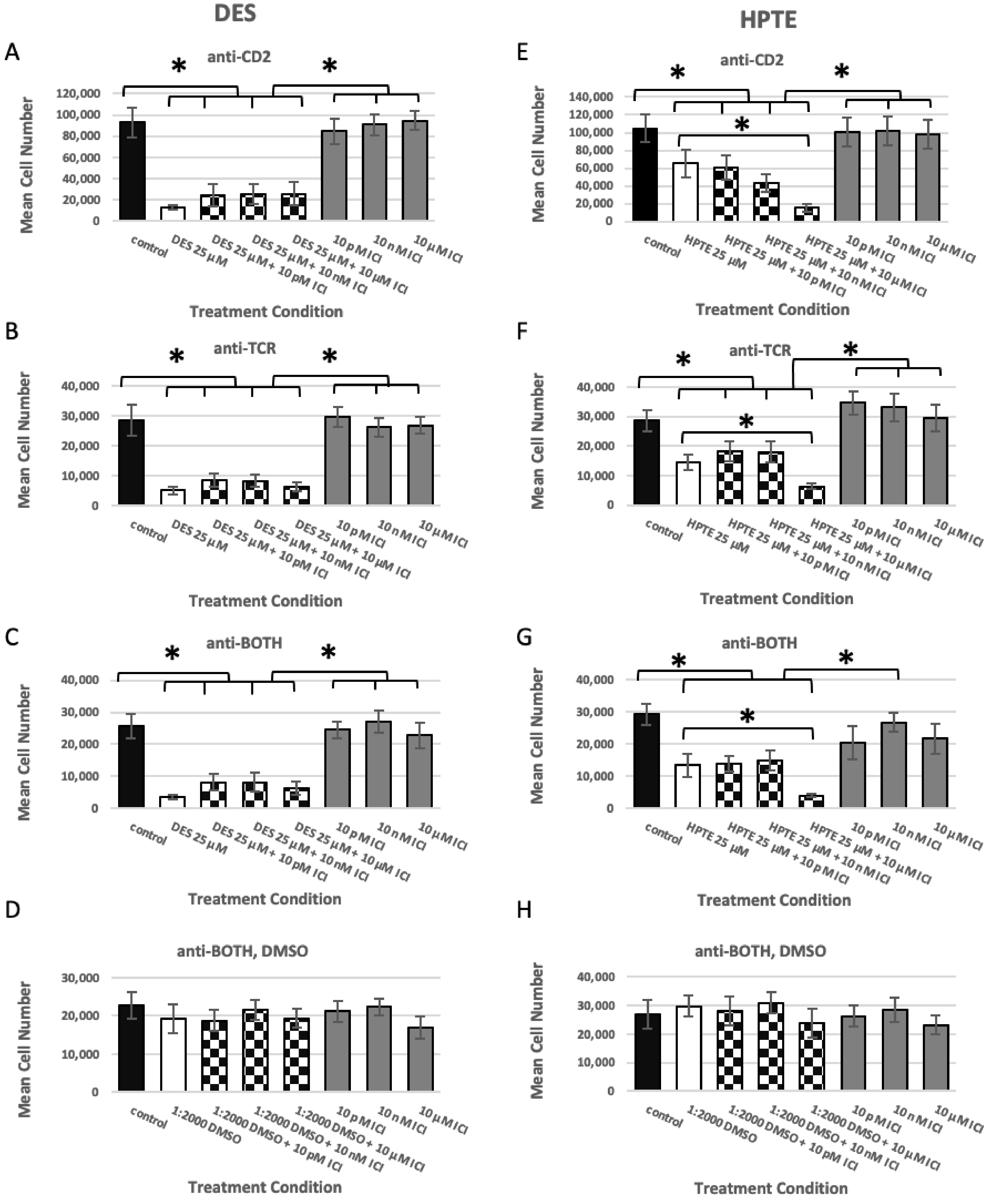
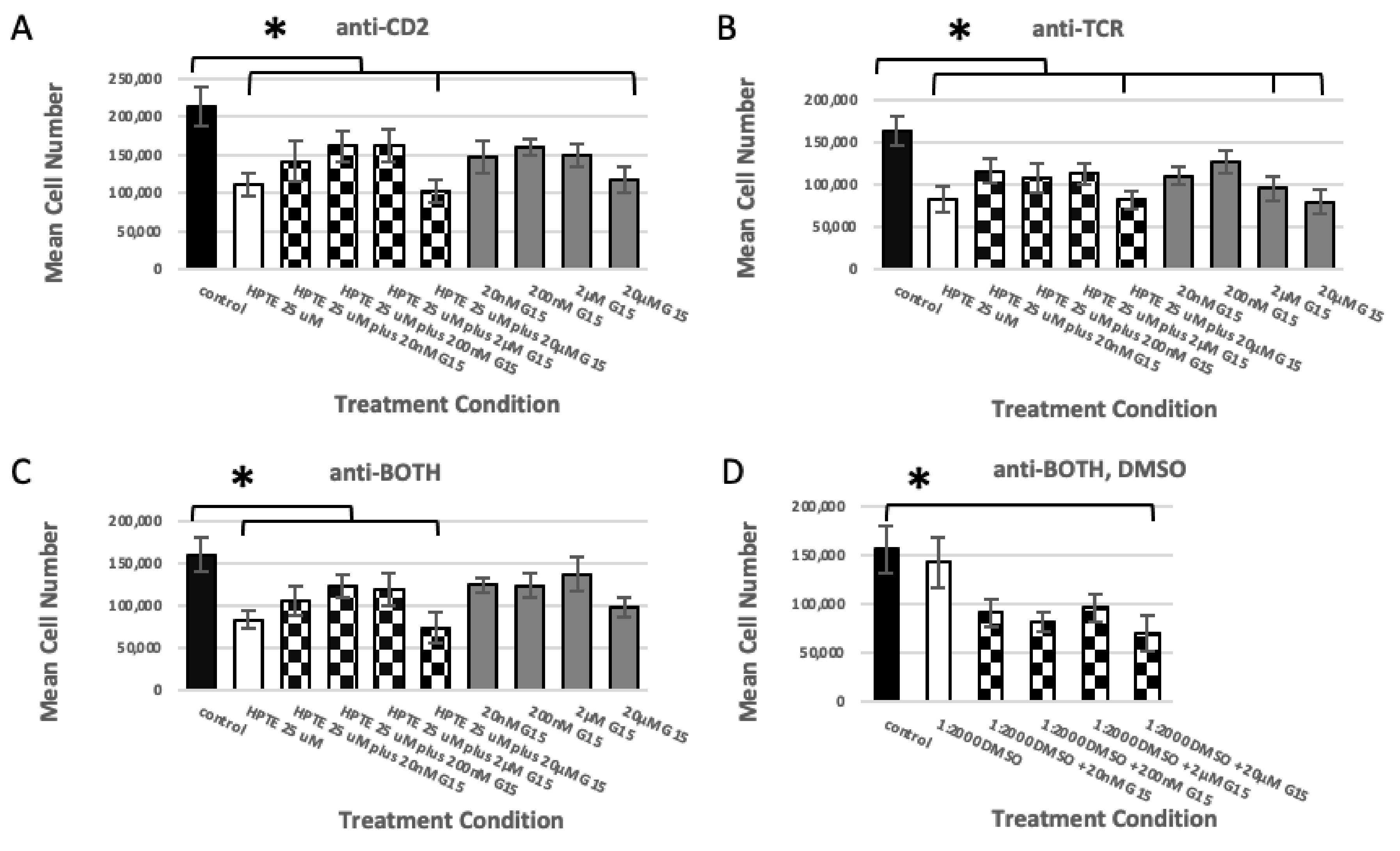
2.1. Estrogen Receptor Usage Study
2.1.1. ER Alpha Inhibition
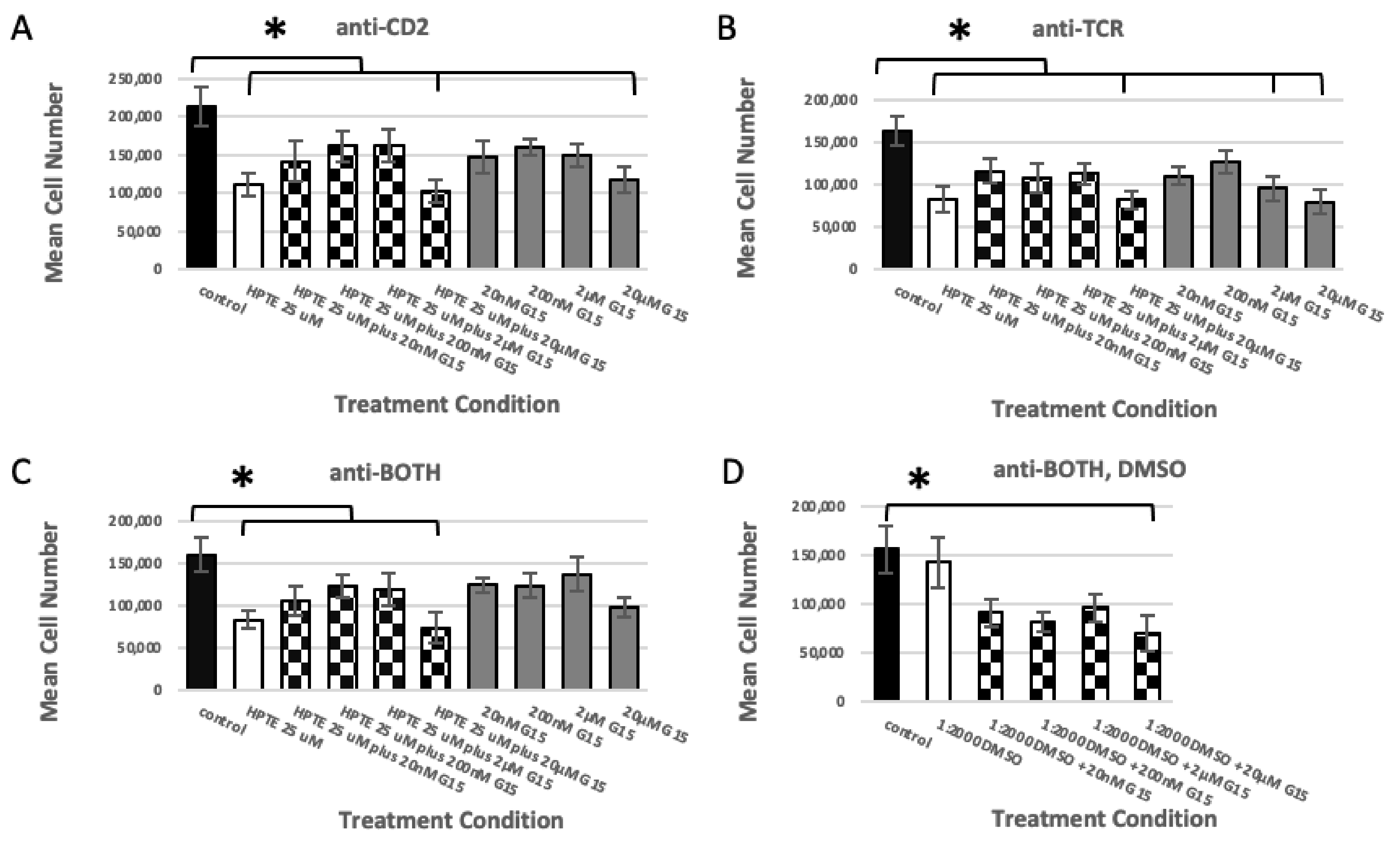
2.1.2. GPER Inhibition
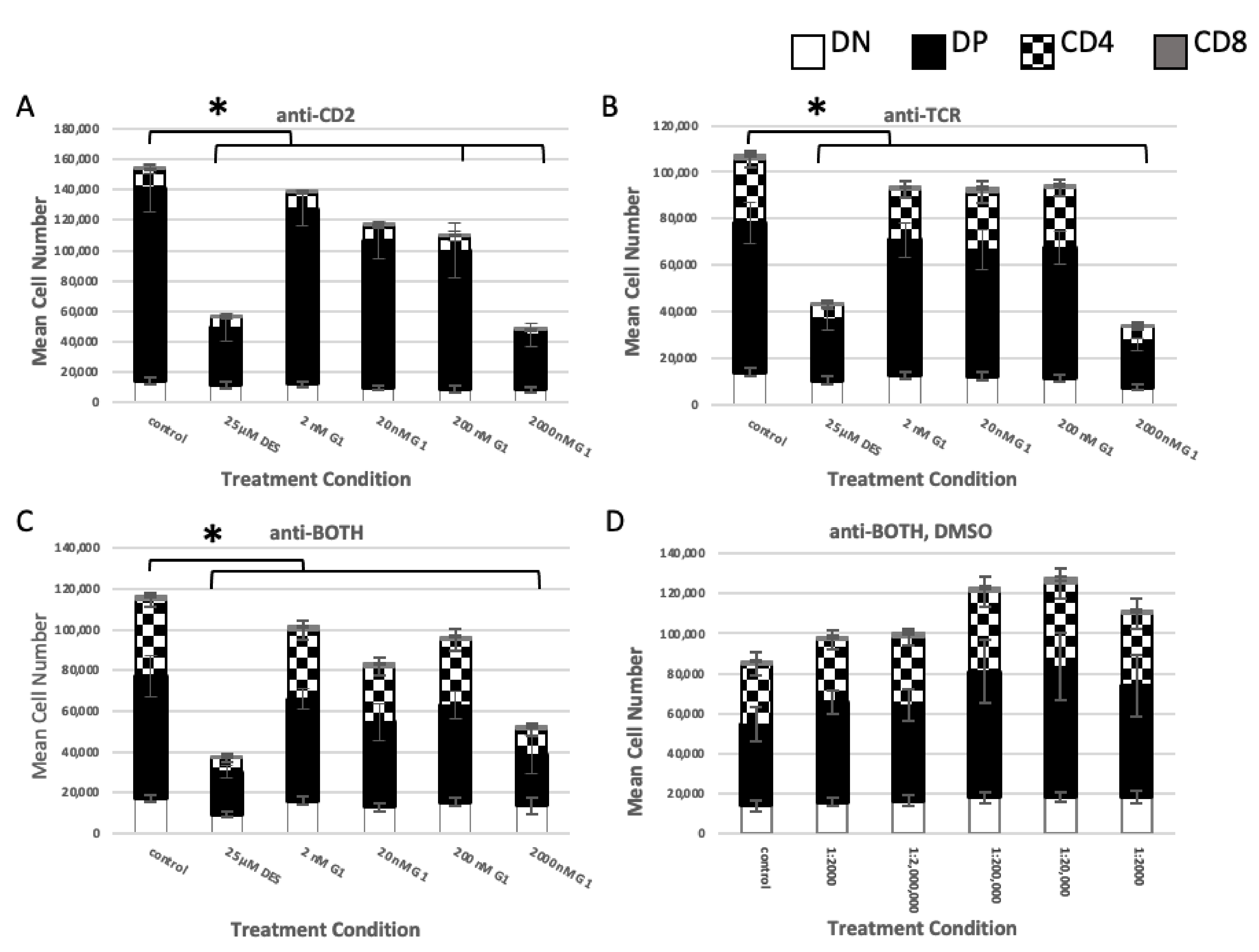
2.1.3. GPER Ligation
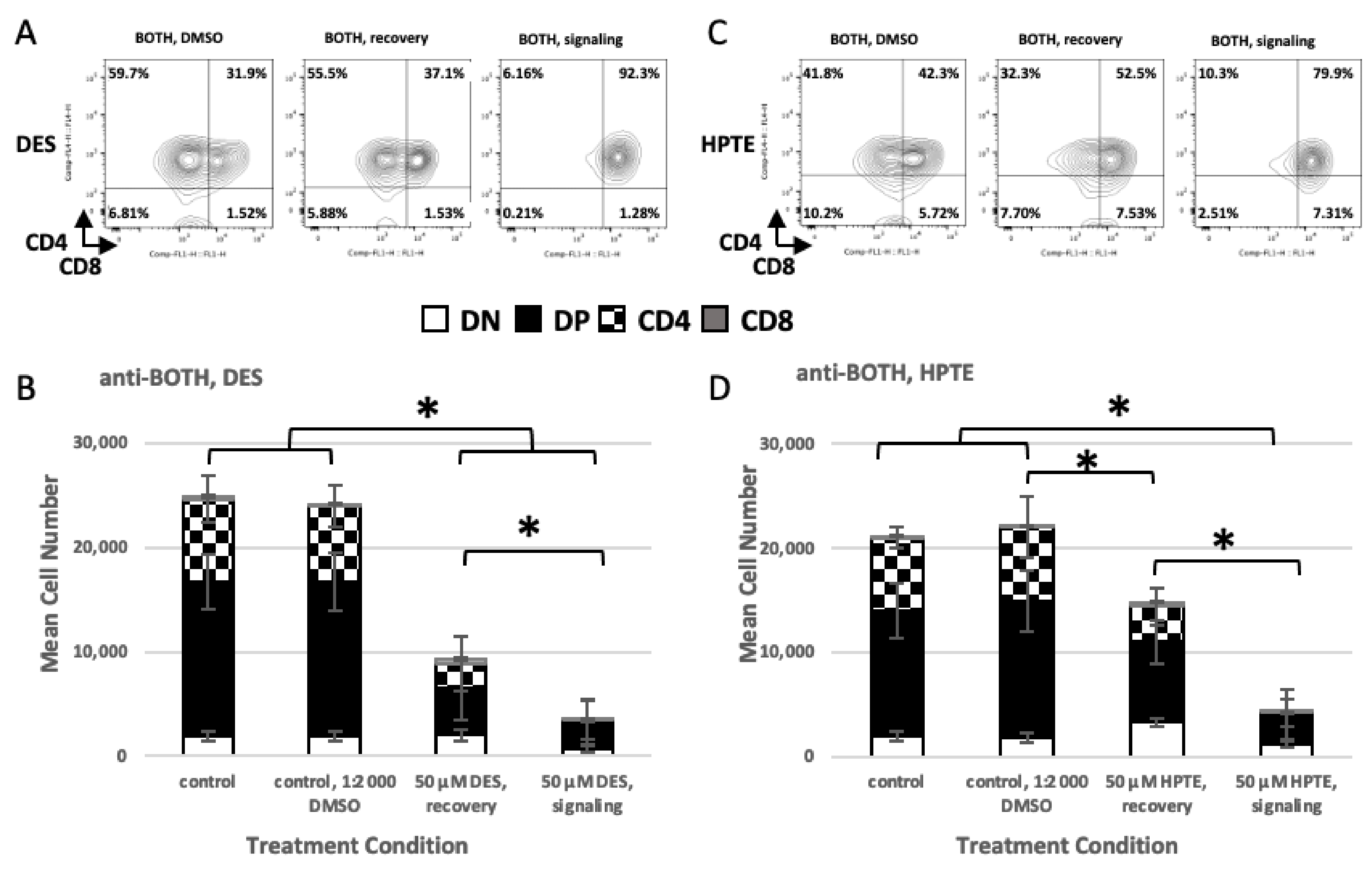
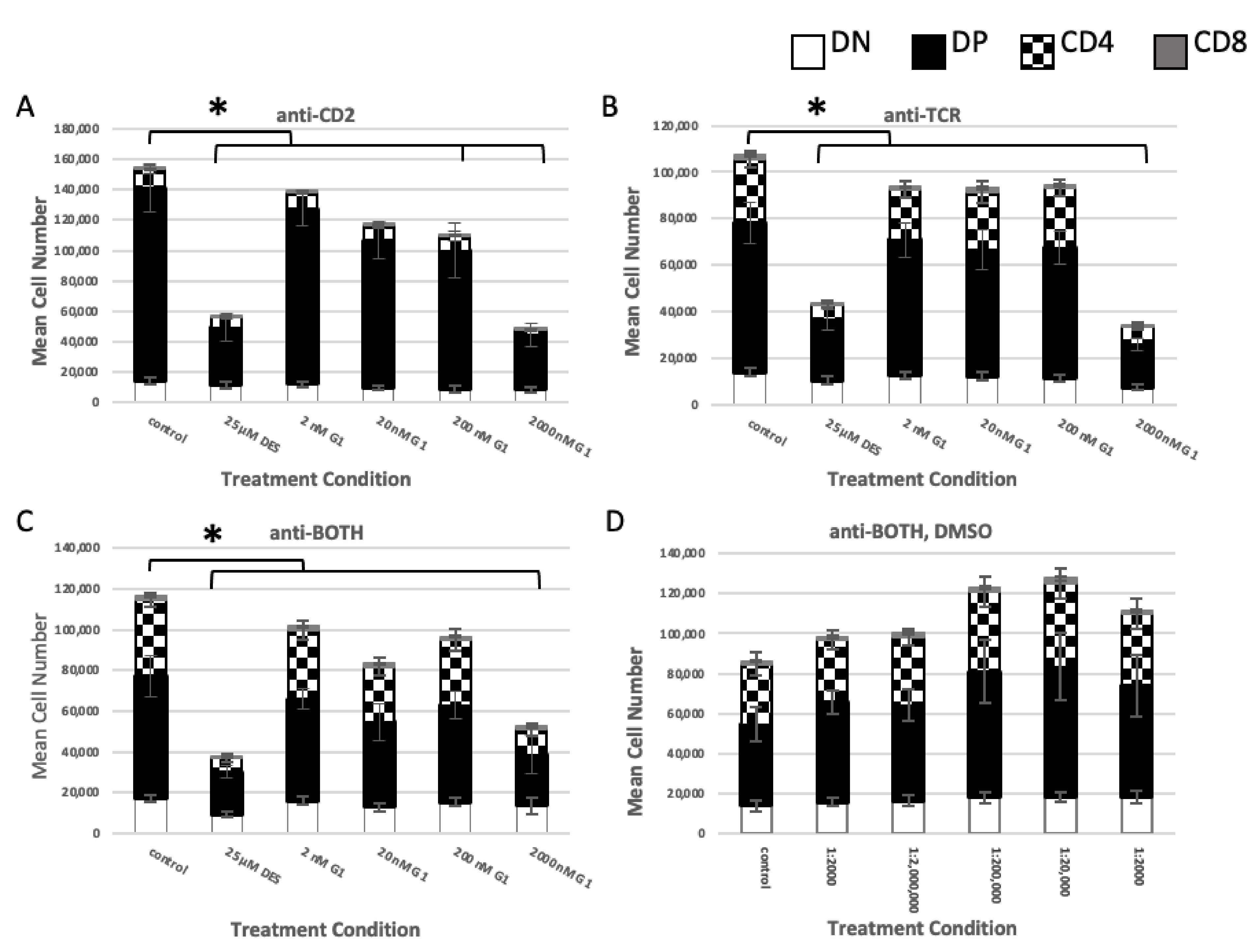
2.2. Role of TCR Signaling
2.2.1. Concurrent versus Sequential Exposure
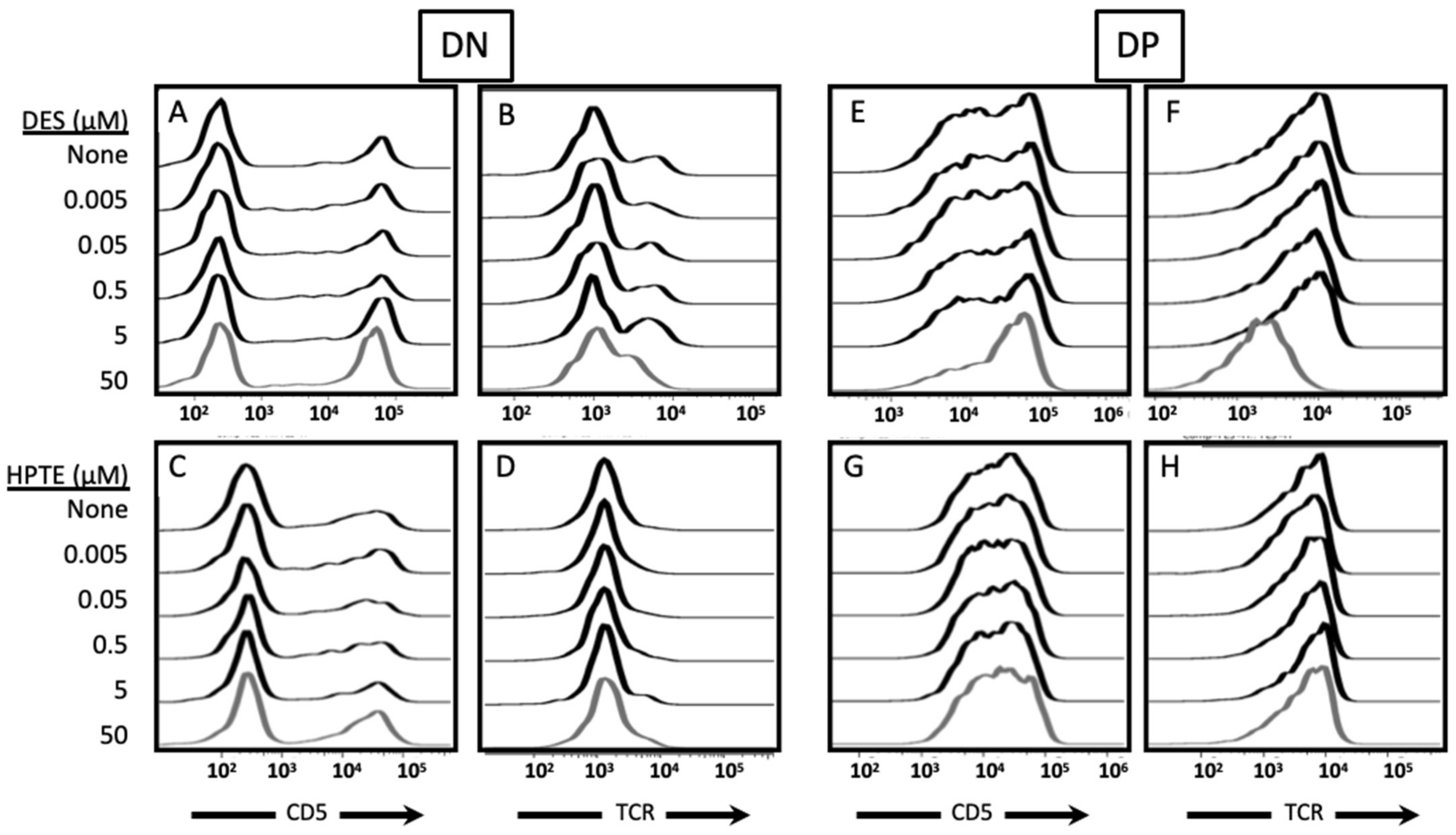
2.2.2. Phenotypic Effects
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Abs and Flow Cytometry Staining, Gating, and Acquisition
4.3. In Vitro Differentiation Assay
4.4. Chemicals and Treatment
4.5. Receptor Inhibition Studies
4.6. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s second scientific statement on endocrine-disrupting chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef]
- Darbre, P.D. The history of endocrine-disrupting chemicals. Curr. Opin. Endocr. Metab. Res. 2019, 7, 26–33. [Google Scholar] [CrossRef]
- Endocrine Society: Introduction to Endocrine Disrupting Chemicals (EDCs). Available online: https://www.endocrine.org/-/media/endosociety/files/advocacy-and-outreach/important-documents/introduction-to-endocrine-disrupting-chemicals.pdf (accessed on 31 July 2021).
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Darbre, P.D. Endocrine Disruption and Human Health; Elsevier: Amsterdam, The Netherlands, 2015; pp. 143–285. [Google Scholar]
- DeWitt, J.C.; Patisaul, H.B. Endocrine disruptors and the developing immune system. Curr. Opin. Toxicol. 2018, 10, 31–36. [Google Scholar] [CrossRef]
- Winans, B.; Humble, M.C.; Lawrence, B.P. Environmental toxicants and the developing immune system: A missing link in the global battle against infectious disease? Reprod. Toxicol. 2011, 31, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Dietert, R.R. Developmental immunotoxicology: Focus on health risks. Chem. Res. Toxicol. 2009, 22, 17–23. [Google Scholar] [CrossRef]
- Medina, K.L. Overview of the immune system. Handb. Clin. Neurol. 2016, 133, 61–76. [Google Scholar] [CrossRef]
- Owen, J.A.; Punt, J.; Stranford, S.A. Kuby Immunology; WH Freeman: New York, NY, USA, 2013; pp. 17–18. [Google Scholar]
- Kurtz, J. Memory in the innate and adaptive immune systems. Microbes Infect. 2004, 6, 1410–1417. [Google Scholar] [CrossRef]
- Germain, R.N. T-cell development and the CD4–CD8 Lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noller, K.; Blair, P.; O’Brien, P.; Melton, L.; Offord, J.; Kaufman, R.; Colton, T. Increased occurrence of autoimmune disease among women exposed in utero to diethylstilbestrol. Fertil. Steril. 1998, 49, 1080–1082. [Google Scholar] [CrossRef]
- Ahmed, S.; Hissong, B.; Verthelyi, D.; Donner, K.; Becker, K.; Karpuzoglu-Sahin, E. Gender and risk of autoimmune diseases: Possible role of estrogenic compounds. Environ. Health Perspect. 1999, 107, 681–686. [Google Scholar]
- Heilmann, C.; Grandjean, P.; Weihe, P.; Nielsen, F.; Budtz-Jørgensen, E. Reduced antibody responses to vaccinations in children exposed to polychlorinated biphenyls. PLoS Med. 2006, 3, e311. [Google Scholar] [CrossRef] [Green Version]
- Besteman, E.G.; Zimmerman, K.L.; Holladay, S.D. Diethylstilbestrol (DES)-Induced Fetal thymic atrophy in C57BL/6 Mice: Inhibited Thymocyte differentiation and increased apoptotic cell death. Int. J. Toxicol. 2005, 24, 231–239. [Google Scholar] [CrossRef]
- Brown, N.; Nagarkatti, M.; Nagarkatti, P.S. Induction of apoptosis in murine fetal thymocytes following perinatal exposure to diethylstilbestrol. Int. J. Toxicol. 2006, 25, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, T.; Kosaka, T.; Tajima, Y.; Hayashi, K.; Shutoh, Y.; Harada, T. Detection of thymocytes apoptosis in mice induced by organochlorine pesticides methoxychlor. Immunopharmacol. Immunotoxicol. 2010, 33, 193–200. [Google Scholar] [CrossRef]
- Lai, Z.-W.; Fiore, N.C.; Gasiewicz, T.A.; Silverstone, A.E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and diethylstilbestrol AFFECT Thymocytes at different stages of development in Fetal thymus Organ Culture. Toxicol. Appl. Pharmacol. 1998, 149, 167–177. [Google Scholar] [CrossRef]
- Okasha, S.A.; Ryu, S.; Do, Y.; McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Evidence for estradiol-induced apoptosis and dysregulated t cell maturation in the thymus. Toxicology 2001, 163, 49–62. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Kosaka, T.; Hayashi, K.; Takeda, M.; Yoshida, T.; Fujisawa, H.; Teramoto, S.; Maita, K.; Harada, T. Thymic atrophy induced by methoxychlor in rat pups. Toxicol. Lett. 2002, 135, 199–207. [Google Scholar] [CrossRef]
- Wang, C.; Dehghani, B.; Magrisso, I.; Rick, E.; Bonhomme, E.; Cody, D.; Elenich, L.; Subramanian, S.; Murphy, S.; Kelly, M.; et al. GPR30 contributes to Estrogen-Induced thymic atrophy. Mol. Endocrinol. 2008, 22, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung-Gurung, L.; Escalante Cobb, P.; Mourad, F.; Zambrano, C.; Muscato, Z.; Sanchez, V.; Godde, K.; Broussard, C. Methoxychlor metabolite HPTE alters viability and differentiation of embryonic thymocytes from C57BL/6 mice. J. Immunotoxicol. 2018, 15, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Nagarkatti, M.; Nagarkatti, P.S. Diethylstilbestrol alters positive and negative selection of t cells in the thymus and modulates t-cell repertoire in the periphery. Toxicol. Appl. Pharmacol. 2006, 212, 119–126. [Google Scholar] [CrossRef]
- Guo, T. Genistein and Methoxychlor modulate the activity of natural killer cells and the expression of Phenotypic markers BY thymocytes And splenocytes IN f0 and F1 generations of Sprague–dawley rats. Toxicology 2002, 172, 205–215. [Google Scholar] [CrossRef]
- Yellayi, S.; Teuscher, C.; Woods, J.A.; Welsh, T.H.; Tung, K.S.; Nakai, M.; Rosenfeld, C.S.; Lubahn, D.B.; Cooke, P.S. Normal development of thymus in male and female mice requires estrogen/estrogen receptor-α signaling pathway. Endocrine 2000, 12, 207–213. [Google Scholar] [CrossRef]
- Erlandsson, M.C.; Ohlsson, C.; Gustafsson, J.-A.; Carlsten, H. Role of oestrogen receptors α and β in immune organ development and in oestrogen-mediated effects on thymus. Immunology 2001, 103, 17–25. [Google Scholar] [CrossRef]
- Staples, J.E.; Gasiewicz, T.A.; Fiore, N.C.; Lubahn, D.B.; Korach, K.S.; Silverstone, A.E. Estrogen receptor alpha is necessary in thymic development and estradiol-induced thymic alterations. J. Immunol. 1999, 163, 4168–4174. [Google Scholar]
- Staples, J.E.; Fiore, N.C.; Frazier, D.E., Jr.; Gasiewicz, T.A.; Silverstone, A.E. Overexpression of the anti-apoptotic oncogene, bcl-2, in the thymus does not prevent thymic atrophy induced by estradiol or 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 1998, 151, 200–210. [Google Scholar] [CrossRef]
- Cibotti, R.; Punt, J.A.; Dash, K.S.; Sharrow, S.O.; Singer, A. Surface molecules that drive T cell development in vitro in the absence of thymic epithelium and in the absence of lineage-specific signals. Immunity 1997, 6, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.X.; Liu, J.; Gu, L.P.; Huang, B.; Pan, X.J. Biological effects of xenoestrogens and the functional mechanisms via genomic and nongenomic pathways. Environ. Rev. 2017, 25, 306–322. [Google Scholar] [CrossRef]
- Watson, C.S.; Bulayeva, N.N.; Wozniak, A.L.; Finnerty, C.C. Signaling from the membrane via membrane estrogen receptor-α: Estrogens, xenoestrogens, and phytoestrogens. Steroids 2005, 70, 364–371. [Google Scholar] [CrossRef]
- Watson, C.S.; Jeng, Y.J.; Kochukov, M.Y. Nongenomic signaling pathways of estrogen toxicity. Toxicol. Sci. 2010, 115, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kiyama, R.; Wada-Kiyama, Y. Estrogenic endocrine disruptors: Molecular mechanisms of action. Environ. Int. 2015, 83, 11–40. [Google Scholar] [CrossRef] [PubMed]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Levin, E.R.; Hammes, S.R. Nuclear receptors outside the nucleus: Extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 2016, 17, 783–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.P.; Gupta, R.K.; Flaws, J.A. Methoxychlor metabolites may cause ovarian toxicity through estrogen-regulated pathways. Toxicol. Sci. 2006, 93, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Shanle, E.K.; Xu, W. Endocrine disrupting Chemicals targeting estrogen Receptor Signaling: Identification and mechanisms of action. Chem. Res. Toxicol. 2011, 24, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Björnström, L.; Sjöberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, V.; Bunick, D.; Finnigan-Bunick, C.; Johnson, R.W.; Wang, H.; Liu, L.; Cooke, P.S. Gene expression profiling of 17β-estradiol and genistein effects on mouse thymus. Toxicol. Sci. 2005, 87, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Frawley, R.; White Jr, K.; Brown, R.; Musgrove, D.; Walker, N.; Germolec, D. Gene expression alterations in immune system pathways in the thymus after exposure to immunosuppressive chemicals. Environ. Health Perspect. 2011, 119, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Folmar, L.C.; Hemmer, M.J.; Denslow, N.D.; Kroll, K.; Chen, J.; Cheek, A.; Richman, H.; Meredith, H.; Grau, E.G. A comparison of the estrogenic potencies of estradiol, ethynylestradiol, diethylstilbestrol, nonylphenol and methoxychlor in vivo and in vitro. Aquat. Toxicol. 2002, 60, 101–110. [Google Scholar] [CrossRef]
- Gaido, K.W.; Leonard, L.S.; Maness, S.C.; Hall, J.M.; McDonnell, D.P.; Saville, B.; Safe, S. Differential interaction of the methoxychlor metabolite 2, 2-bis-(p-hydroxyphenyl)-1, 1, 1-trichloroethane with estrogen receptors α and β. Endocrinology 1999, 140, 5746–5753. [Google Scholar] [CrossRef]
- Wu, F.; Khan, S.; Wu, Q.; Barhoumi, R.; Burghardt, R.; Safe, S. Ligand structure-dependent activation of estrogen receptor α/Sp by estrogens and xenoestrogens. J. Steroid Biochem. Mol. Biol. 2008, 110, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Bolger, R.; Wiese, T.E.; Ervin, K.; Nestich, S.; Checovich, W. Rapid screening of environmental chemicals for estrogen receptor binding capacity. Environ. Health Perspect. 1998, 106, 551–557. [Google Scholar] [CrossRef]
- Nikov, G.N.; Eshete, M.; Rajnarayanan, R.V.; Alworth, W.L. Interactions of synthetic estrogens with human estrogen receptors. J. Endocrinol. 2001, 170, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Sotoca Covaleda, A.M.; van den Berg, H.; Vervoort, J.; van der Saag, P.; Ström, A.; Gustafsson, J.Å.; Rietjens, I.M.; Murk, A.J. Influence of cellular ERα/ERβ ratio on the ERα-agonist induced proliferation of human T47D breast cancer cells. Toxicol. Sci. 2008, 105, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Gustafsson, J.Å. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Adam, A.H.B.; de Haan, L.H.; Estruch, I.M.; Hooiveld, G.J.; Louisse, J.; Rietjens, I.M. Estrogen receptor alpha (ERα)–mediated coregulator binding and gene expression discriminates the toxic ERα agonist diethylstilbestrol (DES) from the endogenous ERα agonist 17β-estradiol (E2). Cell Biol. Toxicol. 2020, 36, 417. [Google Scholar] [CrossRef] [Green Version]
- Romano, S.N.; Gorelick, D.A. Crosstalk between nuclear and g protein-coupled estrogen receptors. Gen. Comp. Endocrinol. 2018, 261, 190–197. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.I.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedram, A.; Razandi, M.; Levin, E.R. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006, 20, 1996–2009. [Google Scholar] [CrossRef]
- Li, L.; Haynes, M.P.; Bender, J.R. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4807–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A variant of estrogen receptor-α, hER-α36: Transduction of estrogen-and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef] [Green Version]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Yoon, K.; Kwack, S.J.; Kim, H.S.; Lee, B.-M. Estrogenic Endocrine-Disrupting Chemicals: Molecular mechanisms of actions on Putative human diseases. J. Toxicol. Environ. Health Part B 2014, 17, 127–174. [Google Scholar] [CrossRef] [PubMed]
- Song, R.X.D.; Santen, R.J. Membrane initiated estrogen signaling in breast cancer. Biol. Reprod. 2006, 75, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Weng, J.R.; Hu, J.L.; Wang, D.; Sargeant, A.M.; Chiu, C.F. G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem. Biol. Interact. 2013, 206, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Courtney, A.H.; Lo, W.L.; Weiss, A. TCR signaling: Mechanisms of initiation and propagation. Trends Biochem. Sci. 2018, 43, 108–123. [Google Scholar] [CrossRef]
- Gascoigne, N.R.; Rybakin, V.; Acuto, O.; Brzostek, J. TCR signal strength and T cell development. Annu. Rev. Cell Dev. Biol. 2016, 32, 327–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punt, J.A.; Osborne, B.A.; Takahama, Y.; Sharrow, S.O.; Singer, A. Negative selection of CD4+ CD8+ thymocytes by T cell receptor-induced apoptosis requires a costimulatory signal that can be provided by CD28. J. Exp. Med. 1994, 179, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.W.; Fiore, N.C.; Hahn, P.J.; Gasiewicz, T.A.; Silverstone, A.E. Differential effects of diethylstilbestrol and 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin on thymocyte differentiation, proliferation, and apoptosis in bcl-2 transgenic mouse fetal thymus organ culture. Toxicol. Appl. Pharmacol. 2000, 168, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.Y.; Yan, L.I.; Chen, W.F. Kinetics of thymocyte developmental process in fetal and neonatal mice. Cell Res. 2003, 13, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.; Manley, N.R. Mechanisms of thymus organogenesis and morphogenesis. Development 2011, 138, 3865–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011.
- Berthois, Y.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Phenol red in tissue culture media is a weak estrogen: Implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. USA 1986, 83, 2496–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, R.L.; Kapoor, I.P.; Nystrom, R.F.; Sangha, G.K. Comparative metabolism of methoxychlor, methiochlor, and DDT in mouse, insects, and in a model ecosystem. J. Agric. Food Chem. 1970, 18, 1145–1152. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef] [Green Version]
- Altman, N.; Krzywinski, M. P-values and the search for significance. Nat. Methods 2017, 14, 3–4. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Endocrine Disruptor | Condition | F-Value Treatment | p-Value Treatment | F-Value Experiment Number | p-Value Experiment Number |
|---|---|---|---|---|---|---|
| ER Alpha Inhibition | ||||||
| DES | ||||||
| anti-CD2 | 19.68 | <0.0001 | 7.59 | <0.0001 | ||
| anti-TCR | 15.13 | <0.0001 | 6.86 | <0.0001 | ||
| anti-BOTH | 20.76 | <0.0001 | 4.95 | 0.0001 | ||
| anti-BOTH, DMSO | 1.32 | 0.264827 | 13.92 | <0.0001 | ||
| HPTE | ||||||
| anti-CD2 | 16.08 | <0.0001 | 19.14 | <0.0001 | ||
| anti-TCR | 32.03 | <0.0001 | 15.23 | <0.0001 | ||
| anti-BOTH | 13.13 | <0.0001 | 6.15 | <0.0001 | ||
| anti-BOTH, DMSO | 0.95 | 0.480282 | 3.28 | 0.008185 | ||
| GPER Inhibition | ||||||
| HPTE | ||||||
| anti-CD2 | 3.387 | 0.00163 | 8.398 | <0.0001 | ||
| anti-TCR | 5.106 | <0.0001 | 8.783 | <0.0001 | ||
| anti-BOTH | 4.637 | <0.0001 | 5.619 | <0.0001 | ||
| anti-BOTH, DMSO | 3.372 | 0.0018 | 11.123 | <0.0001 | ||
| GPER Ligation | ||||||
| DES | ||||||
| anti-CD2 * | 18.433 | <0.0001 | 8.551 | <0.0001 | ||
| anti-TCR | 18.234 | <0.0001 | 7.168 | <0.0001 | ||
| anti-BOTH | 8.311 | <0.0001 | 2.951 | 0.0154 | ||
| anti-BOTH, DMSO | 1.201 | 0.329158 | 5.067 | 0.000482 | ||
| Role of TCR Signaling | ||||||
| DES | ||||||
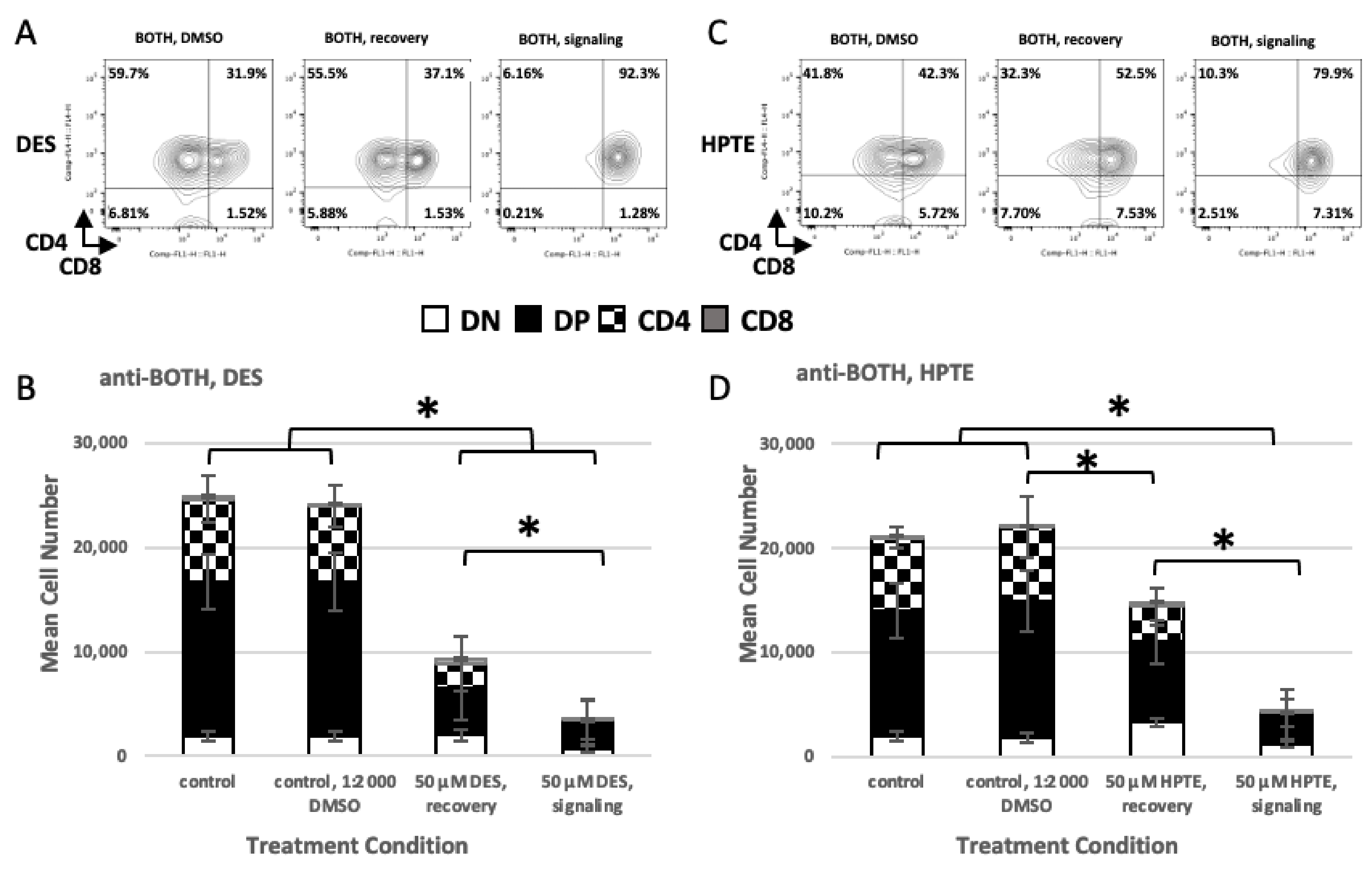
| anti-BOTH | 110.83 | <0.0001 | 19.52 | <0.0001 | ||
| HPTE | ||||||
| anti-BOTH | 56.442 | <0.0001 | 6.984 | 0.000112 | ||
| DN | DP | CD4 Intermediate | CD8 | |||||
|---|---|---|---|---|---|---|---|---|
| Comparison | Difference/F-Value | p-Value | Difference/ F-Value | p-Value | Difference/ F-Value | p-Value | Difference/F-Value | p-Value |
| CD2 | ||||||||
| ANOVA | 2.62 (5.31) | 0.0409 (<0.0001) | 14.46 (7.05) | <0.0001 (<0.0001) | 9.98 (9.84) | <0.0001 (<0.0001) | 3.75 (8.35) | 0.0080 (<0.0001) |
| 25 µM DES—medium | ** | ** | −1.2916 | 0.0001 | −0.6251 | 0.1424 | −0.1043 | 0.9973 |
| 2 µM G1—medium | ** | ** | −1.4876 | <0.0001 | −1.4826 | <0.0001 | −0.6912 | 0.0492 |
| 2 µM G1—25 µM DES | ** | ** | −0.1960 | 0.9623 | −0.8575 | 0.0161 | −0.5869 | 0.1335 |
| TCR | ||||||||
| ANOVA | 3.55 (4.61) | 0.0106 (0.001) | 20.64 (9.06) | <0.0001 (<0.0001) | 13.31 (3.55) | <0.0001 (0.0055) | 6.27 (17.27) | 0.0003 (<0.0001) |
| 25 µM DES—medium | −0.3080 | 0.4260 | −0.8942 | <0.0001 | −1.4969 | <0.0001 | −0.7549 | 0.0148 |
| 2 µM G1—medium | −0.6148 | 0.0074 | −1.2401 | <0.0001 | −1.3609 | 0.0001 | −0.8667 | 0.0036 |
| 2 µM G1—25 µM DES | −0.3068 | 0.4305 | −0.3459 | 0.2945 | 0.1359 | 0.9948 | −0.1118 | 0.9950 |
| BOTH | ||||||||
| ANOVA | 2.02 (2.37) | 0.1001 (0.0428) | 7.62 (4.01) | <0.0001 (<0.0001) | 16.22 (3.3) | <0.0001 (0.0085) | 8.48 (14.95) | <0.0001 (<0.0001) |
| 25 µM DES—medium | ** | ** | −1.0104 | 0.0020 | −1.8813 | <0.0001 | −1.2053 | 0.0002 |
| 2 µM G1—medium | ** | ** | −1.0655 | 0.0010 | −1.2553 | 0.0004 | −0.7800 | 0.0274 |
| 2 µM G1—25 µM DES | ** | ** | −0.0551 | 0.9999 | 0.6260 | 0.1887 | 0.4253 | 0.4917 |
| BOTH, DMSO ** | ||||||||
| ANOVA | 0.84 (4.89) | 0.5316 (0.0006) | 1.37 (8.46) | 0.261 (<0.0001) | 1.1 (6.18) | 0.378 (<0.0001) | 1.03 (17.64) | 0.418 (<0.0001) |
| diluent a—medium | ** | ** | ** | ** | ** | ** | ** | ** |
| 2 µM G1—medium | ** | ** | ** | ** | ** | ** | ** | ** |
| 2 µM G1—diluent | ** | ** | ** | ** | ** | ** | ** | ** |
| DN | DP | CD4 Intermediate | CD8 | |||||
|---|---|---|---|---|---|---|---|---|
| Comparison | Difference/F-Value | p-Value | Difference/F-Value | p-Value | Difference/F-Value | p-Value | Difference/F-Value | p-Value |
| DES | ||||||||
| ANOVA | 30.61 (23.02) | <0.0001 (<0.0001) | 70.02 (11.96) | <0.0001 (<0.0001) | 76.09 (9.43) | <0.0001 (<0.0001) | 27.79 (19.17) | <0.0001 (<0.0001) |
| medium—diluent a | 0.0335 | 0.9941 | 0.0009 | 0.9999 | 0.0414 | 0.9988 | −0.1504 | 0.9090 |
| recovery 50 µM—diluent | 0.1305 | 0.7559 | −1.2581 | <0.0001 | −1.2267 | 0.0005 | −0.0105 | 0.9999 |
| signal 50 µM—diluent | −0.9708 | <0.0001 | −1.9822 | <0.0001 | −3.7025 | <0.0001 | −1.8412 | <0.0001 |
| recovery 50 µM—medium | 0.0970 | 0.8820 | −1.2590 | <0.0001 | −1.2680 | 0.0003 | 0.1398 | 0.9251 |
| signal 50 µM—medium | −1.0044 | <0.0001 | −1.9831 | <0.0001 | −3.7439 | <0.0001 | −1.6908 | <0.0001 |
| signal 50 µM—recovery 50 µM | −1.1014 | <0.0001 | −0.7241 | 0.0006 | −2.4759 | <0.0001 | −1.8306 | <0.0001 |
| HPTE | ||||||||
| ANOVA | 14.10 (11.01) | <0.0001 (<0.0001) | 27.86 (8.51) | <0.0001 (<0.0001) | 121.47 (5.23) | <0.0001 (0.0008) | 7.19 (4.74) | 0.0014 (0.0016) |
| medium—diluent | 0.0294 | 0.9982 | −0.0690 | 0.9778 | 0.0147 | 0.9999 | 0.1267 | 0.9839 |
| recovery 50 µM—diluent | 0.5518 | 0.0193 | −0.5056 | 0.0350 | −0.8879 | 0.0045 | 0.9787 | 0.0502 |
| signal 50 µM—diluent | −0.6033 | 0.0124 | −1.4530 | <0.0001 | −3.9733 | <0.0001 | −0.7007 | 0.2472 |
| recovery 50 µM—medium | 0.5223 | 0.0283 | −0.4367 | 0.0810 | −0.9026 | 0.0039 | 0.8520 | 0.1037 |
| signal 50 µM—medium | −0.6327 | 0.0084 | −1.3840 | <0.0001 | −3.9880 | <0.0001 | −0.8274 | 0.1351 |
| signal 50 µM—recovery 50 µM | −1.1551 | <0.0001 | −0.9474 | 0.0001 | −3.0854 | <0.0001 | −1.6794 | 0.0007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avellaneda, E.; Lim, A.; Moeller, S.; Marquez, J.; Escalante Cobb, P.; Zambrano, C.; Patel, A.; Sanchez, V.; Godde, K.; Broussard, C. HPTE-Induced Embryonic Thymocyte Death and Alteration of Differentiation Is Not Rescued by ERα or GPER Inhibition but Is Exacerbated by Concurrent TCR Signaling. Int. J. Mol. Sci. 2021, 22, 10138. https://doi.org/10.3390/ijms221810138
Avellaneda E, Lim A, Moeller S, Marquez J, Escalante Cobb P, Zambrano C, Patel A, Sanchez V, Godde K, Broussard C. HPTE-Induced Embryonic Thymocyte Death and Alteration of Differentiation Is Not Rescued by ERα or GPER Inhibition but Is Exacerbated by Concurrent TCR Signaling. International Journal of Molecular Sciences. 2021; 22(18):10138. https://doi.org/10.3390/ijms221810138
Chicago/Turabian StyleAvellaneda, Eddie, Atalie Lim, Sara Moeller, Jacqueline Marquez, Priscilla Escalante Cobb, Cristina Zambrano, Aaditya Patel, Victoria Sanchez, K. Godde, and Christine Broussard. 2021. "HPTE-Induced Embryonic Thymocyte Death and Alteration of Differentiation Is Not Rescued by ERα or GPER Inhibition but Is Exacerbated by Concurrent TCR Signaling" International Journal of Molecular Sciences 22, no. 18: 10138. https://doi.org/10.3390/ijms221810138
APA StyleAvellaneda, E., Lim, A., Moeller, S., Marquez, J., Escalante Cobb, P., Zambrano, C., Patel, A., Sanchez, V., Godde, K., & Broussard, C. (2021). HPTE-Induced Embryonic Thymocyte Death and Alteration of Differentiation Is Not Rescued by ERα or GPER Inhibition but Is Exacerbated by Concurrent TCR Signaling. International Journal of Molecular Sciences, 22(18), 10138. https://doi.org/10.3390/ijms221810138