
Metformin and Dichloroacetate Suppress Proliferation of Liver Cancer Cells by Inhibiting mTOR Complex 1

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
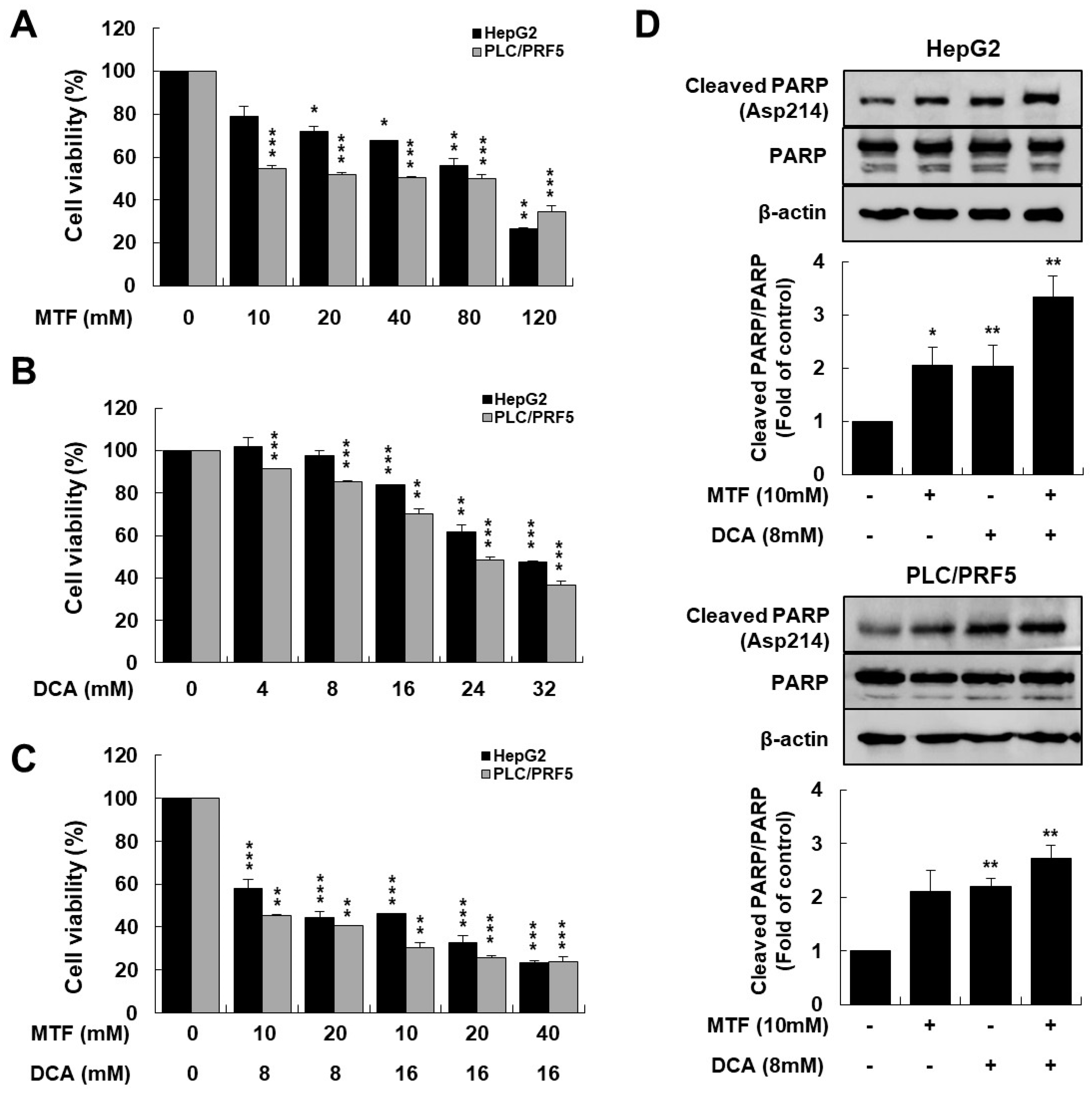
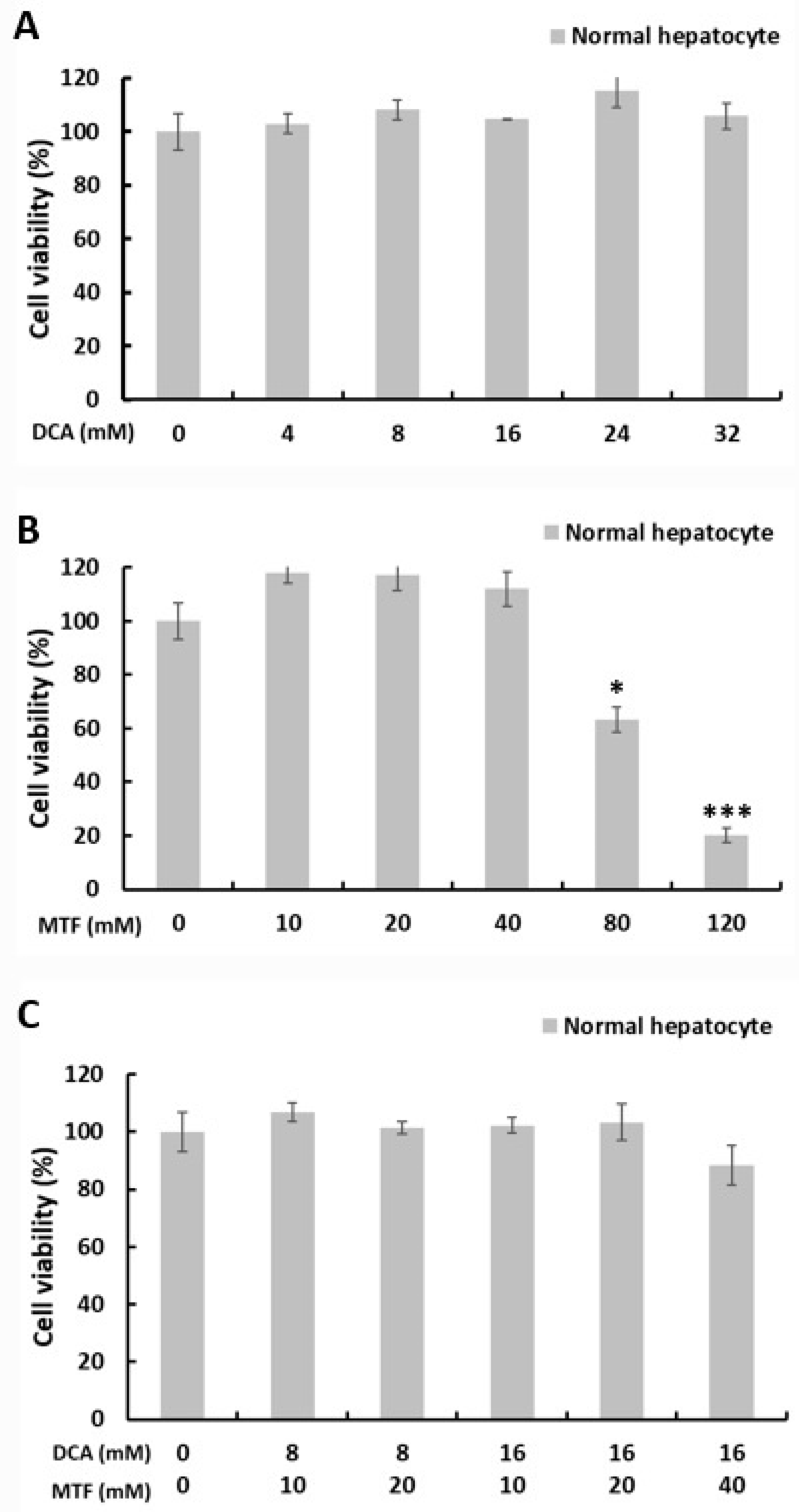
2.1. Metformin and DCA Effectively Induced Apoptosis in Liver Cancer Cells
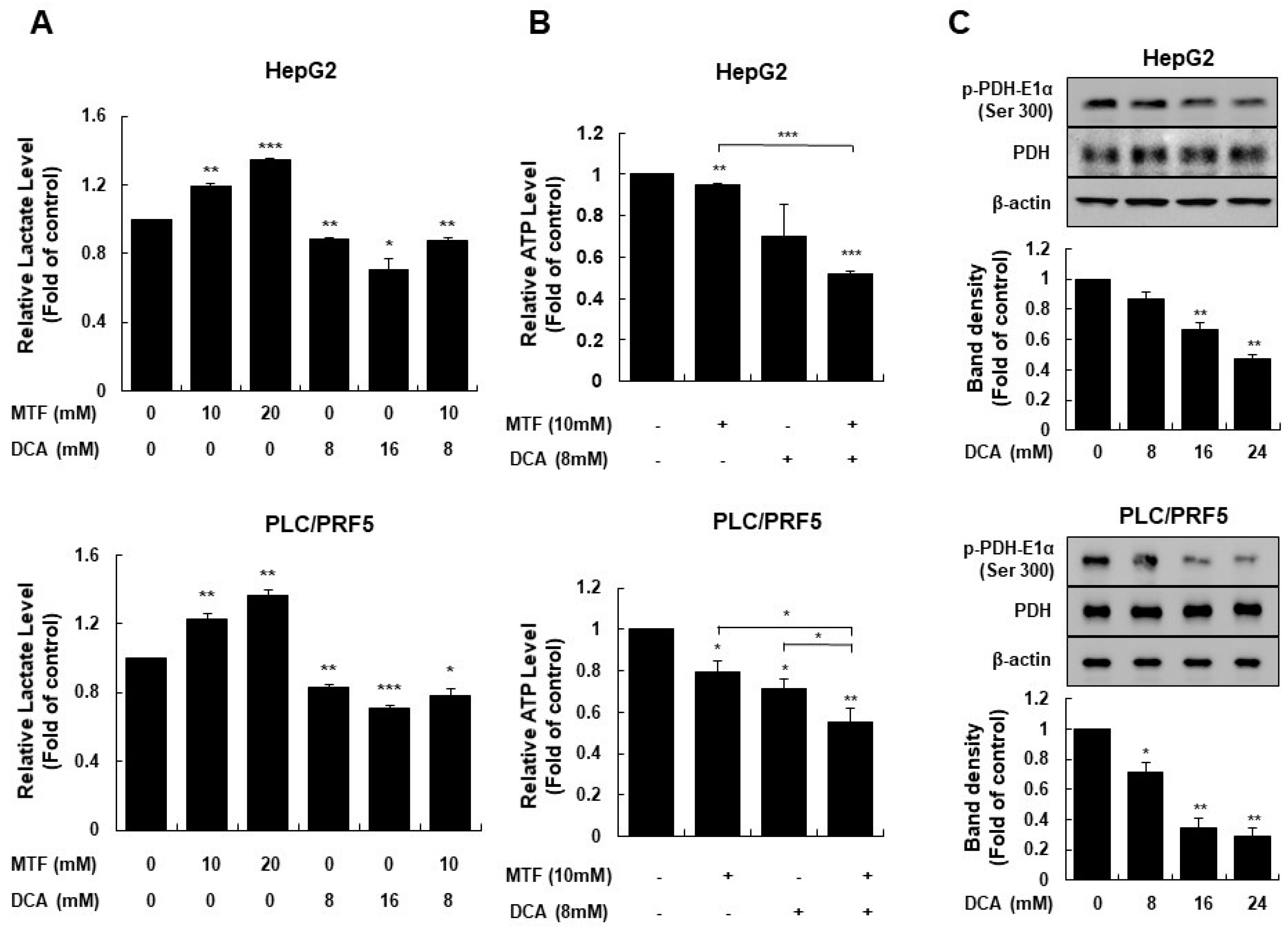
2.2. Metformin Sensitized Liver Cancer Cells to DCA through Increasing Glycolysis
2.3. Metformin and DCA Effectively Suppressed mTORC1 Expression by Inhibiting PI3K/Akt/mTORC1 Signaling and Upregulating REDD1
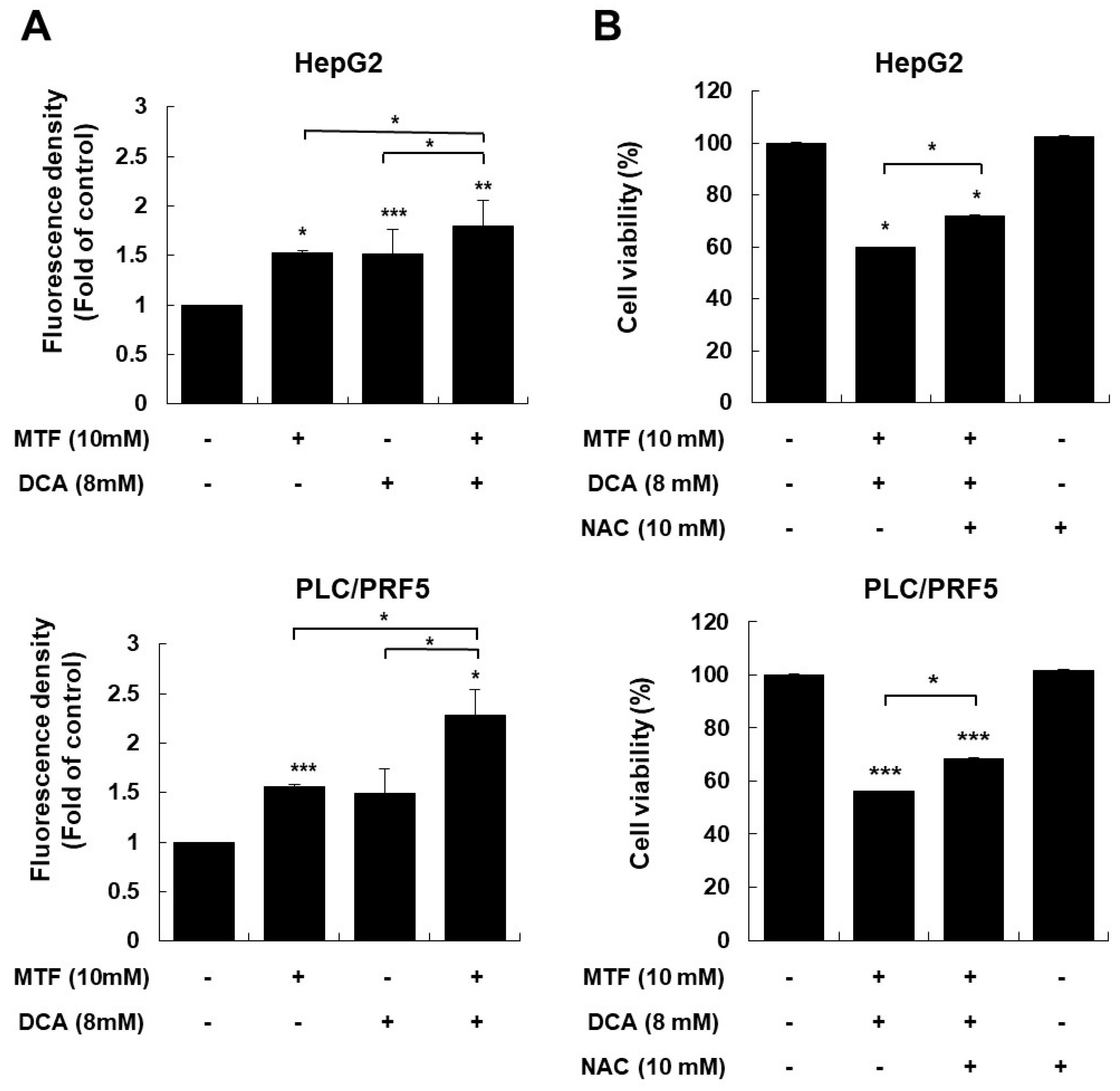
2.4. Modulating ROS Is Important for Anticancer Effects of the Metformin and DCA Treatment
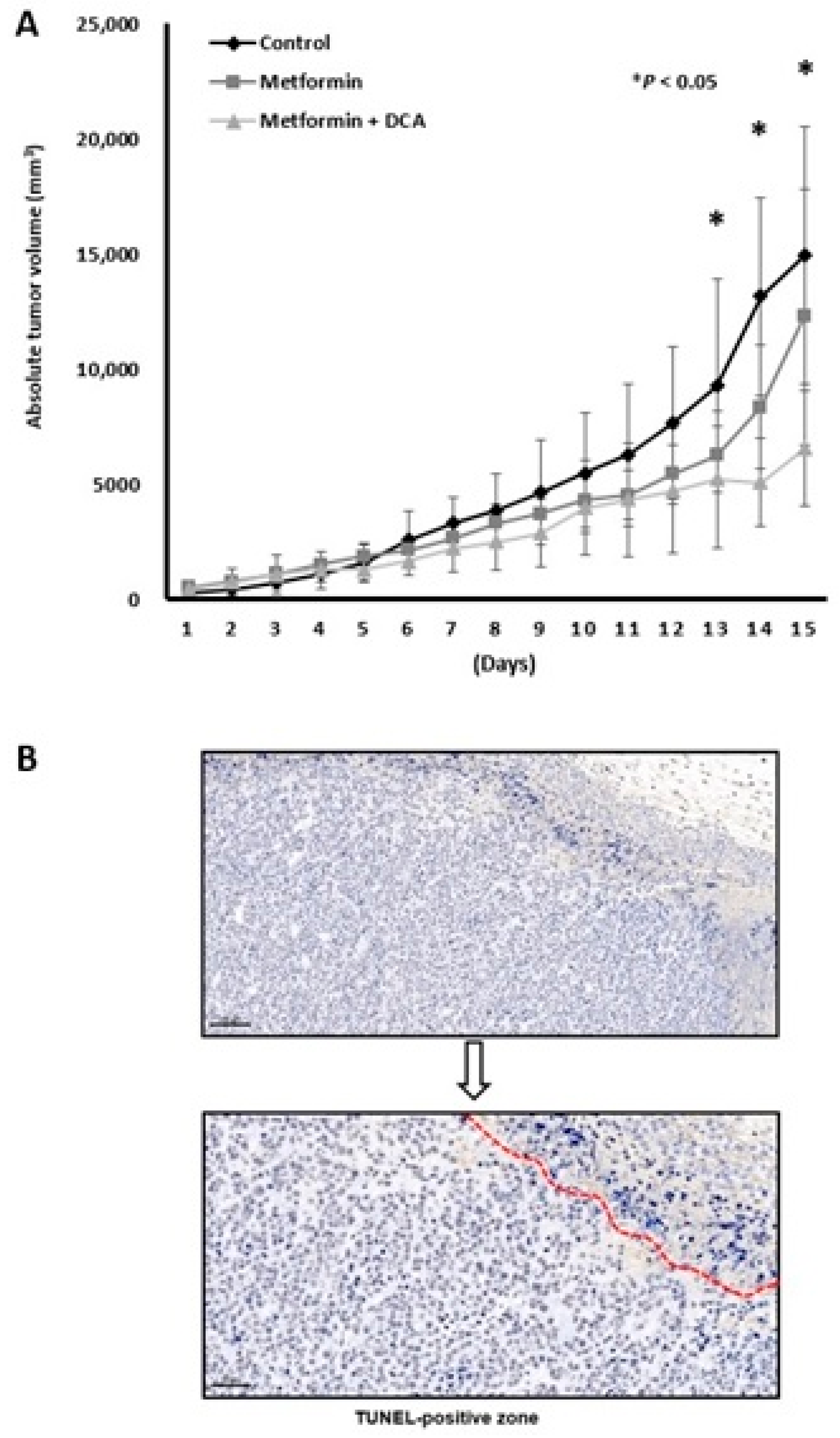
2.5. Anticancer Effect of a Combination Treatment of Metformin and DCA in Murine Models
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Chemicals
4.2. Cell Viability Assay
4.3. Western Blot Analysis
4.4. Flow Cytometry with Annexin V-FITC Staining
4.5. Extracellular Lactate Assay and Intracellular ATP Assay
4.6. Assessment of Intracellular and Mitochondrial Reactive Oxygen Species Generation
4.7. In Vivo Subcutaneous Xenograft Model
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yoon, S.M.; Kim, S.Y.; Lim, Y.S.; Kim, K.M.; Shim, J.H.; Lee, D.; An, J.; Jung, J.; Kim, J.H.; Lee, H.C. Stereotactic body radiation therapy for small (≤5 cm) hepatocellular carcinoma not amenable to curative treatment: Results of a single-arm, phase II clinical trial. Clin. Mol. Hepatol. 2020, 26, 506–515. [Google Scholar] [CrossRef]
- Ahn, K.S.; Kang, K.J. Appropriate treatment modality for solitary small hepatocellular carcinoma: Radiofrequency ablation vs. resection vs. transplantation? Clin. Mol. Hepatol. 2019, 25, 354–359. [Google Scholar] [CrossRef] [Green Version]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, S.S.; Chang, D.R.; Kim, H.; Kim, M.J. Comparison of LI-RADS 2018 and KLCA-NCC 2018 for noninvasive diagnosis of hepatocellular carcinoma using magnetic resonance imaging. Clin. Mol. Hepatol. 2020, 26, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y. Diagnosis of hepatocellular carcinoma: Which MRI contrast agent? Which diagnostic criteria? Clin. Mol. Hepatol. 2020, 26, 309–311. [Google Scholar] [CrossRef]
- Yang, J.D. Detect or not to detect very early stage hepatocellular carcinoma? The western perspective. Clin. Mol. Hepatol. 2019, 25, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanouneh, I.A.; Alkhouri, N.; Singal, A.G. Hepatocellular carcinoma surveillance in the 21st century: Saving lives or causing harm? Clin. Mol. Hepatol. 2019, 25, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Kim, S.Y.; Tang, A.; Lee, J.M. Comparison of international guidelines for noninvasive diagnosis of hepatocellular carcinoma: 2018 update. Clin. Mol. Hepatol. 2019, 25, 245–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.Y.; Park, M.S.; Aljoqiman, K.S.; Choi, J.Y.; Kim, M.J. Gadoxetic acid-enhanced magnetic resonance imaging: Hepatocellular carcinoma and mimickers. Clin. Mol. Hepatol. 2019, 25, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Na, S.K.; Song, B.C. Development and surveillance of hepatocellular carcinoma in patients with sustained virologic response after antiviral therapy for chronic hepatitis C. Clin. Mol. Hepatol. 2019, 25, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.H. Should you advocate for hepatocellular carcinoma surveillance in patients with alcohol-related liver disease or non-alcoholic fatty liver disease? Clin. Mol. Hepatol. 2020, 26, 183–184. [Google Scholar] [CrossRef]
- Miyayama, S. Ultraselective conventional transarterial chemoembolization: When and how? Clin. Mol. Hepatol. 2019, 25, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Sohn, W.; Lee, H.W.; Lee, S.; Lim, J.H.; Lee, M.W.; Park, C.H.; Yoon, S.K. Obesity and the risk of primary liver cancer: A systematic review and meta-analysis. Clin. Mol. Hepatol. 2021, 27, 157–174. [Google Scholar] [CrossRef]
- Esfeh, J.M.; Hajifathalian, K.; Ansari-Gilani, K. Sensitivity of ultrasound in detecting hepatocellular carcinoma in obese patients compared to explant pathology as the gold standard. Clin. Mol. Hepatol. 2020, 26, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N. Metabolic disease as a risk of hepatocellular carcinoma. Clin. Mol. Hepatol. 2021, 27, 87–90. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, B.K.; Kim, S.U.; Park, J.Y.; Ahn, S.H.; Seong, J.S.; Han, K.H.; Kim, D.Y. A survey on transarterial chemoembolization refractoriness and a real-world treatment pattern for hepatocellular carcinoma in Korea. Clin. Mol. Hepatol. 2020, 26, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Kim, D.Y. Optimal sequence of systemic therapy after sorafenib failure in patients with hepatocellular carcinoma. Clin. Mol. Hepatol. 2020, 26, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, Y.B.; Kim, M.A.; Jang, H.; Oh, H.; Kim, S.W.; Cho, E.J.; Lee, K.H.; Lee, J.H.; Yu, S.J.; et al. Effectiveness of nivolumab versus regorafenib in hepatocellular carcinoma patients who failed sorafenib treatment. Clin. Mol. Hepatol. 2020, 26, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Bannangkoon, K.; Hongsakul, K.; Tubtawee, T.; Piratvisuth, T. Safety margin of embolized area can reduce local recurrence of hepatocellular carcinoma after superselective transarterial chemoembolization. Clin. Mol. Hepatol. 2019, 25, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Torimura, T.; Iwamoto, H. Optimizing the management of intermediate-stage hepatocellular carcinoma: Current trends and prospects. Clin. Mol. Hepatol. 2021, 27, 236–245. [Google Scholar] [CrossRef]
- Ogasawara, S.; Ooka, Y.; Koroki, K.; Maruta, S.; Kanzaki, H.; Kanayama, K.; Kobayashi, K.; Kiyono, S.; Nakamura, M.; Kanogawa, N.; et al. Switching to systemic therapy after locoregional treatment failure: Definition and best timing. Clin. Mol. Hepatol. 2020, 26, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Plank, L.D.; Suk, K.T.; Park, Y.E.; Lee, J.; Choi, J.H.; Heo, N.Y.; Park, J.; Kim, T.O.; Moon, Y.S.; et al. Trends in the prevalence of chronic liver disease in the Korean adult population, 1998–2017. Clin. Mol. Hepatol. 2020, 26, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Jung, Y.W.; Kim, B.K.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Han, K.H.; Kim, Y.Y.; Choi, J.Y.; Kim, S.U. Risk assessment of hepatocellular carcinoma development for indeterminate hepatic nodules in patients with chronic hepatitis B. Clin. Mol. Hepatol. 2019, 25, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N. Long-term prognosis and management of hepatocellular carcinoma after curative treatment. Clin. Mol. Hepatol. 2020, 26, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H. How long should physicians follow up with patients after curative treatment for hepatocellular carcinoma? Clin. Mol. Hepatol. 2019, 26, 579–581. [Google Scholar] [CrossRef]
- Kim, J.; Kang, W.; Sinn, D.H.; Gwak, G.Y.; Paik, Y.H.; Choi, M.S.; Lee, J.H.; Koh, K.C.; Paik, S.W. Substantial risk of recurrence even after 5 recurrence-free years in early-stage hepatocellular carcinoma patients. Clin. Mol. Hepatol. 2020, 26, 516–528. [Google Scholar] [CrossRef]
- Moriya, K.; Namisaki, T.; Sato, S.; Furukawa, M.; Douhara, A.; Kawaratani, H.; Kaji, K.; Shimozato, N.; Sawada, Y.; Saikawa, S.; et al. Bi-monthly hepatic arterial infusion chemotherapy as a novel strategy for advanced hepatocellular carcinoma in decompensated cirrhotic patients. Clin. Mol. Hepatol. 2019, 25, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Bae, S.H. Phosphoinositide 3-kinase inhibitors are effective therapeutic drugs for the treatment of hepatocellular carcinoma? Clin. Mol. Hepatol. 2020, 26, 577–578. [Google Scholar] [CrossRef]
- Lee, S.; Choi, E.J.; Cho, E.J.; Lee, Y.B.; Lee, J.H.; Yu, S.J.; Yoon, J.H.; Kim, Y.J. Inhibition of PI3K/Akt signaling suppresses epithelial-to-mesenchymal transition in hepatocellular carcinoma through the Snail/GSK-3/beta-catenin pathway. Clin. Mol. Hepatol. 2020, 26, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Song, J. Targeting epithelial-mesenchymal transition pathway in hepatocellular carcinoma. Clin. Mol. Hepatol. 2020, 26, 484–486. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.; Chen, Q.; Jiang, F.; Yu, D.; Mao, Q.; Xia, W.; Shi, R.; Wang, J.; Xu, L. Diisopropylamine dichloroacetate enhances radiosensitization in esophageal squamous cell carcinoma by increasing mitochondria-derived reactive oxygen species levels. Oncotarget 2016, 7, 68170–68178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Xiong, X.; Huang, G.; Liu, J.; Sheng, S.; Wang, H.; Qin, W. Dichloroacetate enhances adriamycin-induced hepatoma cell toxicity in vitro and in vivo by increasing reactive oxygen species levels. PLoS ONE 2014, 9, e92962. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.P.; Poff, A.M.; Koutnik, A.P.; D’Agostino, D.P. Complex I inhibition augments dichloroacetate cytotoxicity through enhancing oxidative stress in VM-M3 glioblastoma cells. PLoS ONE 2017, 12, e0180061. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Ni, Z.; Zhang, Y.; Zeng, Y.; Yan, X.; Huang, Y.; He, J.; Lyu, X.; Wu, Y.; et al. Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells. Oncotarget 2016, 7, 59458–59470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Park, K.G. Metabolic roles of AMPK and metformin in cancer cells. Mol. Cells 2013, 36, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, E.A.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutendra, G.; Michelakis, E.D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 2013, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Troncone, M.; Cargnelli, S.M.; Villani, L.A.; Isfahanian, N.; Broadfield, L.A.; Zychla, L.; Wright, J.; Pond, G.; Steinberg, G.R.; Tsakiridis, T. Targeting metabolism and AMP-activated kinase with metformin to sensitize non-small cell lung cancer (NSCLC) to cytotoxic therapy: Translational biology and rationale for current clinical trials. Oncotarget 2017, 8, 57733–57754. [Google Scholar] [CrossRef] [Green Version]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.Y.; Storz, P. Detecting reactive oxygen species by immunohistochemistry. Methods Mol. Biol. 2015, 1292, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, N.C.; Vega-Rubin-de-Celis, S.; Xie, X.J.; Castrillon, D.H.; Kabbani, W.; Brugarolas, J. Cell-type-dependent regulation of mTORC1 by REDD1 and the tumor suppressors TSC1/TSC2 and LKB1 in response to hypoxia. Mol. Cell Biol. 2011, 31, 1870–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Chao, X.; Qian, H.; Wang, S.; Fulte, S.; Ding, W.X. Autophagy and liver cancer. Clin. Mol. Hepatol. 2020, 26, 606–617. [Google Scholar] [CrossRef]
- Wang, H.; Xu, L.; Zhu, X.; Wang, P.; Chi, H.; Meng, Z. Activation of phosphatidylinositol 3-kinase/Akt signaling mediates sorafenib-induced invasion and metastasis in hepatocellular carcinoma. Oncol. Rep. 2014, 32, 1465–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ombrato, L.; Malanchi, I. The EMT universe: Space between cancer cell dissemination and metastasis initiation. Crit. Rev. Oncog. 2014, 19, 349–361. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial-mesenchymal transition: Molecular basis and therapeutic strategy. Sign. Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [Green Version]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, T.S.; Lee, M.; Park, M.; Kim, S.Y.; Shim, M.S.; Lee, C.Y.; Choi, D.H.; Cho, Y. Metformin and Dichloroacetate Suppress Proliferation of Liver Cancer Cells by Inhibiting mTOR Complex 1. Int. J. Mol. Sci. 2021, 22, 10027. https://doi.org/10.3390/ijms221810027
Kim TS, Lee M, Park M, Kim SY, Shim MS, Lee CY, Choi DH, Cho Y. Metformin and Dichloroacetate Suppress Proliferation of Liver Cancer Cells by Inhibiting mTOR Complex 1. International Journal of Molecular Sciences. 2021; 22(18):10027. https://doi.org/10.3390/ijms221810027
Chicago/Turabian StyleKim, Tae Suk, Minjong Lee, Minji Park, Sae Yun Kim, Min Suk Shim, Chea Yeon Lee, Dae Hee Choi, and Yuri Cho. 2021. "Metformin and Dichloroacetate Suppress Proliferation of Liver Cancer Cells by Inhibiting mTOR Complex 1" International Journal of Molecular Sciences 22, no. 18: 10027. https://doi.org/10.3390/ijms221810027
APA StyleKim, T. S., Lee, M., Park, M., Kim, S. Y., Shim, M. S., Lee, C. Y., Choi, D. H., & Cho, Y. (2021). Metformin and Dichloroacetate Suppress Proliferation of Liver Cancer Cells by Inhibiting mTOR Complex 1. International Journal of Molecular Sciences, 22(18), 10027. https://doi.org/10.3390/ijms221810027