
Aggregation Condition–Structure Relationship of Mouse Prion Protein Fibrils

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
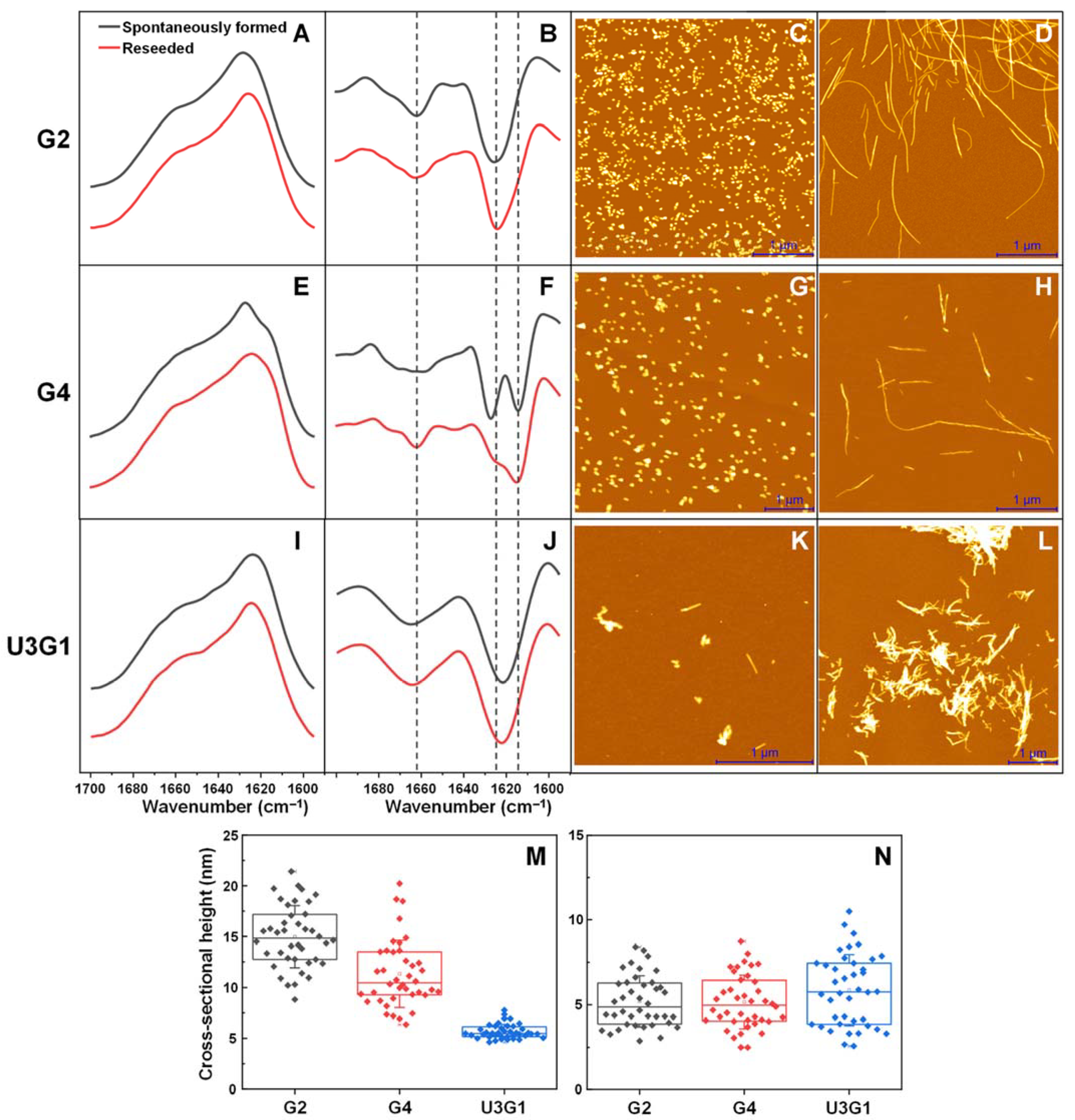
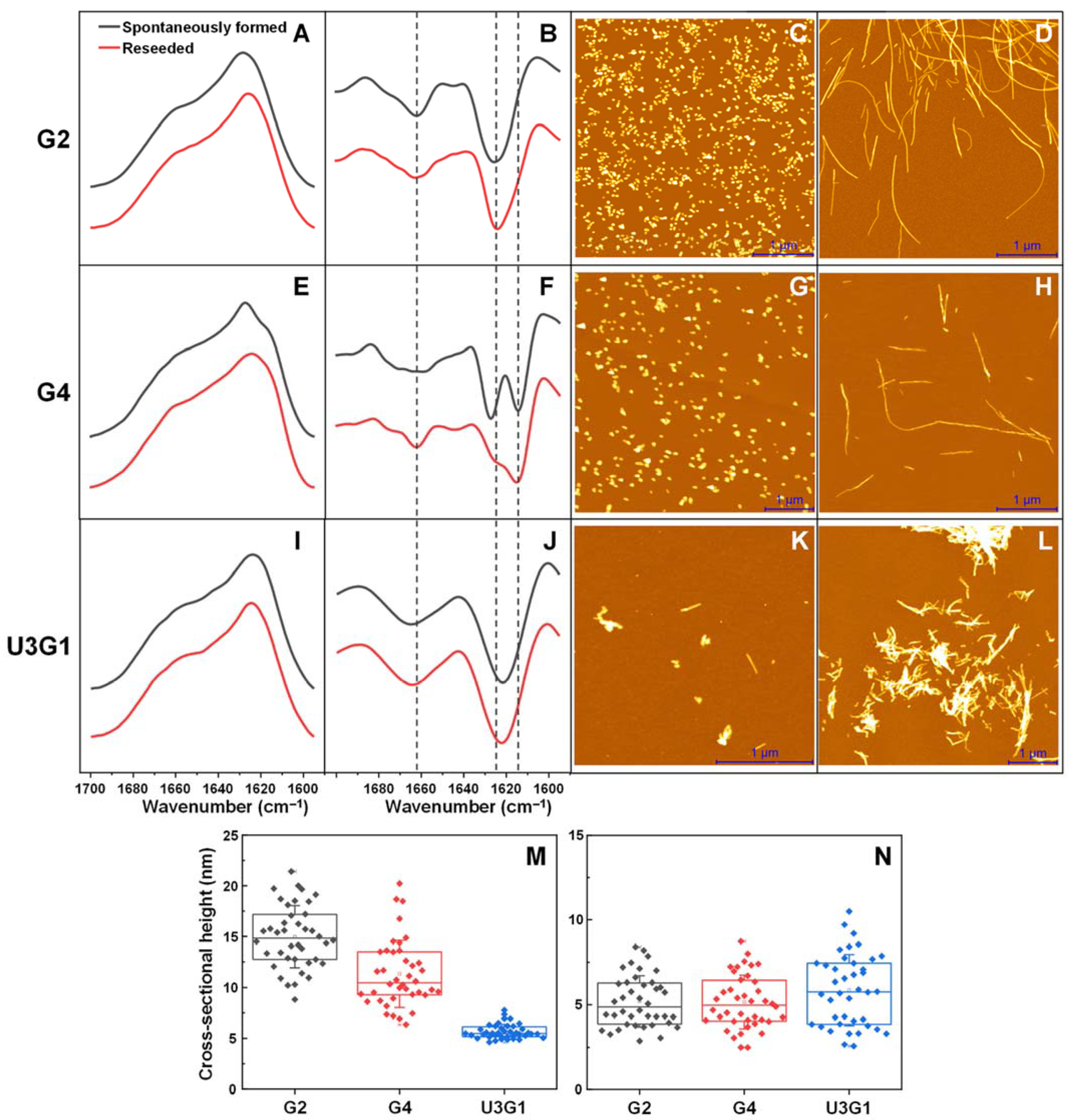
2.1. Fibril Secondary Structure and Morphology Assessed by FTIR and AFM
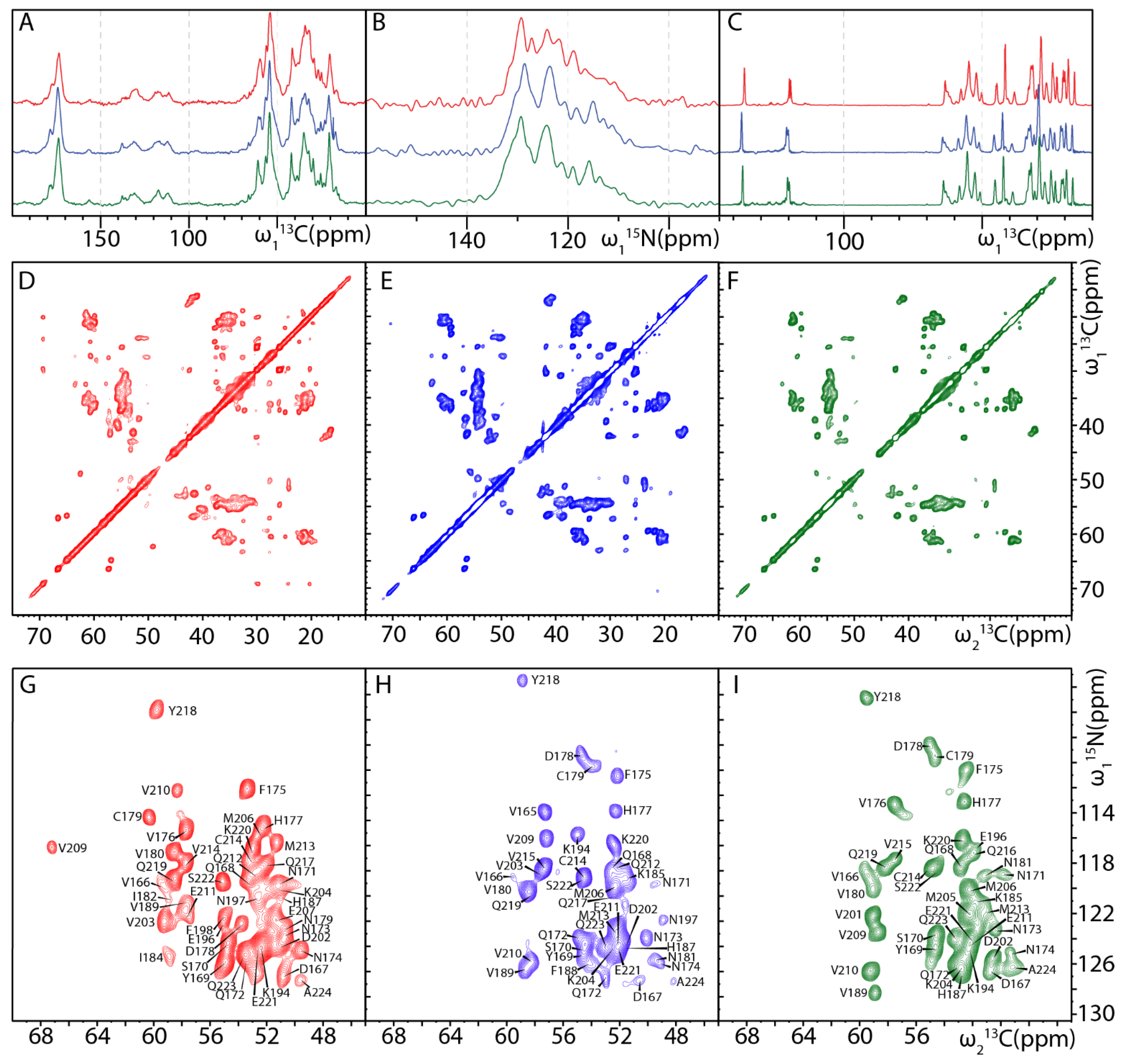
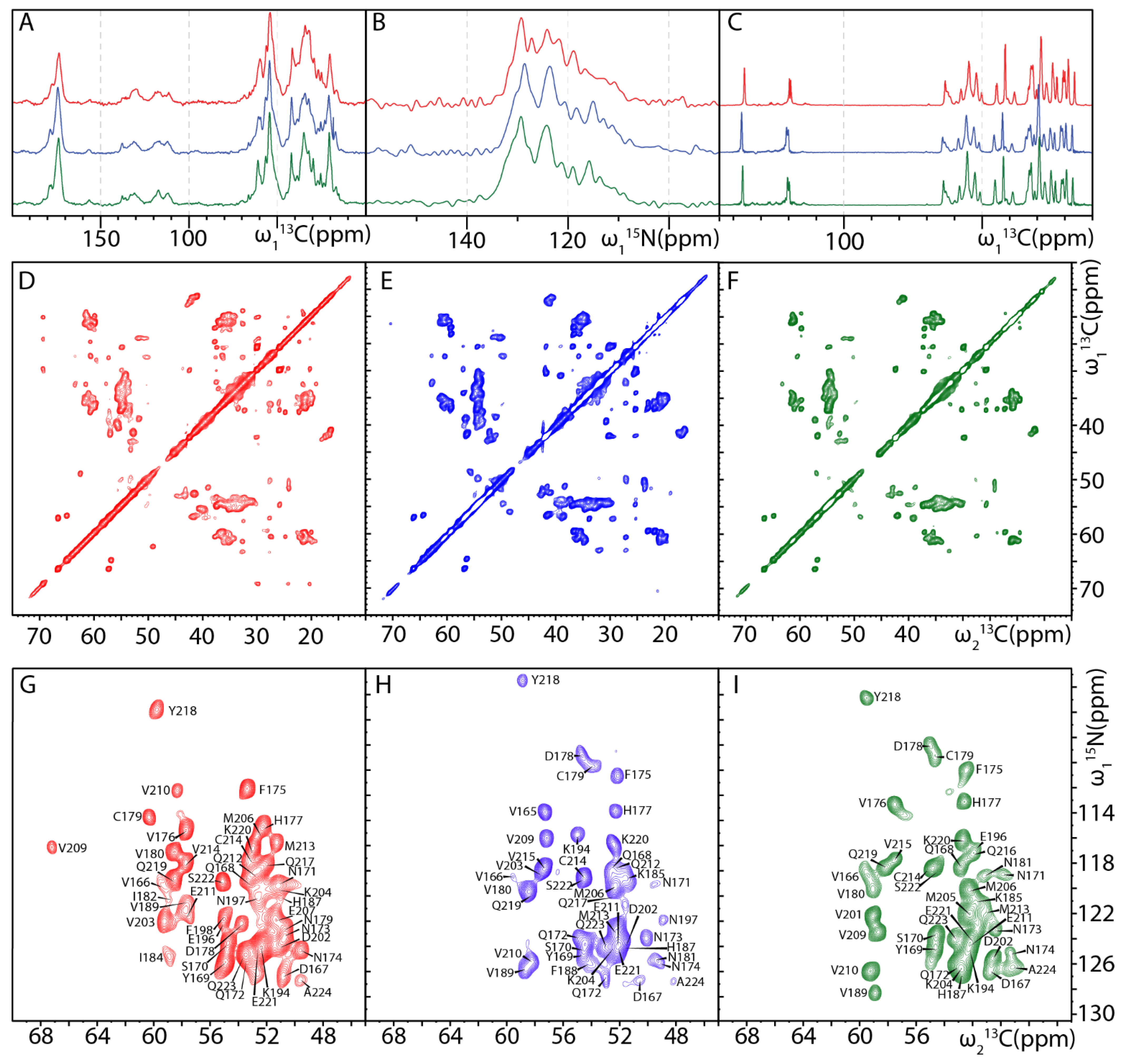
2.2. MAS NMR Fingerprints
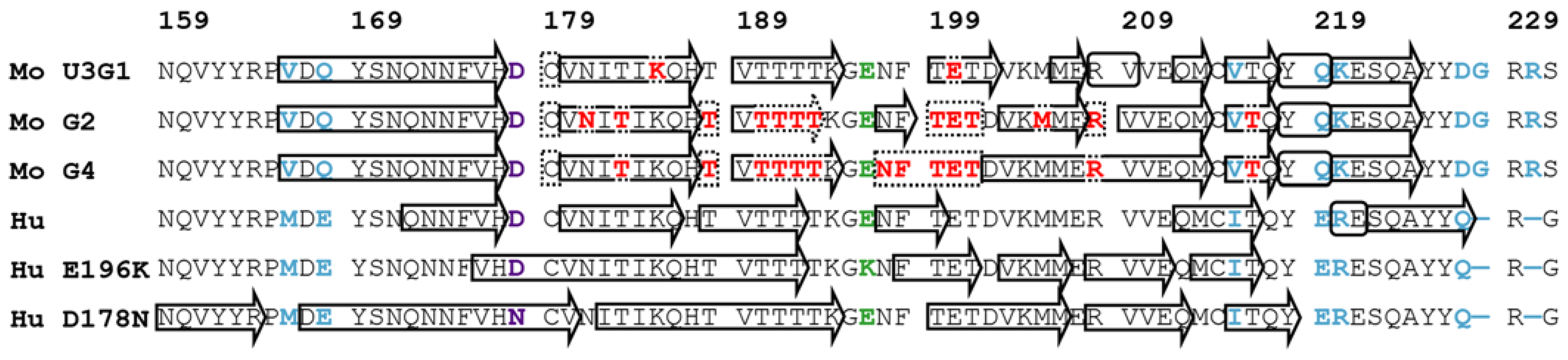
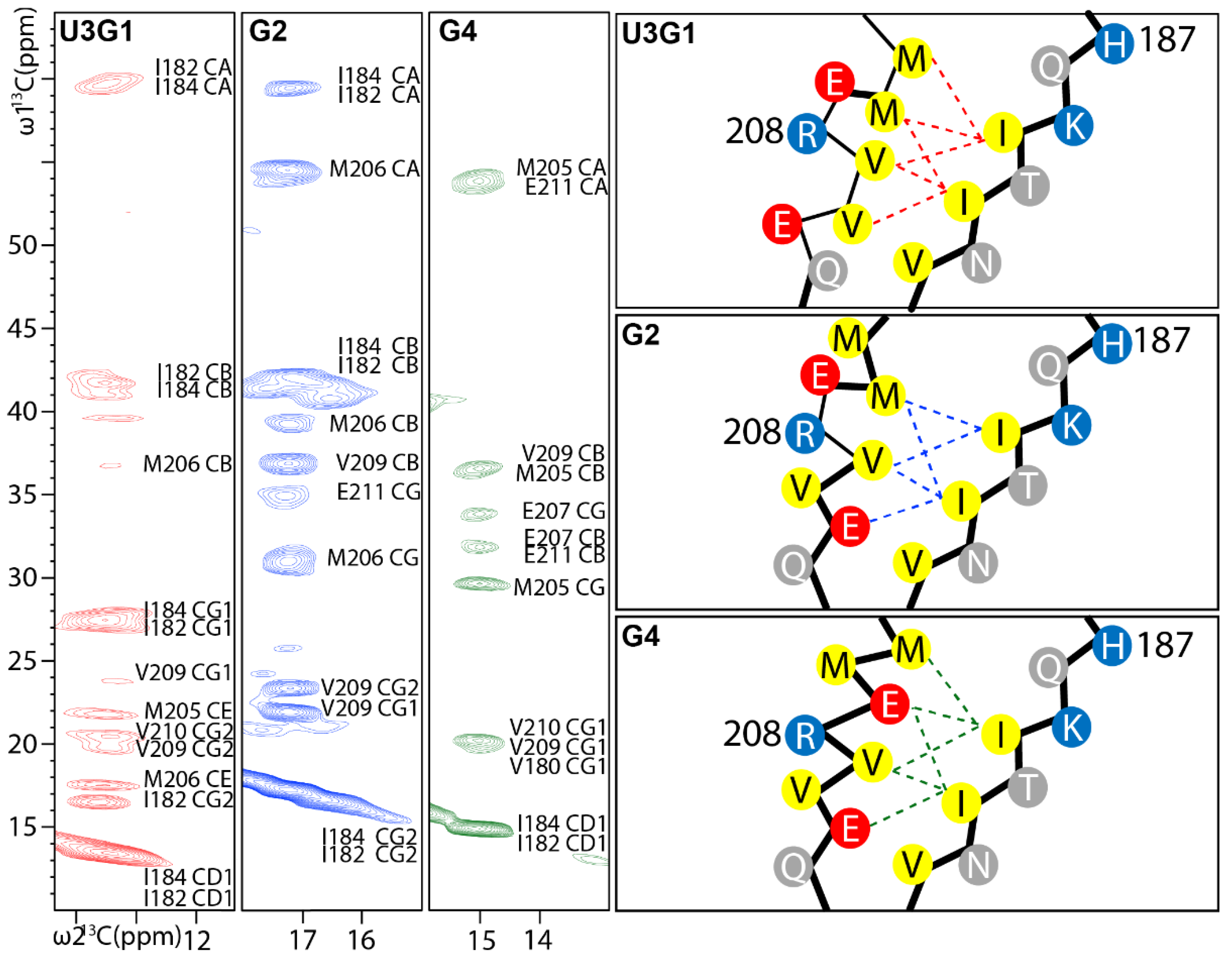
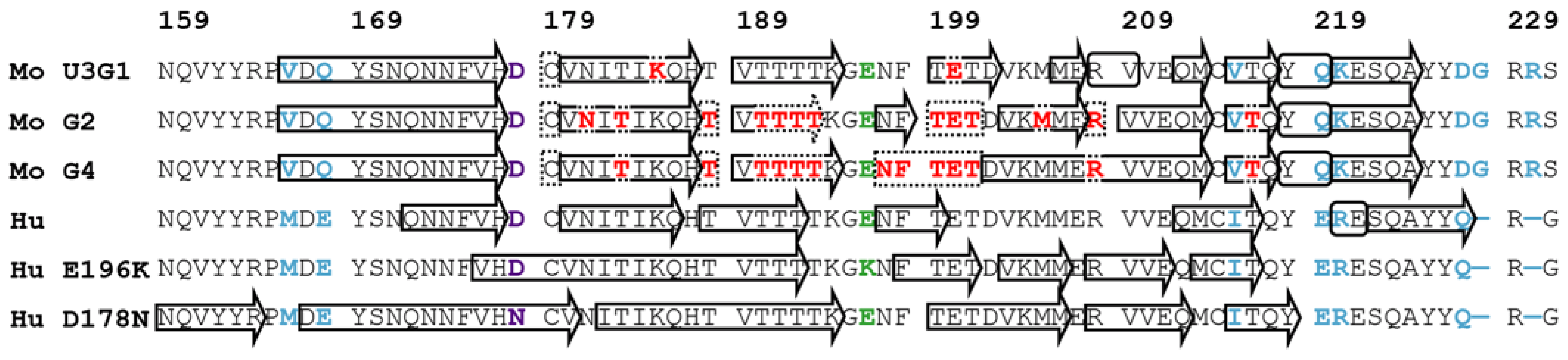
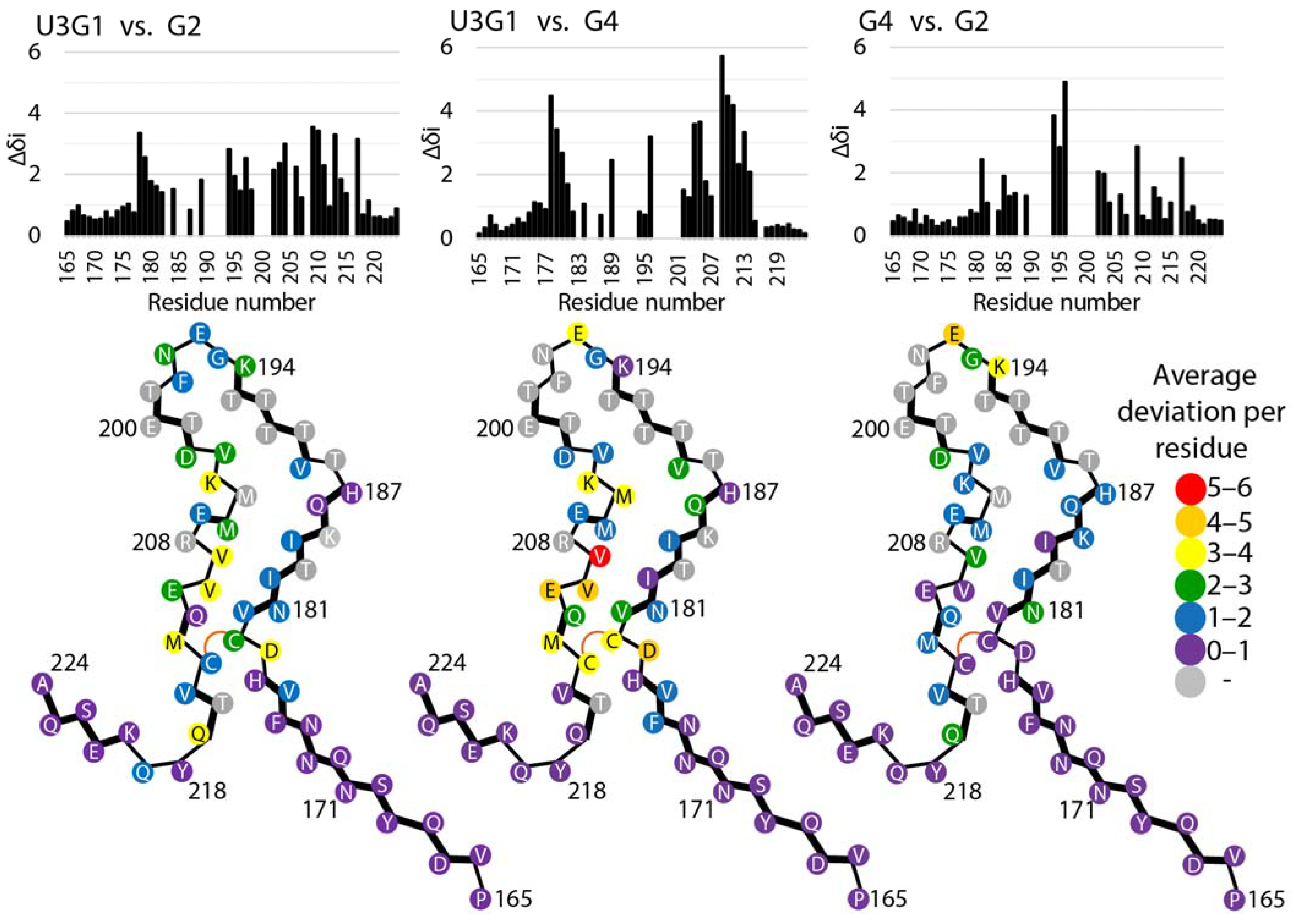
2.3. Locations of Secondary Structure Elements
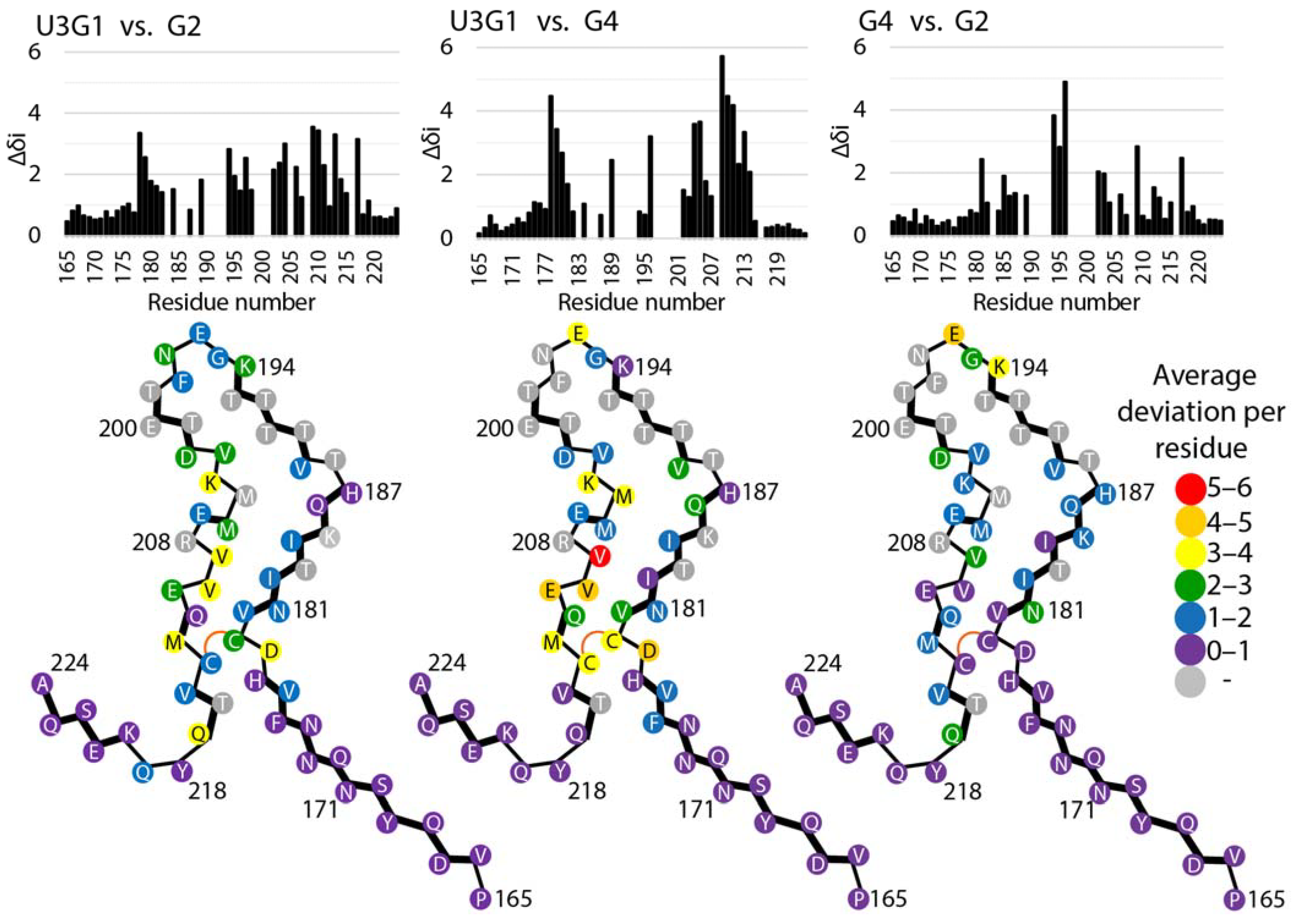
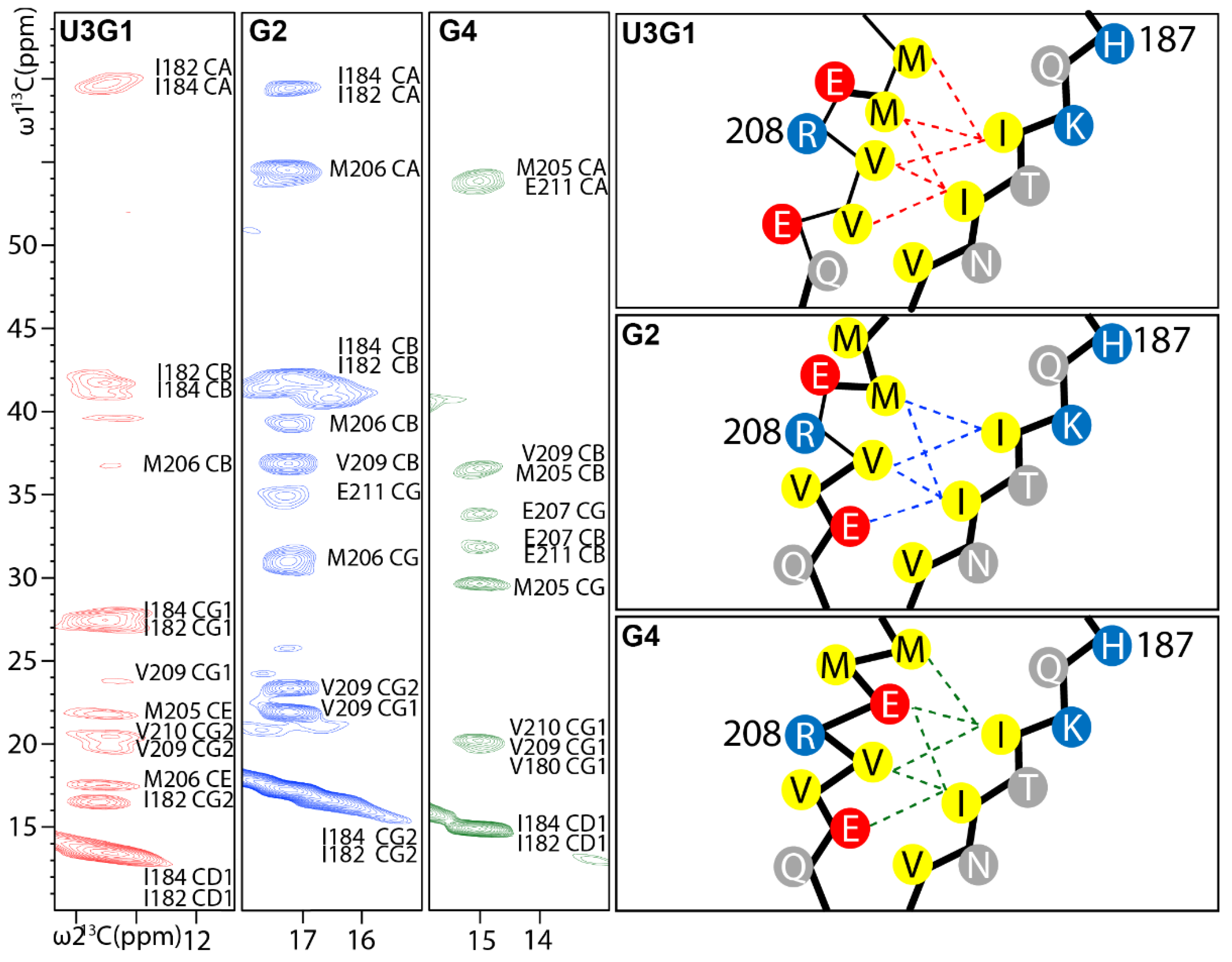
2.4. Tertiary Structure Analysis
3. Materials and Methods
3.1. Production of 13C, 15N Labeled Protein
3.2. Aggregation of Labeled MoPrP(89-230) into Fibrils
3.3. Fourier-Transform Infrared (FTIR) Analysis of Aggregates
3.4. AFM Imaging of Aggregates
3.5. MAS NMR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic Structure and Hierarchical Assembly of a Cross-β Amyloid Fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, K.R.; Rice, L. The Amyloidoses: Clinical Features, Diagnosis and Treatment. Methodist Debakey Cardiovasc. J. 2012, 8, 3–7. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Marshall, K.E.; Marchante, R.; Xue, W.-F.; Serpell, L.C. The Relationship between Amyloid Structure and Cytotoxicity. Prion 2014, 8, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Chatani, E.; Yamamoto, N. Recent Progress on Understanding the Mechanisms of Amyloid Nucleation. Biophys. Rev. 2018, 10, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisl, G.; Kirkegaard, J.B.; Arosio, P.; Michaels, T.C.T.; Vendruscolo, M.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Molecular Mechanisms of Protein Aggregation from Global Fitting of Kinetic Models. Nat. Protoc. 2016, 11, 252–272. [Google Scholar] [CrossRef]
- Meisl, G.; Rajah, L.; Cohen, S.A.I.; Pfammatter, M.; Šarić, A.; Hellstrand, E.; Buell, A.K.; Aguzzi, A.; Linse, S.; Vendruscolo, M.; et al. Scaling Behaviour and Rate-Determining Steps in Filamentous Self-Assembly. Chem. Sci. 2017, 8, 7087–7097. [Google Scholar] [CrossRef] [Green Version]
- Fändrich, M.; Meinhardt, J.; Grigorieff, N. Structural Polymorphism of Alzheimer Abeta and Other Amyloid Fibrils. Prion 2009, 3, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsbury, C.; Frey, P.; Olivieri, V.; Aebi, U.; Müller, S.A. Multiple Assembly Pathways Underlie Amyloid-Beta Fibril Polymorphisms. J. Mol. Biol. 2005, 352, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.-M.; Mattson, M.P.; Tycko, R. Self-Propagating, Molecular-Level Polymorphism in Alzheimer’s β-Amyloid Fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sakalauskas, A.; Smirnovas, V. Identifying Insulin Fibril Conformational Differences by Thioflavin-T Binding Characteristics. Biomacromolecules 2020, 21, 4989–4997. [Google Scholar] [CrossRef]
- Mocanu, M.-M.; Ganea, C.; Siposova, K.; Filippi, A.; Demjen, E.; Marek, J.; Bednarikova, Z.; Antosova, A.; Baran, I.; Gazova, Z. Polymorphism of Hen Egg White Lysozyme Amyloid Fibrils Influences the Cytotoxicity in LLC-PK1 Epithelial Kidney Cells. Int. J. Biol. Macromol. 2014, 65, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why Do Trials for Alzheimer’s Disease Drugs Keep Failing? A Discontinued Drug Perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s Disease Drug Development Pipeline: 2020. Alzheimers Dement. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Hu, P.P.; Duran-Aniotz, C.; Moda, F.; Diaz-Espinoza, R.; Chen, B.; Bravo-Alegria, J.; Makarava, N.; Baskakov, I.V.; Soto, C. Strain-Dependent Profile of Misfolded Prion Protein Aggregates. Sci. Rep. 2016, 6, 20526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.T. Prion Diseases. Lancet Neurol. 2005, 4, 635–642. [Google Scholar] [CrossRef]
- Tixador, P.; Herzog, L.; Reine, F.; Jaumain, E.; Chapuis, J.; Le Dur, A.; Laude, H.; Béringue, V. The Physical Relationship between Infectivity and Prion Protein Aggregates Is Strain-Dependent. PLoS Pathog. 2010, 6, e1000859. [Google Scholar] [CrossRef] [Green Version]
- Langenfeld, K.A.; Shikiya, R.A.; Kincaid, A.E.; Bartz, J.C. Incongruity between Prion Conversion and Incubation Period Following Coinfection. J. Virol. 2016, 90, 5715–5723. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Collins, S.R.; Toyama, B.H.; Weissman, J.S. The Physical Basis of How Prion Conformations Determine Strain Phenotypes. Nature 2006, 442, 585–589. [Google Scholar] [CrossRef]
- Marchante, R.; Beal, D.M.; Koloteva-Levine, N.; Purton, T.J.; Tuite, M.F.; Xue, W.-F. The Physical Dimensions of Amyloid Aggregates Control Their Infective Potential as Prion Particles. Elife 2017, 6, e27109. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Avalos, R.; King, C.-Y.; Wall, J.; Simon, M.; Caspar, D.L.D. Strain-Specific Morphologies of Yeast Prion Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 10165–10170. [Google Scholar] [CrossRef] [Green Version]
- Kurouski, D.; Van Duyne, R.P.; Lednev, I.K. Exploring the Structure and Formation Mechanism of Amyloid Fibrils by Raman Spectroscopy: A Review. Analyst 2015, 140, 4967–4980. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Baskakov, I.V. The Same Primary Structure of the Prion Protein Yields Two Distinct Self-Propagating States. J. Biol. Chem. 2008, 283, 15988–15996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnirov, V.V.; Dergalev, A.A.; Alexandrov, A.I. Proteinase K Resistant Cores of Prions and Amyloids. Prion 2020, 14, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groveman, B.R.; Dolan, M.A.; Taubner, L.M.; Kraus, A.; Wickner, R.B.; Caughey, B. Parallel In-Register Intermolecular β-Sheet Architectures for Prion-Seeded Prion Protein (PrP) Amyloids. J. Biol. Chem. 2014, 289, 24129–24142. [Google Scholar] [CrossRef] [Green Version]
- Sneideris, T.; Darguzis, D.; Botyriute, A.; Grigaliunas, M.; Winter, R.; Smirnovas, V. PH-Driven Polymorphism of Insulin Amyloid-Like Fibrils. PLoS ONE 2015, 10, e0136602. [Google Scholar] [CrossRef]
- Sakalauskas, A.; Ziaunys, M.; Smirnovas, V. Concentration-Dependent Polymorphism of Insulin Amyloid Fibrils. PeerJ 2019, 7, e8208. [Google Scholar] [CrossRef]
- Jain, S.; Udgaonkar, J.B. Salt-Induced Modulation of the Pathway of Amyloid Fibril Formation by the Mouse Prion Protein. Biochemistry 2010, 49, 7615–7624. [Google Scholar] [CrossRef]
- Cobb, N.J.; Apostol, M.I.; Chen, S.; Smirnovas, V.; Surewicz, W.K. Conformational Stability of Mammalian Prion Protein Amyloid Fibrils Is Dictated by a Packing Polymorphism within the Core Region. J. Biol. Chem. 2014, 289, 2643–2650. [Google Scholar] [CrossRef] [Green Version]
- Sneideris, T.; Milto, K.; Smirnovas, V. Polymorphism of Amyloid-like Fibrils Can Be Defined by the Concentration of Seeds. PeerJ 2015, 3, e1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Characterization of Tau Fibrillization in Vitro. Alzheimers Dement. 2010, 6, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, V.L.; Webb, W.W. Transmission Electron Microscopy Characterization of Fluorescently Labelled Amyloid β 1-40 and α-Synuclein Aggregates. BMC Biotechnol. 2011, 11, 125. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, F.S.; Šneideris, T.; Vendruscolo, M.; Knowles, T.P.J. Atomic Force Microscopy for Single Molecule Characterisation of Protein Aggregation. Arch. Biochem. Biophys. 2019, 664, 134–148. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Portaccio, M.; Irace, G.; Sirangelo, I. Insights into Insulin Fibril Assembly at Physiological and Acidic PH and Related Amyloid Intrinsic Fluorescence. Int. J. Mol. Sci. 2017, 18, 2551. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.D.; McGlinchey, R.P.; Walker, R.L.; Lee, J.C. Structural Features of α-Synuclein Amyloid Fibrils Revealed by Raman Spectroscopy. J. Biol. Chem. 2018, 293, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Tycko, R.; Savtchenko, R.; Ostapchenko, V.G.; Makarava, N.; Baskakov, I.V. The α-Helical C-Terminal Domain of Full-Length Recombinant PrP Converts to an in-Register Parallel β-Sheet Structure in PrP Fibrils: Evidence from Solid State Nuclear Magnetic Resonance. Biochemistry 2010, 49, 9488–9497. [Google Scholar] [CrossRef] [Green Version]
- Cobb, N.J.; Sönnichsen, F.D.; McHaourab, H.; Surewicz, W.K. Molecular Architecture of Human Prion Protein Amyloid: A Parallel, in-Register Beta-Structure. Proc. Natl. Acad. Sci. USA 2007, 104, 18946–18951. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-Q.; Zhao, K.; Yuan, H.-Y.; Wang, Q.; Guan, Z.; Tao, J.; Li, X.-N.; Sun, Y.; Yi, C.-W.; Chen, J.; et al. Cryo-EM Structure of an Amyloid Fibril Formed by Full-Length Human Prion Protein. Nat. Struct. Mol. Biol. 2020, 27, 598–602. [Google Scholar] [CrossRef]
- Wang, L.-Q.; Zhao, K.; Yuan, H.-Y.; Li, X.-N.; Dang, H.-B.; Ma, Y.; Wang, Q.; Wang, C.; Sun, Y.; Chen, J.; et al. Familial Prion Disease-Related Mutation E196K Displays a Novel Amyloid Fibril Structure Revealed by Cryo-EM. bioRxiv 2021. [Google Scholar] [CrossRef]
- Leffers, K.-W.; Wille, H.; Stöhr, J.; Junger, E.; Prusiner, S.B.; Riesner, D. Assembly of Natural and Recombinant Prion Protein into Fibrils. Biol. Chem. 2005, 386, 569–580. [Google Scholar] [CrossRef]
- Milto, K.; Michailova, K.; Smirnovas, V. Elongation of Mouse Prion Protein Amyloid-like Fibrils: Effect of Temperature and Denaturant Concentration. PLoS ONE 2014, 9, e94469. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, X.; Orrú, C.D.; Groveman, B.R.; Surewicz, K.; Abskharon, R.; Imamura, M.; Yokoyama, T.; Kim, Y.-S.; Vander Stel, K.J.; et al. Self-Propagating, Protease-Resistant, Recombinant Prion Protein Conformers with or without in Vivo Pathogenicity. PLoS Pathog. 2017, 13, e1006491. [Google Scholar] [CrossRef]
- Dutta, A.; Chen, S.; Surewicz, W.K. The Effect of Β2-A2 Loop Mutation on Amyloidogenic Properties of the Prion Protein. FEBS Lett. 2013, 587, 2918–2923. [Google Scholar] [CrossRef] [Green Version]
- Baskakov, I.; Disterer, P.; Breydo, L.; Shaw, M.; Gill, A.; James, W.; Tahiri-Alaoui, A. The Presence of Valine at Residue 129 in Human Prion Protein Accelerates Amyloid Formation. FEBS Lett. 2005, 579, 2589–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sneideris, T.; Ziaunys, M.; Chu, B.K.-Y.; Chen, R.P.-Y.; Smirnovas, V. Self-Replication of Prion Protein Fragment 89-230 Amyloid Fibrils Accelerated by Prion Protein Fragment 107-143 Aggregates. Int. J. Mol. Sci. 2020, 21, 7410. [Google Scholar] [CrossRef] [PubMed]
- Bocharova, O.V.; Breydo, L.; Parfenov, A.S.; Salnikov, V.V.; Baskakov, I.V. In Vitro Conversion of Full-Length Mammalian Prion Protein Produces Amyloid Form with Physical Properties of PrP(Sc). J. Mol. Biol. 2005, 346, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Barth, A. Infrared Spectroscopy of Proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Smirnovas, V.; Baron, G.S.; Offerdahl, D.K.; Raymond, G.J.; Caughey, B.; Surewicz, W.K. Structural Organization of Brain-Derived Mammalian Prions Examined by Hydrogen-Deuterium Exchange. Nat. Struct. Mol. Biol. 2011, 18, 504–506. [Google Scholar] [CrossRef] [Green Version]
- Baral, P.K.; Yin, J.; Aguzzi, A.; James, M.N.G. Transition of the Prion Protein from a Structured Cellular Form (PrPC) to the Infectious Scrapie Agent (PrPSc). Protein Sci. 2019, 28, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Sneideris, T.; Sakalauskas, A.; Sternke-Hoffmann, R.; Peduzzo, A.; Ziaunys, M.; Buell, A.K.; Smirnovas, V. The Environment Is a Key Factor in Determining the Anti-Amyloid Efficacy of EGCG. Biomolecules 2019, 9, 855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziaunys, M.; Sneideris, T.; Smirnovas, V. Formation of Distinct Prion Protein Amyloid Fibrils under Identical Experimental Conditions. Sci. Rep. 2020, 10, 4572. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, F.S.; Vieweg, S.; Cendrowska, U.; Longo, G.; Chiki, A.; Lashuel, H.A.; Dietler, G. Nanoscale Studies Link Amyloid Maturity with Polyglutamine Diseases Onset. Sci. Rep. 2016, 6, 31155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, R.L.J. The Computer Aided Resonance Assignment Tutorial, 1st ed.; CANTINA Verlag: Goldau, Switzerland, 2004. [Google Scholar]
- Kjaergaard, M.; Poulsen, F.M. Sequence Correction of Random Coil Chemical Shifts: Correlation between Neighbor Correction Factors and Changes in the Ramachandran Distribution. J. Biomol. NMR 2011, 50, 157–165. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fridmanis, J.; Toleikis, Z.; Sneideris, T.; Ziaunys, M.; Bobrovs, R.; Smirnovas, V.; Jaudzems, K. Aggregation Condition–Structure Relationship of Mouse Prion Protein Fibrils. Int. J. Mol. Sci. 2021, 22, 9635. https://doi.org/10.3390/ijms22179635
Fridmanis J, Toleikis Z, Sneideris T, Ziaunys M, Bobrovs R, Smirnovas V, Jaudzems K. Aggregation Condition–Structure Relationship of Mouse Prion Protein Fibrils. International Journal of Molecular Sciences. 2021; 22(17):9635. https://doi.org/10.3390/ijms22179635
Chicago/Turabian StyleFridmanis, Jēkabs, Zigmantas Toleikis, Tomas Sneideris, Mantas Ziaunys, Raitis Bobrovs, Vytautas Smirnovas, and Kristaps Jaudzems. 2021. "Aggregation Condition–Structure Relationship of Mouse Prion Protein Fibrils" International Journal of Molecular Sciences 22, no. 17: 9635. https://doi.org/10.3390/ijms22179635
APA StyleFridmanis, J., Toleikis, Z., Sneideris, T., Ziaunys, M., Bobrovs, R., Smirnovas, V., & Jaudzems, K. (2021). Aggregation Condition–Structure Relationship of Mouse Prion Protein Fibrils. International Journal of Molecular Sciences, 22(17), 9635. https://doi.org/10.3390/ijms22179635