
Biotransformation of the Phenolic Constituents from Licorice and Cytotoxicity Evaluation of Their Metabolites


Abstract
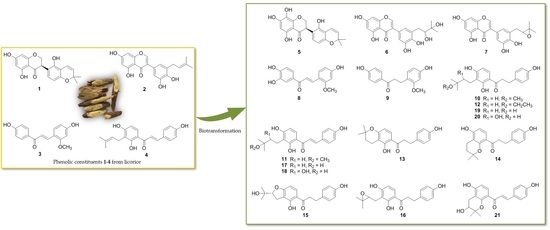
1. Introduction
2. Results and Discussion
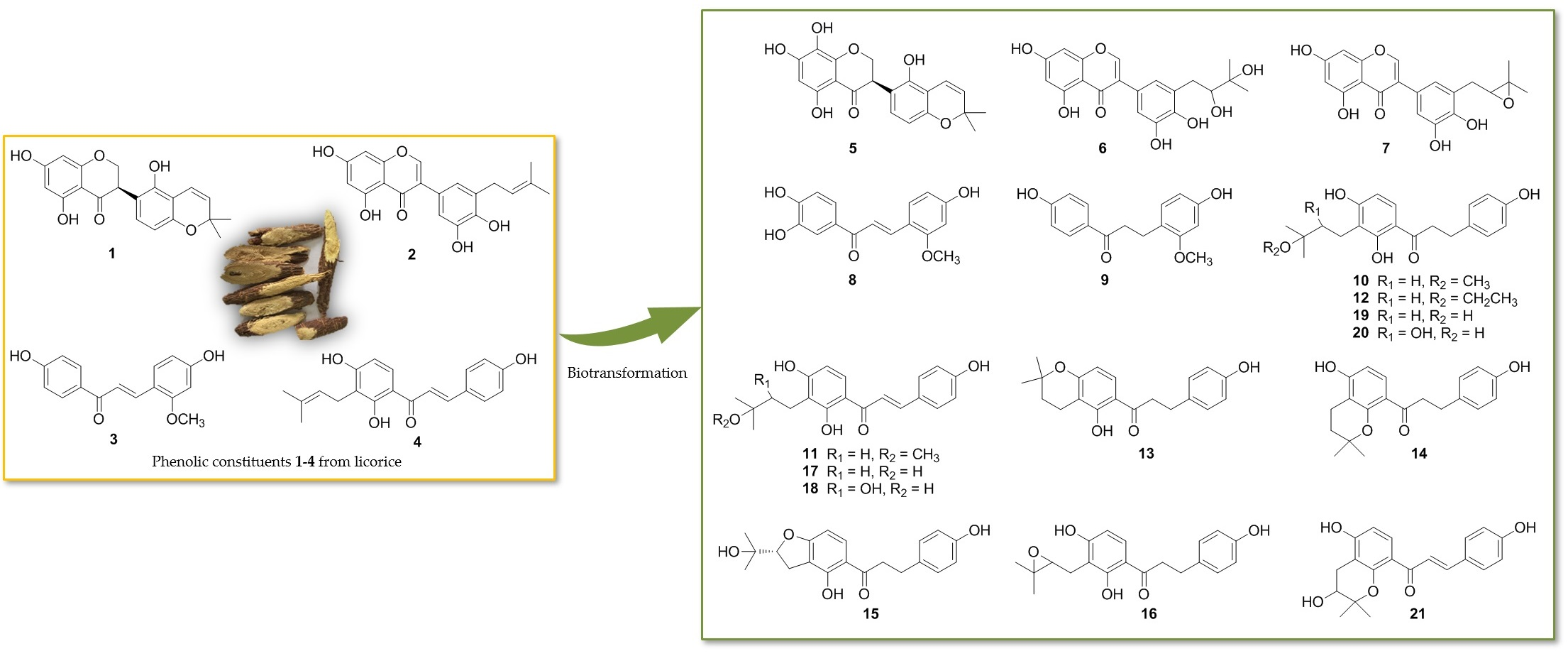
2.1. Biotransformation of Two Isoflavonoids, Licoisoflavanone (1) and Glycyrrhisoflavone (2) with A. niger KCCM 60332
2.2. Biotransformation of Two Chalcones, Echinatin (3) and Isobavachalcone (4) with A. niger KCCM 60332
2.3. Proposed Metabolic Pathways of Isobavachalcone (4) Catalyzed by A. niger KCCM 60332
2.4. Cytotoxicity Evaluation
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation of Substrates 1 and 2
3.4. Synthesis of Substrates 3 and 4
3.5. Microorganisms and Screening for Biostransformation
3.6. Scale-up Fermentation, Extraction, and Isolation of Metabolites 5–21
3.7. Spectroscopic Data of Metabolites 5–21
3.7.1. Spectroscopic Data of the New Compounds 5–7, 10–17, and 19
- Compound 5
- 2.
- Compound 6
- 3.
- Compound 7
- 4.
- Compound 10
- 5.
- Compound 11
- 6.
- Compound 12
- 7.
- Compound 13
- 8.
- Compound 14
- 9.
- Compound 15
- 10.
- Compound 16
- 11.
- Compound 17
- 12.
- Compound 19
3.7.2. 1H-NMR Data of the Compounds 8, 9, 18, 20, and 21
- Compound 8
- 2.
- Compound 9
- 3.
- Compound 18
- 4.
- Compound 20
- 5.
- Compound 21
3.8. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lilly, M.D. Advances in biotransformation processes. Chem. Eng. Sci. 1994, 49, 151–159. [Google Scholar] [CrossRef]
- Hegazy, M.E.F.; Mohamed, T.A.; ElShamy, A.I.; Abou-El-Hamd, H.; Mahalel, U.A.; Reda, E.H.; Shaheen, A.M.; Tawfik, W.A.; Shahat, A.A.; Shams, K.A.; et al. Microbial biotransformation as a tool for drug development based on natural products from mevalonic acid pathway: A review. J. Adv. Res. 2015, 6, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Singh, V.; Katiyar, D.; Khan, M.T.A.; Tripathi, V.; El Enshasy, H.; Pasupuleti, M.; Mishra, B.N. Biotransformation of newly synthesized coumarin derivatives by Candida albicans as potential antibacterial, antioxidant and cytotoxic agents. Process Biochem. 2019, 87, 138–144. [Google Scholar]
- Shanu-Wilson, J.; Evans, L.; Wrigley, S.; Steele, J.; Atherton, J.; Boer, J. Biotransformation: Impact and application of metabolism in drug discovery. ACS Med. Chem. Lett. 2020, 11, 2087–2107. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Lee, I.-S. Microbial transformation of bavachin by Absidia coerulea. Phytochem. Lett. 2016, 18, 136–139. [Google Scholar] [CrossRef]
- Dou, F.; Wang, Z.; Li, G.; Dun, B. Microbial transformation of flavonoids by Isaria fumosorosea ACCC 37814. Molecules 2019, 24, 1028. [Google Scholar] [CrossRef] [PubMed]
- Chokchaisiri, R.; Chaichompoo, W.; Sukcharoen, O.; Suksamrarn, A.; Ganranoo, L. Microbial transformation of isocoronarin D by Cunninghamella echinulata NRRL 1386. Nat. Prod. Res. 2019, 33, 2890–2896. [Google Scholar] [CrossRef]
- Pervaiz, I.; Ahmad, S.; Madni, M.; Ahmad, H.; Khaliq, F. Microbial biotransformation: A tool for drug designing. Appl. Biochem. Microbiol. 2013, 49, 437–450. [Google Scholar] [CrossRef]
- Liu, J.-H.; Yu, B.-Y. Biotransformation of bioactive natural products for pharmaceutical lead compounds. Curr. Org. Chem. 2010, 14, 1400–1406. [Google Scholar] [CrossRef]
- Salter, R.; Beshore, D.C.; Colletti, S.L.; Evans, L.; Gong, Y.; Helmy, R.; Liu, Y.; Maciolek, C.M.; Martin, G.; Pajkovic, N. Microbial biotransformation-an important tool for the study of drug metabolism. Xenobiotica 2019, 49, 877–886. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, P.; Zhao, Y.; Wang, K.; Chang, X.; Bai, Y.; Zhang, D.; Yang, L. A microbial transformation model for simulating mammal metabolism of artemisinin. Molecules 2019, 24, 315. [Google Scholar] [CrossRef]
- Murphy, C.D. Drug metabolism in microorganisms. Biotechnol. Lett. 2015, 37, 19–28. [Google Scholar] [CrossRef]
- Hosseinzadeh, H.; Nassiri-Asl, M. Pharmacological effects of Glycyrrhiza spp. and its bioactive constituents: Update and review. Phytother. Res. 2015, 29, 1868–1886. [Google Scholar] [CrossRef]
- Ji, S.; Li, Z.; Song, W.; Wang, Y.; Liang, W.; Li, K.; Tang, S.; Wang, Q.; Qiao, X.; Zhou, D. Bioactive constituents of Glycyrrhiza uralensis (licorice): Discovery of the effective components of a traditional herbal medicine. J. Nat. Prod. 2016, 79, 281–292. [Google Scholar] [CrossRef]
- McKee, T.C.; Bokesch, H.R.; McCormick, J.L.; Rashid, M.A.; Spielvogel, D.; Gustafson, K.R.; Alavanja, M.M.; Cardellina, J.H.; Boyd, M.R. Isolation and characterization of new anti-HIV and cytotoxic leads from plants, marine, and microbial organisms. J. Nat. Prod. 1997, 60, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Won, D.-H.; Shin, B.-K.; Han, J.-H. Synthesis and the absolute configurations of isoflavanone enantiomers. J. Appl. Biol. Chem. 2008, 51, 17–19. [Google Scholar] [CrossRef][Green Version]
- Tangdenpaisal, K.; Songthammawat, P.; Akkarasereenon, K.; Chuayboonsong, K.; Ruchirawat, S.; Ploypradith, P. Total synthesis of palodesangren B trimethyl ether and D dimethyl ether via a late-stage formation of 2H-pyran-2-one of the tetrahydrobenzo[c]pyranochromenone core. J. Org. Chem. 2019, 84, 13410–13429. [Google Scholar] [CrossRef] [PubMed]
- Meksuriyen, D.; Cordell, G.A. Retrodihydrochalcones from Dracaena loureiri. J. Nat. Prod. 1988, 51, 1129–1135. [Google Scholar] [CrossRef]
- Abegaz, B.M.; Ngadjui, B.T.; Dongo, E.; Ngameni, B.; Nindi, M.N.; Bezabih, M. Chalcones and other constituents of Dorstenia prorepens and Dorstenia zenkeri. Phytochemistry 2002, 59, 877–883. [Google Scholar] [CrossRef]
- Akihisa, T.; Tokuda, H.; Hasegawa, D.; Ukiya, M.; Kimura, Y.; Enjo, F.; Suzuki, T.; Nishino, H. Chalcones and other compounds from the exudates of Angelica keiskei and their cancer chemopreventive effects. J. Nat. Prod. 2006, 69, 38–42. [Google Scholar] [CrossRef]
- Torres, S.L.; Arruda, M.S.P.; Arruda, A.C.; Müller, A.H.; Silva, S.C. Flavonoids from Brosimum acutifolium. Phytochemistry 2000, 53, 1047–1050. [Google Scholar] [CrossRef]
- Won, T.H.; Song, I.-H.; Kim, K.-H.; Yang, W.-Y.; Lee, S.K.; Oh, D.-C.; Oh, W.-K.; Oh, K.-B.; Shin, J. Bioactive metabolites from the fruits of Psoralea corylifolia. J. Nat. Prod. 2015, 78, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yan, G.R.; Pan, S.L.; Wang, H.Y.; Hou, A.J. New Isoprenylated 2-arylbenzofurans and pancreatic lipase inhibitory constituents from Artocarpus nitidus. Chem. Biodivers. 2009, 6, 2209–2216. [Google Scholar] [CrossRef]
- Zhao, M.-B.; Zhou, S.-X.; Zhang, Q.-Y.; Wei, W.-F.; Li, M.-H.; Xing, J.-Y.; Jiang, Y.; Tu, P.-F. Prenylated benzoic acid derivatives from the stem of Euodia lepta. Nat. Prod. Res. 2017, 31, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Takashima, J.; Komiyama, K.; Ishiyama, H.; Kobayashi, J.i.; Ohsaki, A. Brosimacutins JM, four new flavonoids from Brosimum acutifolium and their cytotoxic activity. Planta Med. 2005, 71, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Takashima, J.; Ohsaki, A. Brosimacutins A-I, nine new flavonoids from Brosimum acutifolium. J. Nat. Prod. 2002, 65, 1843–1847. [Google Scholar] [CrossRef]
- Suri, J.; Gupta, G.; Dhar, K.; Atal, C. Bavachromanol: A new chalcone from the seeds of Psoralea corylifolia. Phytochemistry 1980, 19, 336–337. [Google Scholar] [CrossRef]
- Shah, P.A.; Wiemer, D.F. Synthesis of bavachromanol from resorcinol via a tandem cationic cascade/EAS sequence. Tetrahedron Lett. 2018, 59, 1363–1365. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Jiang, M.; Zhao, S.; Yang, S.; Lin, X.; He, X.; Wei, X.; Song, Q.; Li, R.; Fu, C.; Zhang, J. An “essential herbal medicine”-licorice: A review of phytochemicals and its effects in combination preparations. J. Ethnopharmacol. 2020, 249, 112439. [Google Scholar] [CrossRef]
- Chung, W.T.; Lee, S.H.; Dai Kim, J.; Sung, N.S.; Hwang, B.; Lee, S.Y.; Yu, C.Y.; Lee, H.Y. Effect of the extracts from Glycyrrhiza uralensis Fisch on the growth characteristics of human cell lines: Anti-tumor and immune activation activities. Cytotechnology 2001, 37, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Park, K.K.; Lim, S.S.; Park, J.H.Y.; Chung, W.Y. Effects of the licorice extract against tumor growth and cisplatin-induced toxicity in a mouse xenograft model of colon cancer. Biol. Pharm. Bull. 2007, 30, 2191–2195. [Google Scholar] [CrossRef]
- Rathi, S.; Suthar, M.; Patel, P.; Bhaskar, V.; Rajgor, N. In-vitro cytotoxic screening of Glycyrrhiza glabra L. (Fabaceae): A natural anticancer drug. J. Young Pharm. 2009, 1, 239. [Google Scholar] [CrossRef]
- Fukuchi, K.; Okudaira, N.; Adachi, K.; Odai-Ide, R.; Watanabe, S.; Ohno, H.; Yamamoto, M.; Kanamoto, T.; Terakubo, S.; Nakashima, H. Antiviral and antitumor activity of licorice root extracts. In Vivo 2016, 30, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hiramoto, K.; Ma, N.; Yoshikawa, N.; Ohnishi, S.; Murata, M.; Kawanishi, S. Glycyrrhizin attenuates carcinogenesis by inhibiting the inflammatory response in a murine model of colorectal cancer. Int. J. Mol. Sci. 2021, 22, 2609. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R.; Cox, G.M.; Lee, J.Y.; Kauffman, C.A.; De Repentigny, L.; Chapman, S.W.; Morrison, V.A.; Pappas, P.; Hiemenz, J.W.; Stevens, D.A.; et al. The impact of culture isolation of Aspergillus species: A hospital-based survey of aspergillosis. Clin. Infect. Dis. 2001, 33, 1824–1833. [Google Scholar] [CrossRef]
- Chen, G.; Song, Y.; Ge, H.; Ren, J.; Yang, X.; Li, J. Biotransformation of 20(S)-protopanaxatriol by Aspergillus niger and the cytotoxicity of the resulting metabolites. Phytochem. Lett. 2015, 11, 111–115. [Google Scholar] [CrossRef]
- Parshikov, I.A.; Woodling, K.A.; Sutherland, J.B. Biotransformations of organic compounds mediated by cultures of Aspergillus niger. Appl. Microbiol. Biotechnol. 2015, 99, 6971–6986. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, S.; Wang, Y.; Luo, K.; Wang, Y.; Cai, Y. Liquiritigenin inhibits tumor growth and vascularization in a mouse model of HeLa cells. Molecules 2012, 17, 7206–7216. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Chemopreventive effectsof licorice and its components. Curr. Pharmacol. Rep. 2015, 1, 60–71. [Google Scholar] [CrossRef]
- Tang, Z.-H.; Li, T.; Tong, Y.-G.; Chen, X.-J.; Chen, X.-P.; Wang, Y.-T.; Lu, J.-J. A systematic review of the anticancer properties of compounds isolated from licorice (Gancao). Planta Med. 2015, 81, 1670–1687. [Google Scholar] [CrossRef]
- Hatano, T.; Kagawa, H.; Yasuhara, T.; Okuda, T. Two new flavonoids and other constituents in licorice root: Their relative astringency and radical scavenging effects. Chem. Pharm. Bull. 1988, 36, 2090–2097. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.; Paudel, S.; Yoon, G.; Cheon, S.H. Concise synthesis of licochalcone C and its regioisomer, licochalcone H. Arch. Pharm. Res. 2013, 36, 1432–1436. [Google Scholar] [CrossRef]
- Grealis, J.P.; Müller-Bunz, H.; Ortin, Y.; Casey, M.; McGlinchey, M.J. Synthesis of isobavachalcone and some organometallic derivatives. Eur. J. Org. Chem. 2013, 2, 332–347. [Google Scholar] [CrossRef]
- Hou, S.-S.; Chen, S.-Y.; Yang, M.-Z.; Li, X.-M.; Lu, D.-Y.; Zhang, J.; Chen, L. Isolation and identification of echinatin from cultured cells of Glycyrrhiza uralensis. J. Integr. Plant Biol. 1993, 35, 567–571. [Google Scholar]
- Pistelli, L.; Spera, K.; Flamini, G.; Mele, S.; Morelli, I. Isoflavonoids and chalcones from Anthyllis hermanniae. Phytochemistry 1996, 42, 1455–1458. [Google Scholar] [CrossRef]
- Smith, R.V.; Rosazza, J.P. Microbial models of mammalian metabolism. J. Pharm. Sci. 1975, 64, 1737–1759. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 5 | 6 | 7 | |||
|---|---|---|---|---|---|---|
| C/H | δH a (J/Hz) | δC b | δH a (J/Hz) | δC b | δH a (J/Hz) | δC b |
| 2 | 4.65 t (8.3) | 70.5 | 8.04 s | 153.5 | 8.03 s | 153.6 |
| 3 | 4.53 dd (10.0, 5.4) 4.23 dd (10.0, 5.4) | 46.9 | 127.2 | 123.3 | ||
| 4 | 197.9 | 180.9 | 180.8 | |||
| 5 | 156.0 | 162.4 | 162.4 | |||
| 6 | 5.99 s | 95.4 | 6.22 d (2.1) | 98.7 | 6.22 d (2.0) | 98.8 |
| 7 | 157.0 | 164.6 | 164.5 | |||
| 8 | 125.3 | 6.34 d (2.1) | 93.4 | 6.33 d (2.0) | 93.5 | |
| 9 | 149.2 | 158.3 | 158.3 | |||
| 10 | 102.0 | 104.9 | 104.9 | |||
| 1′ | 115.7 | 123.6 | 122.7 | |||
| 2′ | 150.7 | 6.81 d (2.1) | 122.3 | 6.75 d (2.1) | 120.8 | |
| 3′ | 110.9 | 143.9 | 141.2 | |||
| 4′ | 153.3 | 145.1 | 145.3 | |||
| 5′ | 6.34 d (8.3) | 108.5 | 122.0 | 120.6 | ||
| 6′ | 6.91 d (8.3) | 129.3 | 6.92 d (2.1) | 114.2 | 6.86 s | 113.8 |
| 1″ | 6.69 d (10.0) | 116.6 | 2.97 dd (14.0, 1.9) 2.68 dd (14.0, 10.2) | 32.8 | 3.03 dd (16.7, 5.4) 2.74 dd (16.7, 7.3) | 30.7 |
| 2″ | 5.68 d (10.0) | 129.1 | 3.65 dd (10.2, 1.9) | 79.2 | 3.79 m | 69.2 |
| 3″ | 75.1 | 72.5 | 77.4 | |||
| 4″ | 1.40 s | 26.5 | 1.25 s | 24.3 | 1.39 s | 24.4 |
| 5″ | 1.40 s | 26.4 | 1.25 s | 23.6 | 1.31 s | 19.6 |
| No. | 10 a | 11 a | 12 b | 13 b | ||||
|---|---|---|---|---|---|---|---|---|
| C/H | δH c (J/Hz) | δC d | δH c (J/Hz) | δC d | δH c (J/Hz) | δC d | δH c (J/Hz) | δC d |
| α | 3.17 t (7.6) | 38.9 | 7.71 s | 117.3 | 3.18 t (7.4) | 40.0 | 3.17 t (7.4) | 39.9 |
| β | 2.81 t (7.6) | 29.3 | 7.71 s | 143.6 | 2.97 t (7.4) | 29.9 | 2.97 t (7.4) | 29.9 |
| C=O | 203.9 | 191.1 | 204.0 | 203.9 | ||||
| 1 | 131.0 | 125.6 | 133.2 | 133.3 | ||||
| 2,6 | 7.04 d (8.6) | 129.2 | 7.73 d (8.6) | 131.0 | 7.10 d (8.2) | 129.6 | 7.11 d (8.3) | 129.6 |
| 3,5 | 6.66 d (8.6) | 115.0 | 6.84 d (8.6) | 115.9 | 6.77 d (8.2) | 115.5 | 6.77 d (8.3) | 115.5 |
| 4 | 155.5 | 160.5 | 154.2 | 154.1 | ||||
| 1′ | 111.5 | 112.0 | 112.8 | 112.3 | ||||
| 2′ | 162.3 | 163.7 | 162.3 | 163.0 | ||||
| 3′ | 115.2 | 115.4 | 116.8 | 109.3 | ||||
| 4′ | 13.13 s (OH) | 162.3 | 14.05 s (OH) | 163.7 | 13.10 s (OH) | 162.6 | 13.17 s (OH) | 160.8 |
| 5′ | 6.41 d (8.9) | 107.8 | 6.47 d (8.9) | 107.9 | 6.41 d (8.9) | 108.7 | 6.32 d (9.0) | 109.3 |
| 6′ | 7.63 d (8.9) | 129.8 | 7.97 d (8.9) | 129.5 | 7.53 d (8.9) | 129.6 | 7.51 d (9.0) | 128.9 |
| 1″ | 2.50 (overlay) | 16.6 | 2.53 m | 16.7 | 2.74 t (6.8) | 15.5 | 2.69 t (6.8) | 16.4 |
| 2″ | 1.52 m | 37.4 | 1.55 m | 37.4 | 1.77 t (6.8) | 41.6 | 1.81 t (6.8) | 32.0 |
| 3″ | 73.9 | 73.9 | 76.3 | 75.9 | ||||
| 4″ | 1.12 s | 25.0 | 1.14 s | 25.0 | 1.21 s | 24.7 | 1.34 s | 26.8 |
| 5″ | 1.12 s | 25.0 | 1.14 s | 25.0 | 1.21 s | 24.7 | 1.34 s | 26.8 |
| 1′′′ | 3.13 s | 48.4 | 3.15 s | 48.4 | 3.56 q (7.0) | 57.6 | ||
| 2′′′ | 1.29 t (7.0) | 15.6 | ||||||
| No. | 14 | 15 | 16 | 17 | 19 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| C/H | δH a (J/Hz) | δC b | δH a (J/Hz) | δC b | δH a (J/Hz) | δC b | δH a (J/Hz) | δC b | δH a (J/Hz) | δC b |
| α | 3.12 t (7.3) | 44.9 | 3.23 t (7.7) | 39.5 | 3.23 t (7.1) | 39.0 | 7.72 s | 117.5 | 3.17 t (7.7) | 38.9 |
| β | 2.75 t (7.3) | 29.5 | 2.82 t (7.7) | 29.2 | 2.82 t (7.1) | 29.1 | 7.72 s | 143.6 | 2.81 t (7.7) | 29.3 |
| C=O | 198.6 | 204.6 | 204.6 | 191.1 | 203.8 | |||||
| 1 | 131.7 | 130.9 | 130.8 | 125.7 | 131.0 | |||||
| 2,6 | 7.00 d (8.1) | 128.9 | 7.06 d (8.0) | 129.2 | 7.06 d (8.3) | 129.1 | 7.74 d (8.0) | 131.1 | 7.06 d (8.2) | 129.2 |
| 3,5 | 6.66 d (8.1) | 115.0 | 6.67 d (8.0) | 115.1 | 6.67 d (8.3) | 115.0 | 6.84 d (8.0) | 125.7 | 6.67 d (8.2) | 115.0 |
| 4 | 155.3 | 155.6 | 155.5 | 160.4 | 155.5 | |||||
| 1′ | 118.2 | 113.7 | 112.0 | 112.0 | 111.4 | |||||
| 2′ | 155.2 | 159.4 | 162.1 | 163.8 | 162.3 | |||||
| 3′ | 108.1 | 113.3 | 107.8 | 115.8 | 115.6 | |||||
| 4′ | 161.2 | 12.80 s (OH) | 166.8 | 13.18 s (OH) | 159.3 | 14.06 s (OH) | 163.8 | 13.13 s (OH) | 162.3 | |
| 5′ | 6.40 d (8.6) | 107.0 | 6.41 d (8.7) | 101.7 | 6.34 d (8.9) | 108.5 | 6.43 d (9.0) | 108.0 | 6.40 d (8.8) | 107.9 |
| 6′ | 7.38 d (8.6) | 129.1 | 7.81 d (8.7) | 132.9 | 7.73 d (8.9) | 129.7 | 7.98 d (9.0) | 129.5 | 7.63 d (8.8) | 129.7 |
| 1″ | 2.55 t (6.7) | 17.0 | 3.04 d (8.8) | 26.5 | 2.78 dd (17.0, 5.1) 2.46 dd (17.0, 7.0) | 25.2 | 2.57 m | 17.5 | 2.54 m | 17.4 |
| 2″ | 1.76 t (6.7) | 31.1 | 4.71 t (8.8) | 91.2 | 3.67 t (5.1) | 67.0 | 1.50 m | 42.3 | 1.47 m | 42.3 |
| 3″ | 74.7 | 70.1 | 78.4 | 69.0 | 69.0 | |||||
| 4″ | 1.29 s | 26.5 | 1.13 s | 25.8 | 1.27 s | 25.2 | 1.14 s | 29.2 | 1.13 s | 29.1 |
| 5″ | 1.29 s | 26.5 | 1.12 s | 24.8 | 1.20 s | 20.9 | 1.14 s | 29.2 | 1.13 s | 29.1 |
| Compound | IC50 ± SD (μM) | Compound | IC50 ± SD (μM) | ||||
|---|---|---|---|---|---|---|---|
| A375P | HT-29 | MCF-7 | A375P | HT-29 | MCF-7 | ||
| 1 | 8.09 ± 0.35 | 7.54 ± 0.70 | 9.20 ± 0.13 | 12 | 4.35 ± 0.35 | 5.77 ± 0.28 | 10.07 ± 1.11 |
| 2 | 25.48 ± 1.53 | 25.98 ± 0.72 | 26.99 ± 0.77 | 13 | 27.38 ± 0.67 | 60.15 ± 1.15 | 33.87 ± 1.80 |
| 3 | >100 | >100 | >100 | 14 | 31.90 ± 1.27 | 77.65 ± 1.33 | 47.43 ± 1.91 |
| 4 | 5.21 ± 0.39 | 21.34 ± 1.40 | 20.94 ± 0.19 | 15 | 66.57 ± 1.91 | >100 | >100 |
| 5 | >100 | >100 | >100 | 16 | 28.91 ± 1.99 | >100 | 82.77 ± 1.82 |
| 6 | >100 | >100 | >100 | 17 | 42.98 ± 0.62 | >100 | 43.58 ± 1.39 |
| 7 | >100 | >100 | >100 | 18 | 70.05 ± 1.27 | >100 | >100 |
| 8 | 29.41 ± 1.86 | 57.17 ± 4.28 | 59.44 ± 0.39 | 19 | 21.92 ± 2.26 | 70.39 ± 1.53 | 68.26 ± 2.28 |
| 9 | >100 | >100 | >100 | 20 | 57.60 ± 0.67 | >100 | 85.25 ± 1.51 |
| 10 | 14.20 ± 0.40 | 73.39 ± 0.48 | 47.23 ± 1.07 | 21 | >100 | >100 | >100 |
| 11 | 29.38 ± 0.59 | >100 | 61.75 ± 1.57 | DZ | 2.10 ± 0.06 | 10.13 ± 0.27 | 2.33 ± 0.05 |
| Substrate | Substrate Amount (mg/Flask) | Number of Flasks | Total Extract Amount (g) |
|---|---|---|---|
| 1 | 3 | 8 | 0.22 |
| 2 | 3 | 13 | 0.47 |
| 3 | 3 | 15 | 0.54 |
| 4 | 3 | 36 | 1.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Han, F.; Lee, I.-S. Biotransformation of the Phenolic Constituents from Licorice and Cytotoxicity Evaluation of Their Metabolites. Int. J. Mol. Sci. 2021, 22, 10109. https://doi.org/10.3390/ijms221810109
Xiao Y, Han F, Lee I-S. Biotransformation of the Phenolic Constituents from Licorice and Cytotoxicity Evaluation of Their Metabolites. International Journal of Molecular Sciences. 2021; 22(18):10109. https://doi.org/10.3390/ijms221810109
Chicago/Turabian StyleXiao, Yina, Fubo Han, and Ik-Soo Lee. 2021. "Biotransformation of the Phenolic Constituents from Licorice and Cytotoxicity Evaluation of Their Metabolites" International Journal of Molecular Sciences 22, no. 18: 10109. https://doi.org/10.3390/ijms221810109
APA StyleXiao, Y., Han, F., & Lee, I.-S. (2021). Biotransformation of the Phenolic Constituents from Licorice and Cytotoxicity Evaluation of Their Metabolites. International Journal of Molecular Sciences, 22(18), 10109. https://doi.org/10.3390/ijms221810109