
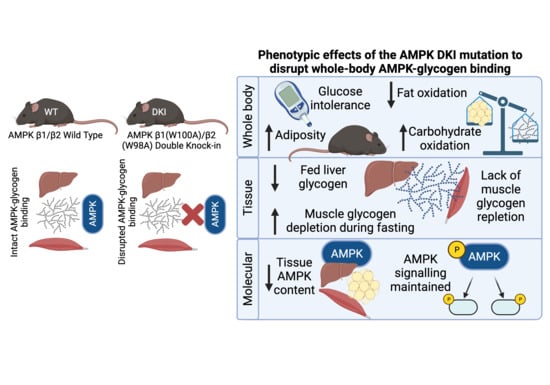
Mice with Whole-Body Disruption of AMPK-Glycogen Binding Have Increased Adiposity, Reduced Fat Oxidation and Altered Tissue Glycogen Dynamics

 , ,
, ,  ,
,
Abstract
:
1. Introduction
2. Results
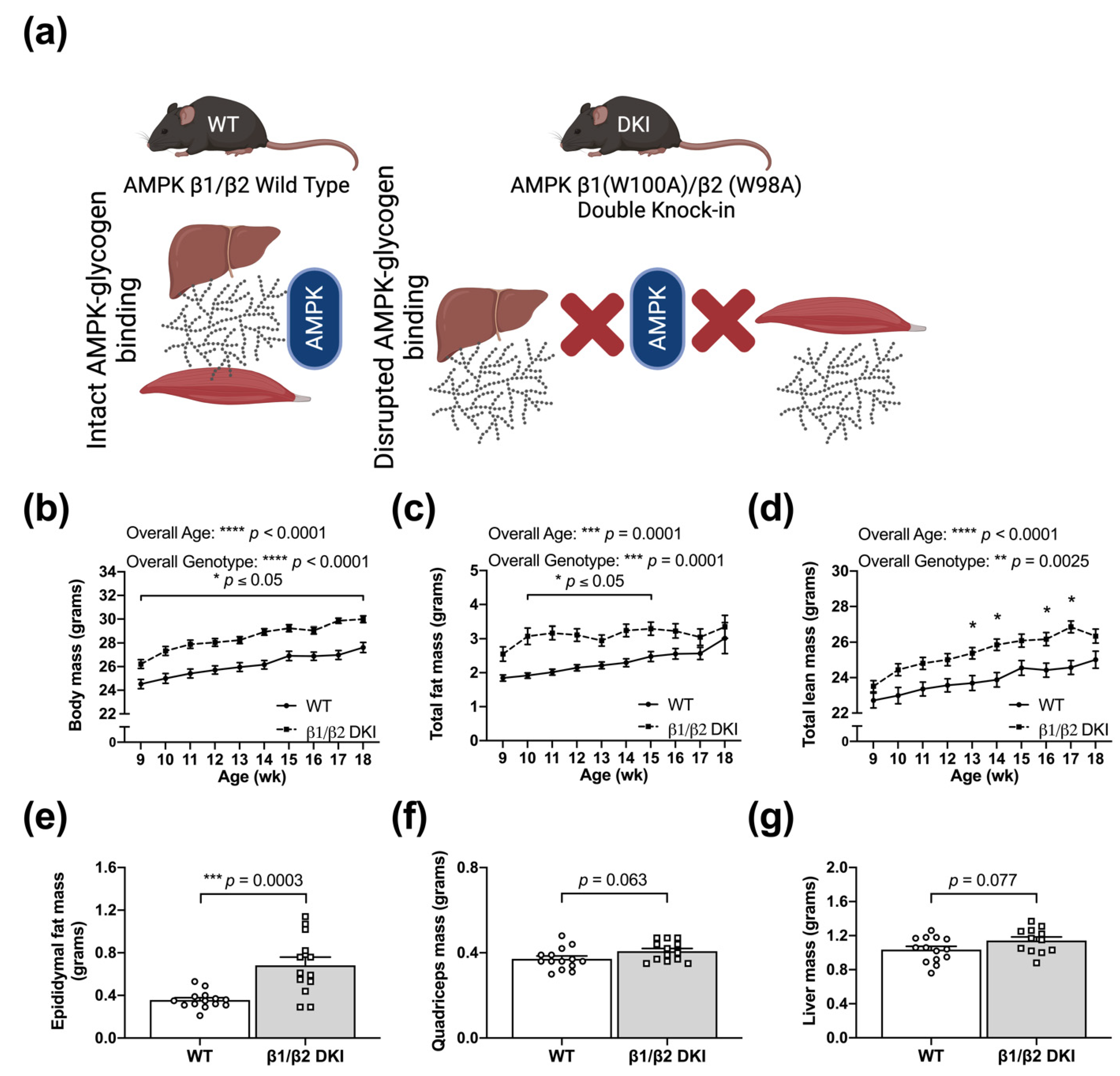
2.1. DKI Mice Display Increased Body Mass Associated with Increased Adiposity
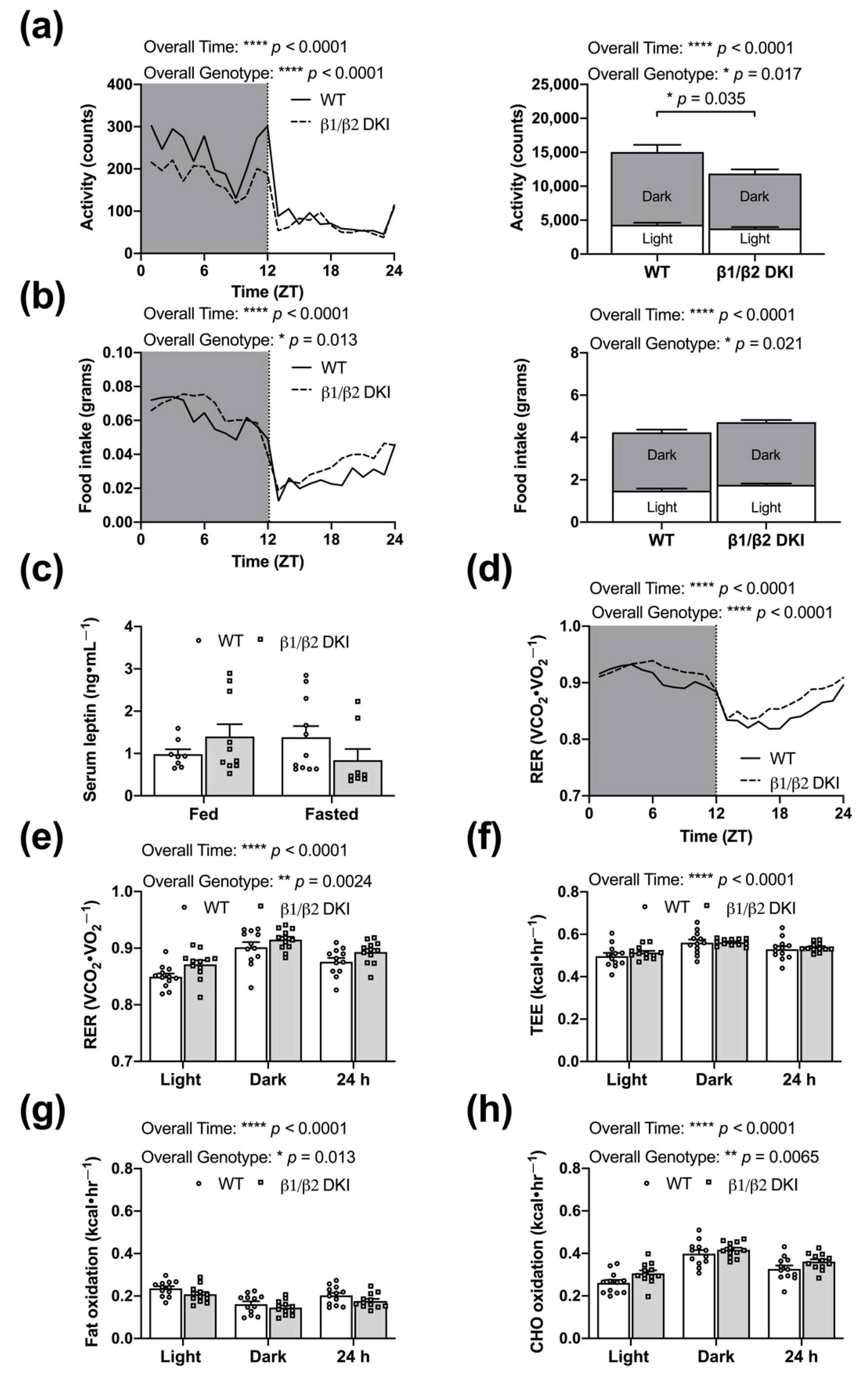
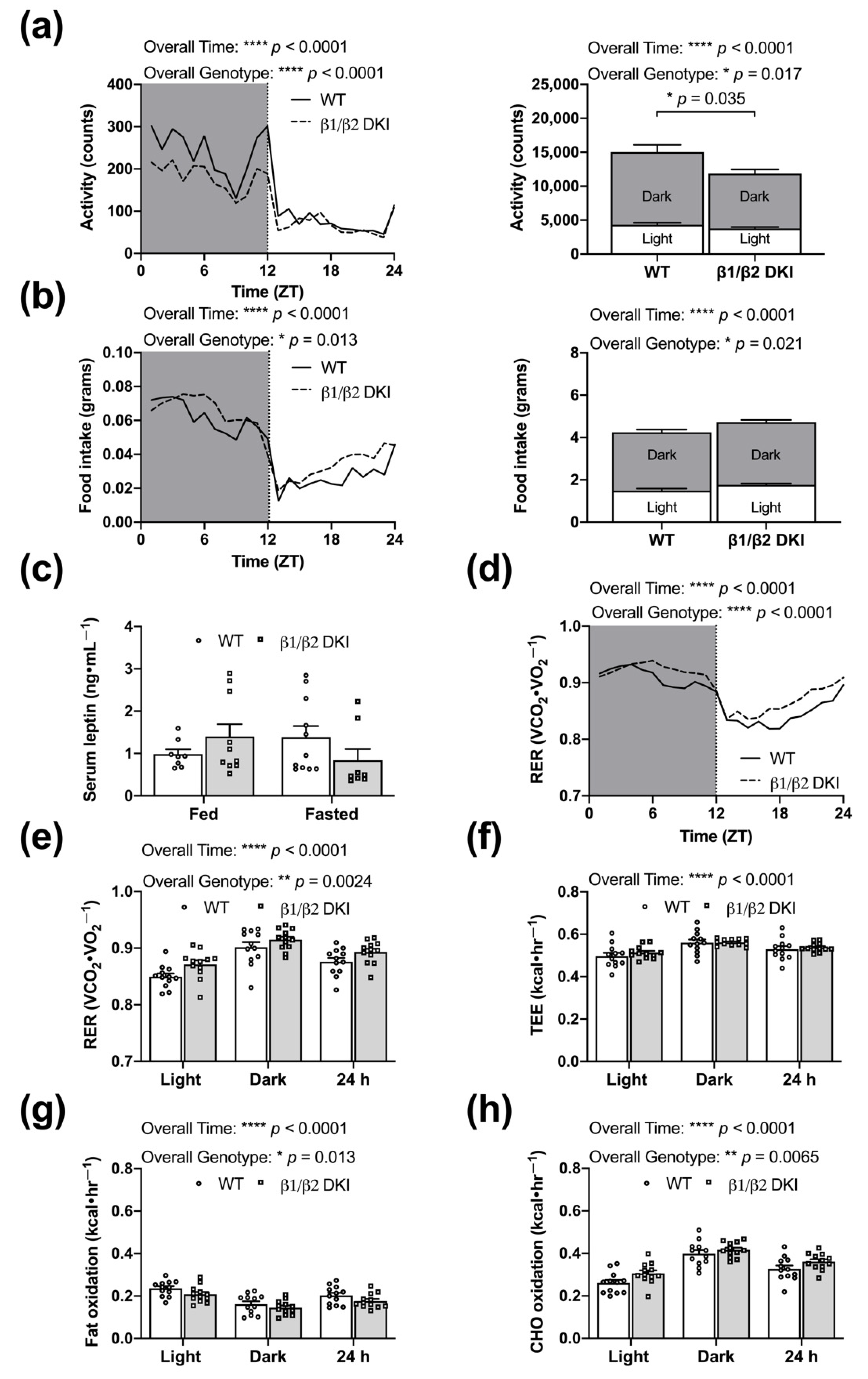
2.2. DKI Mice Have Reduced Voluntary Physical Activity, Increased Rates of CHO Oxidation, and Reduced Rates of Fat Oxidation
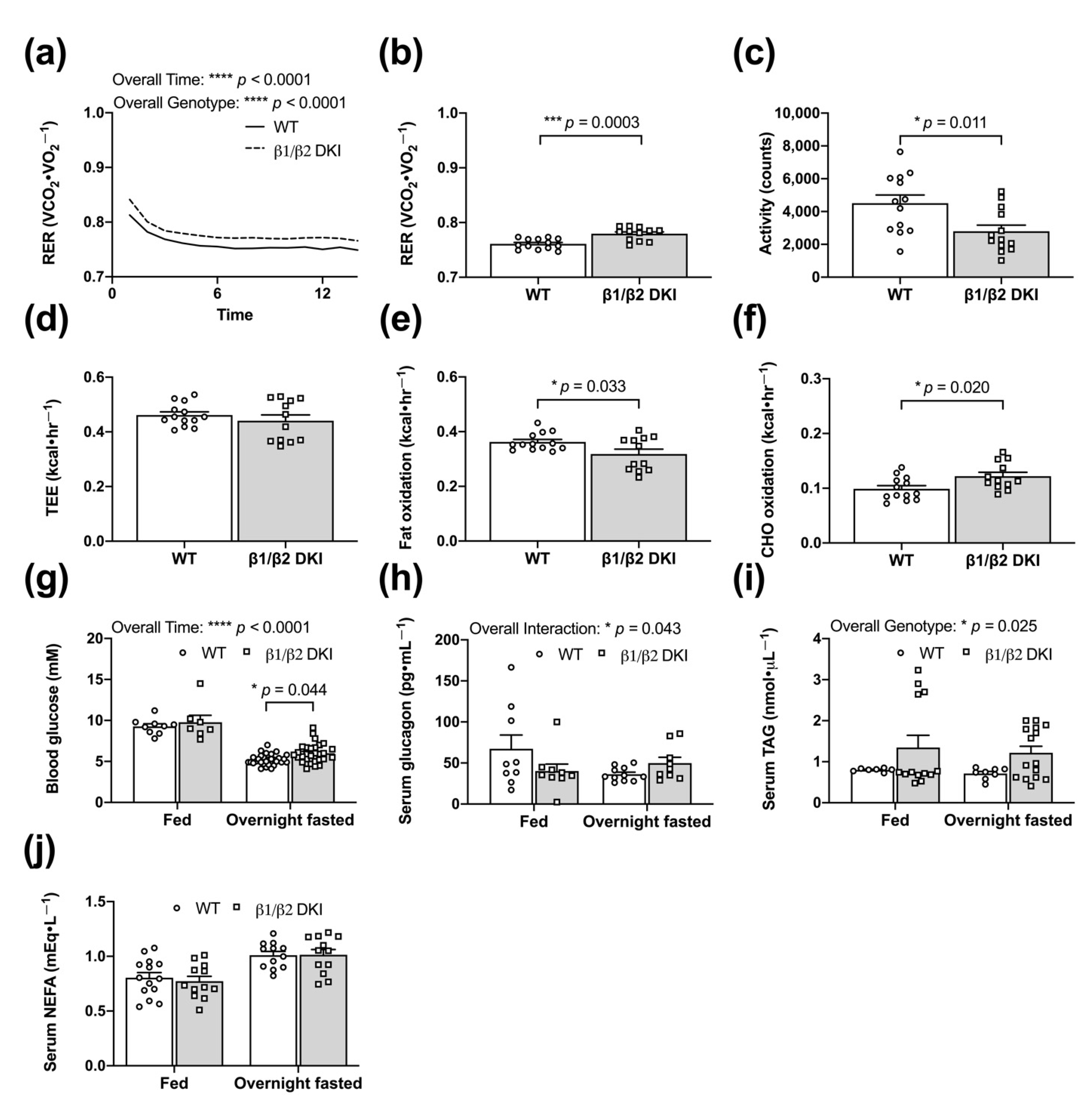
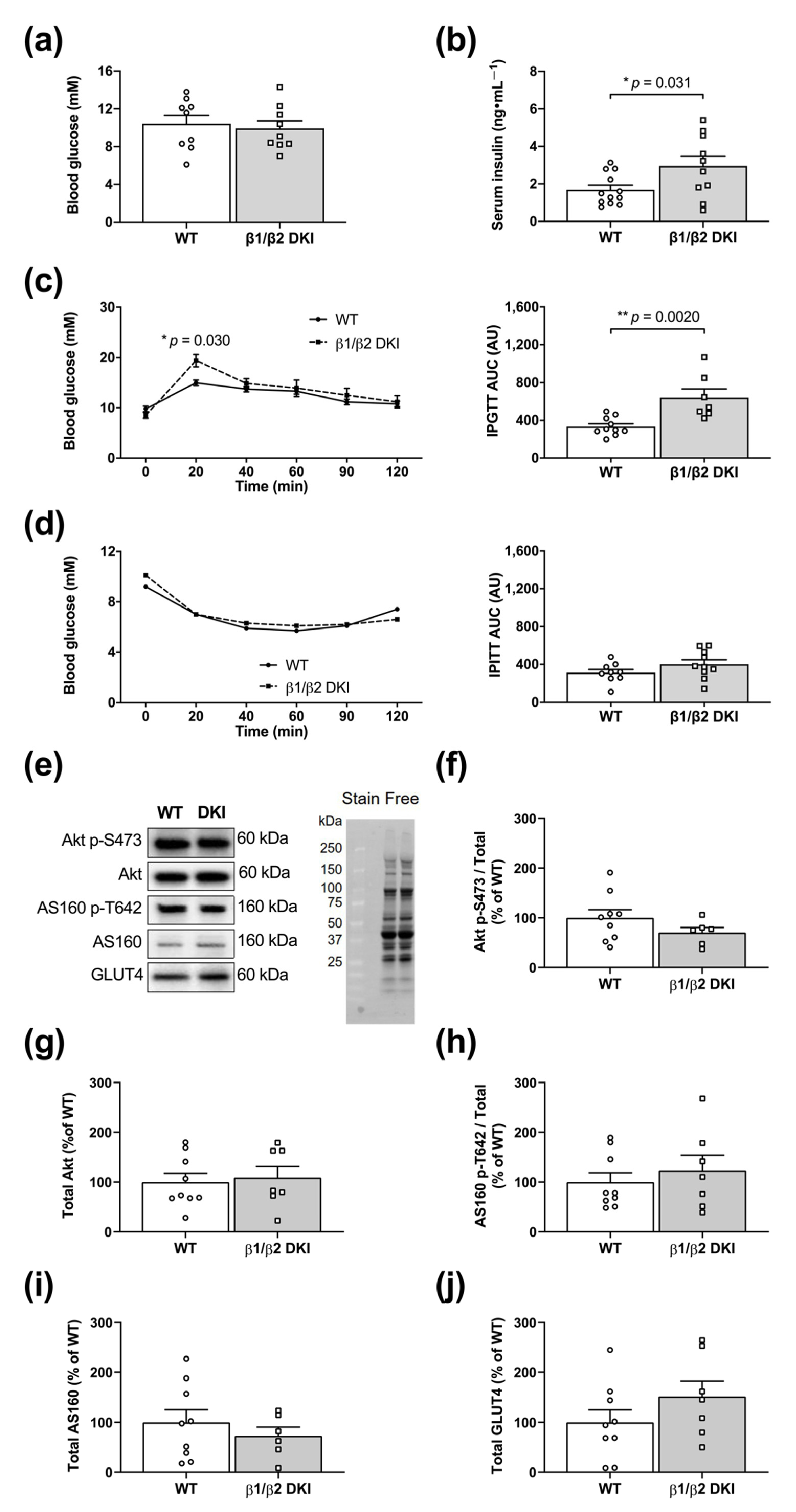
2.3. DKI Mice Display Hyperinsulinemia and Glucose Intolerance but Normal Whole-Body Insulin Sensitivity
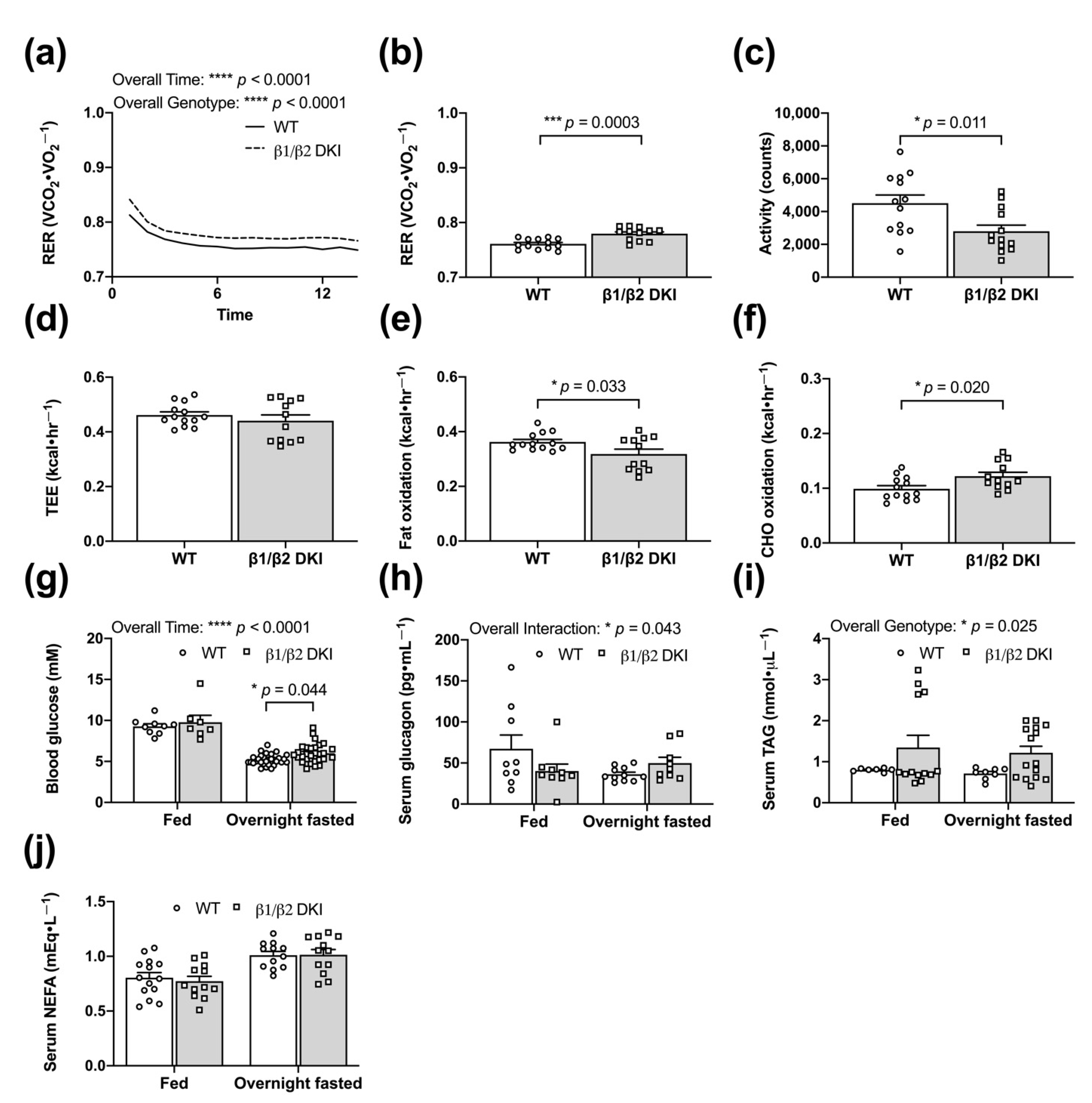
2.4. DKI Mice Have Decreased Voluntary Ambulatory Activity and Decreased Rates of Fat Oxidation during Overnight Fasting
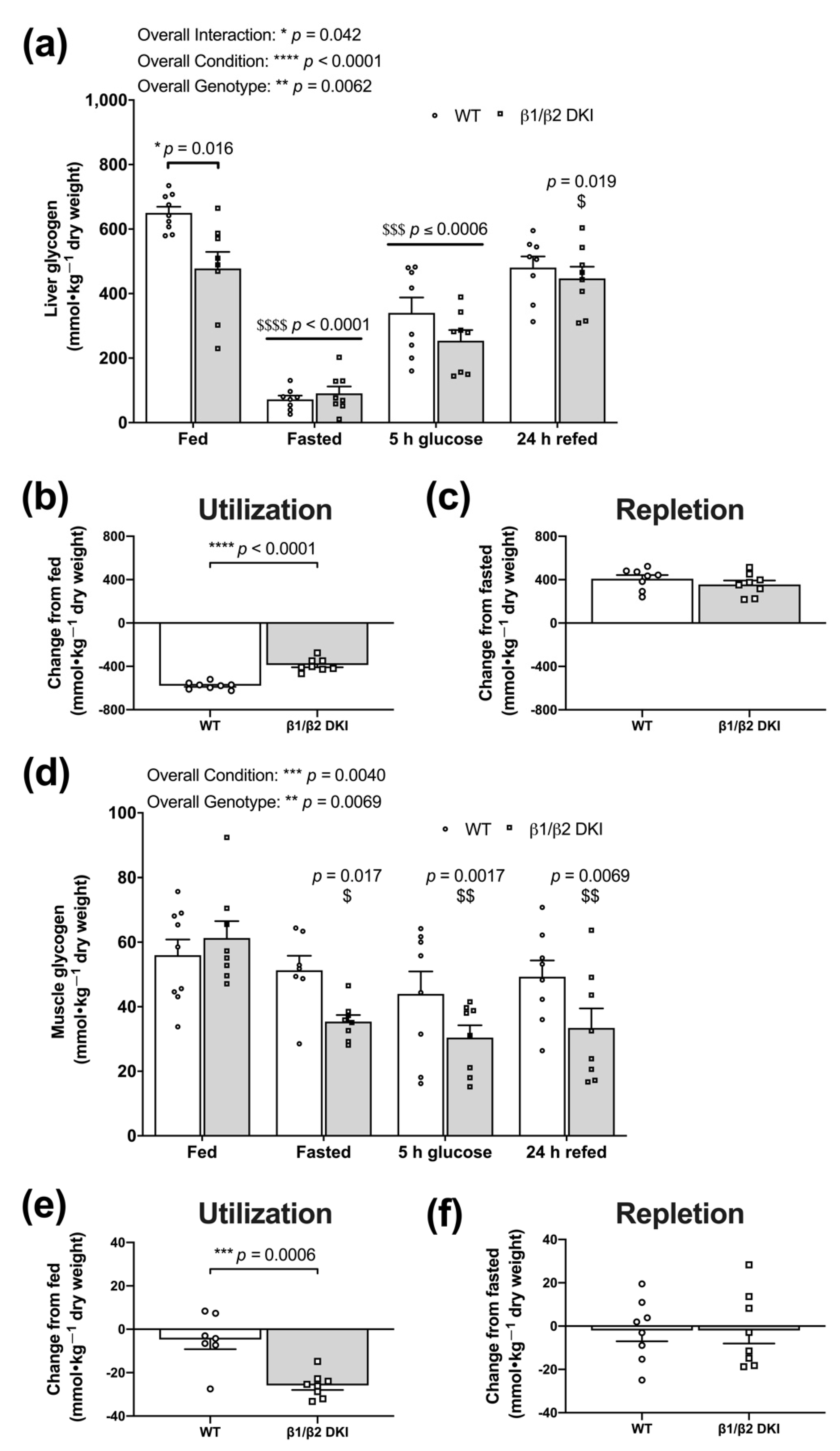
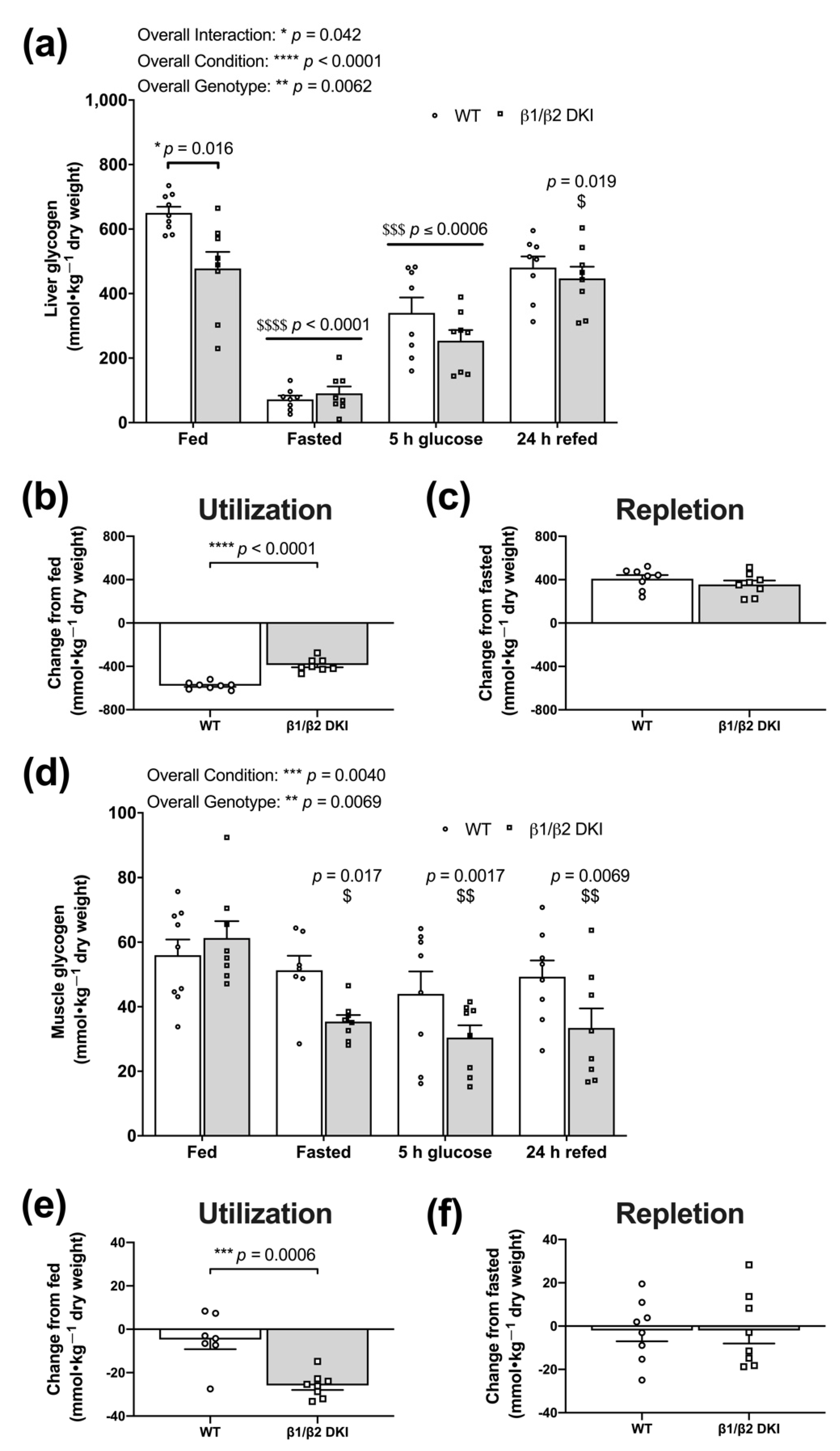
2.5. DKI Mice Display Reduced Liver Glycogen Concentration in the Fed State and Altered Fasting-Induced Skeletal Muscle Glycogen Depletion
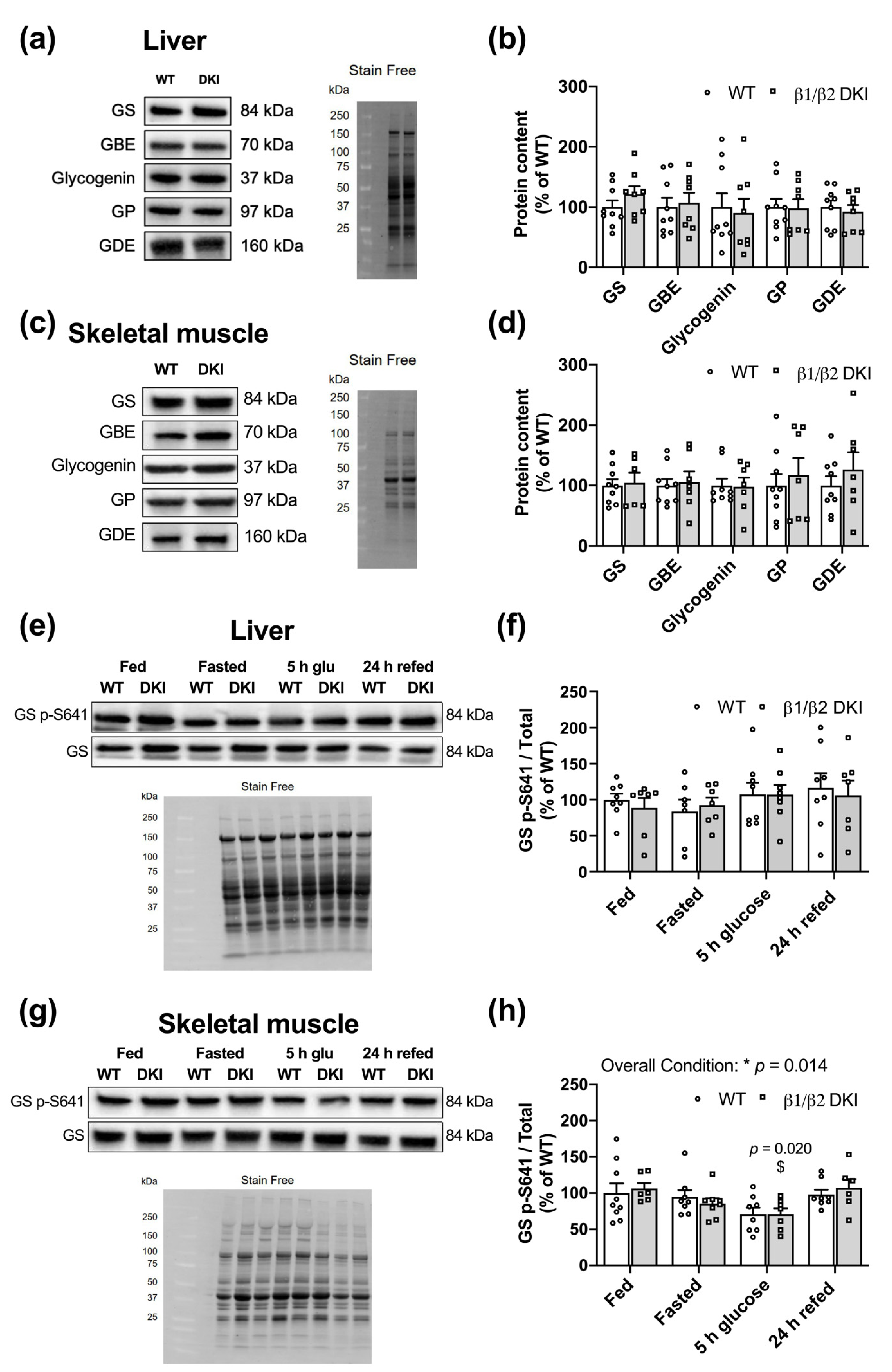
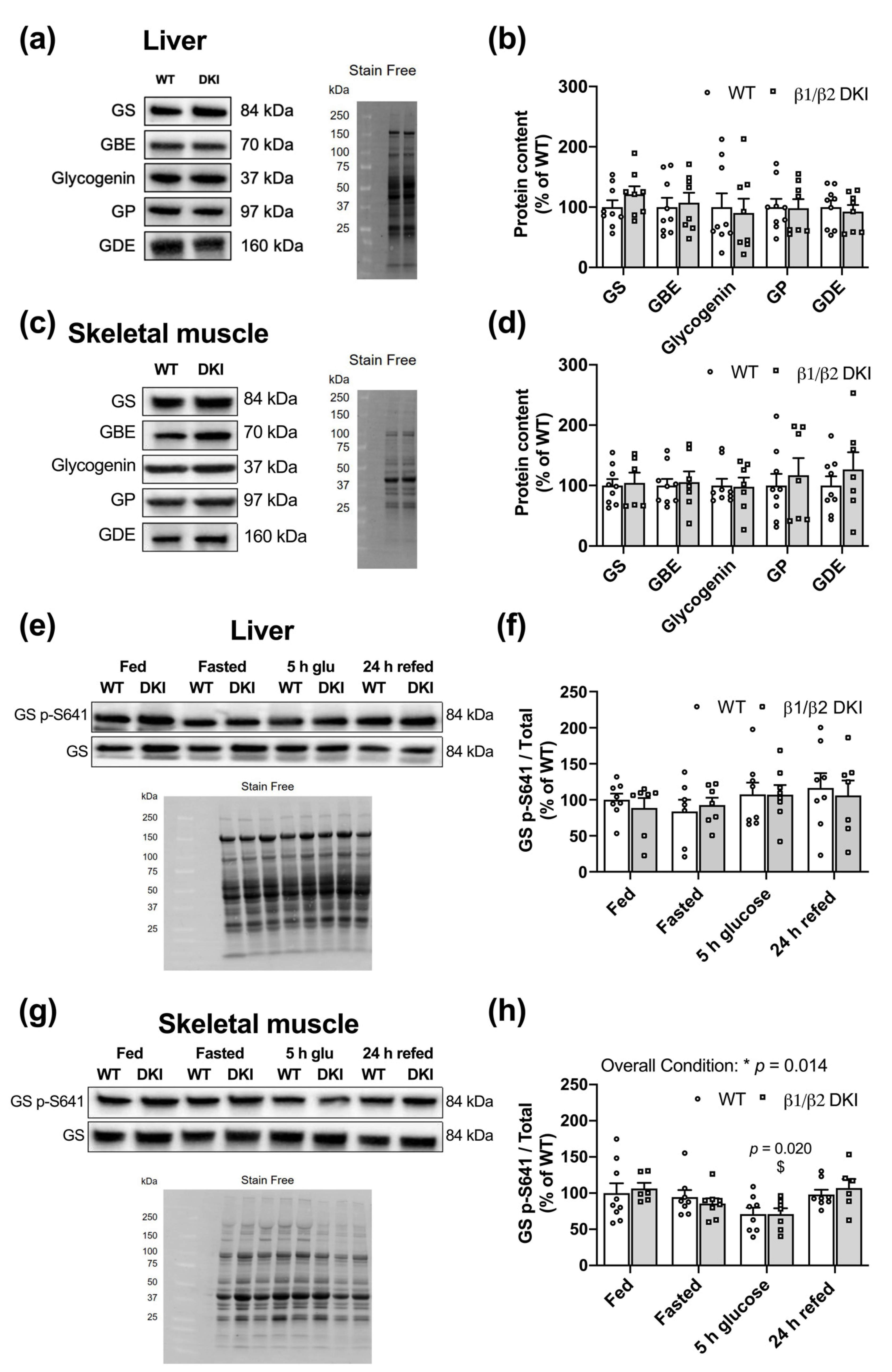
2.6. Liver and Muscle from DKI Mice Display No Changes in Glycogen Associated Proteins Relative to WT
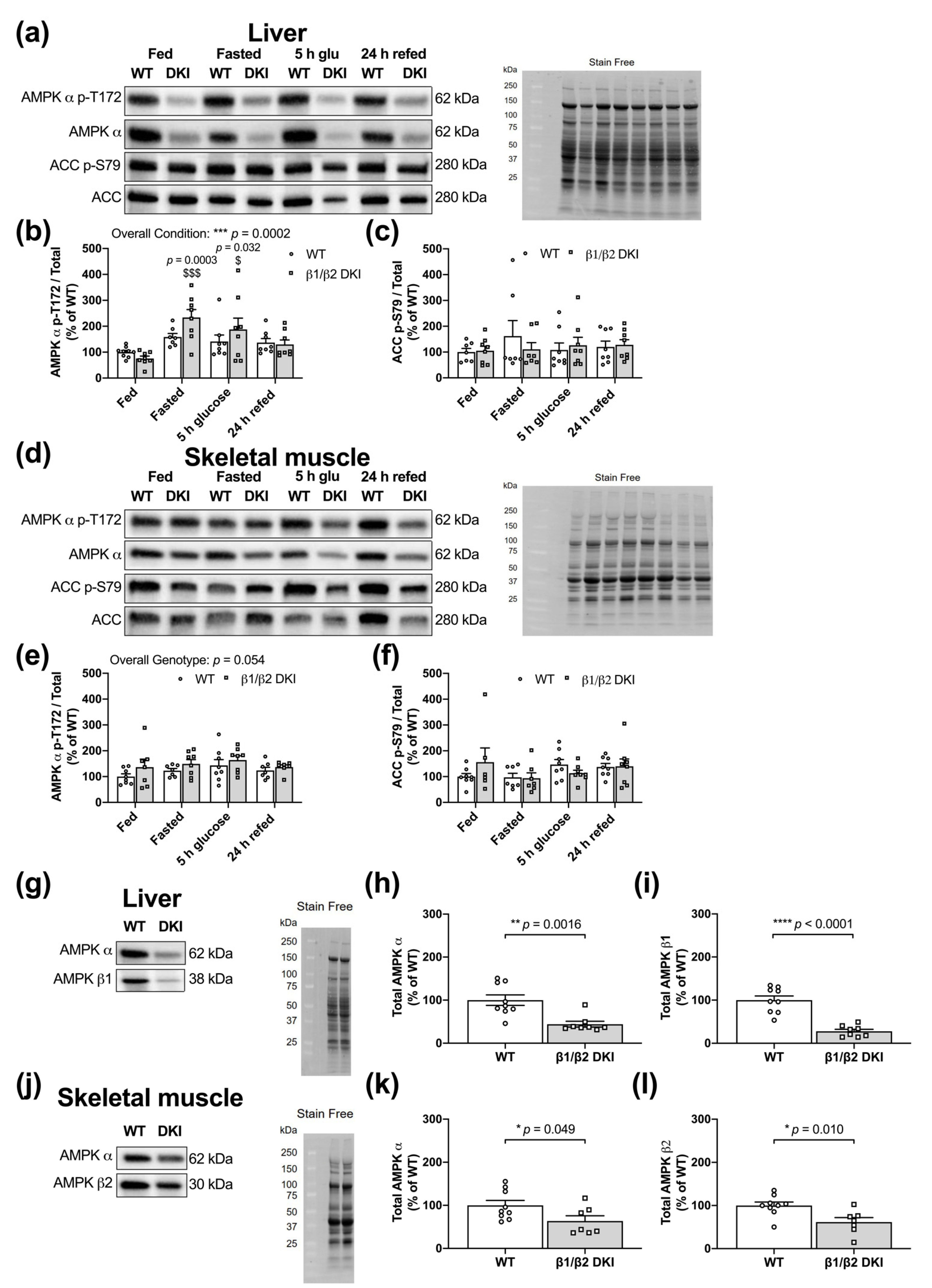
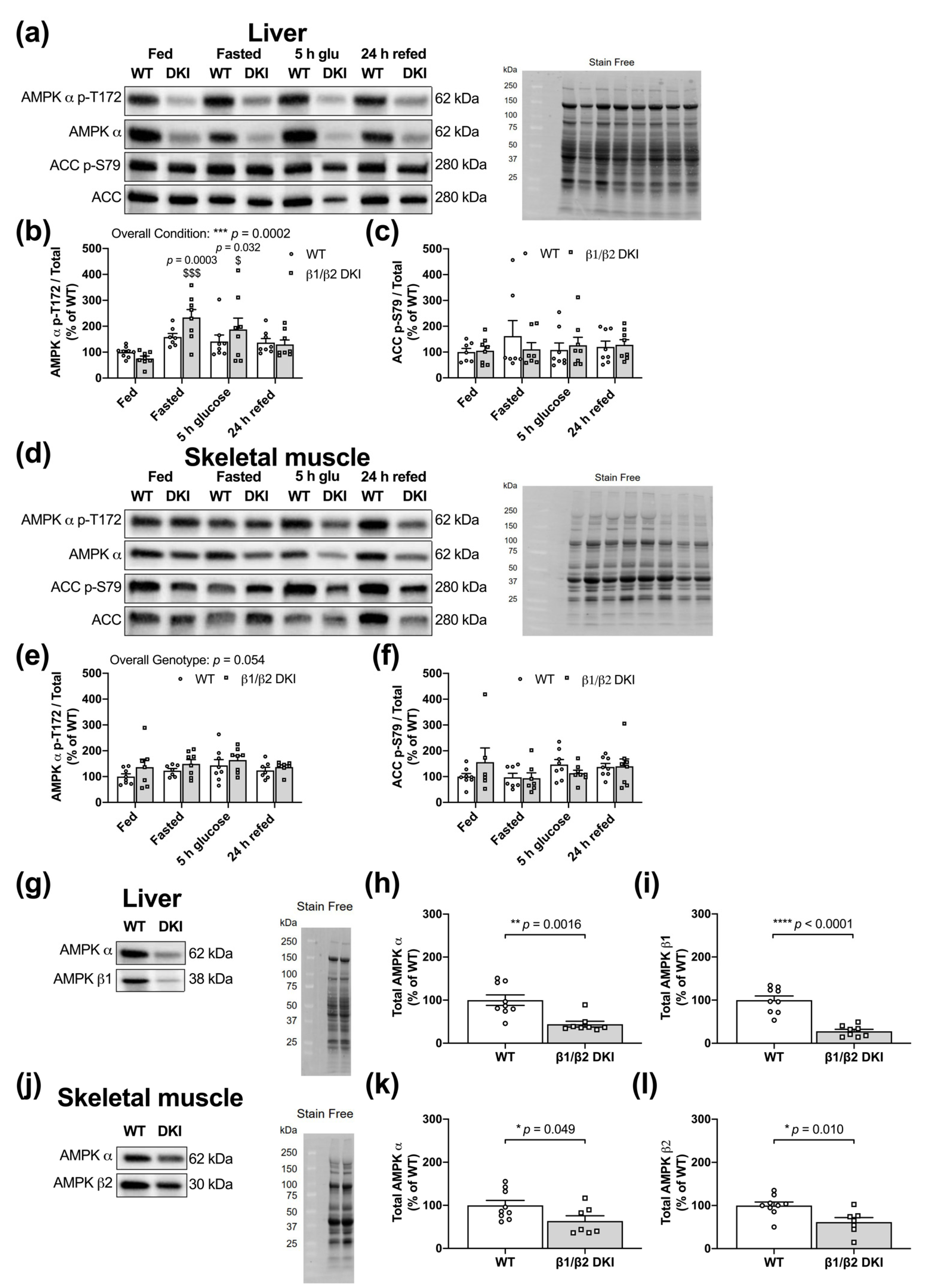
2.7. DKI Mice Have Intact AMPK and ACC Phosphorylation in Liver and Skeletal Muscle
2.8. DKI Mice Have Reduced AMPK Protein Content in Liver and Skeletal Muscle
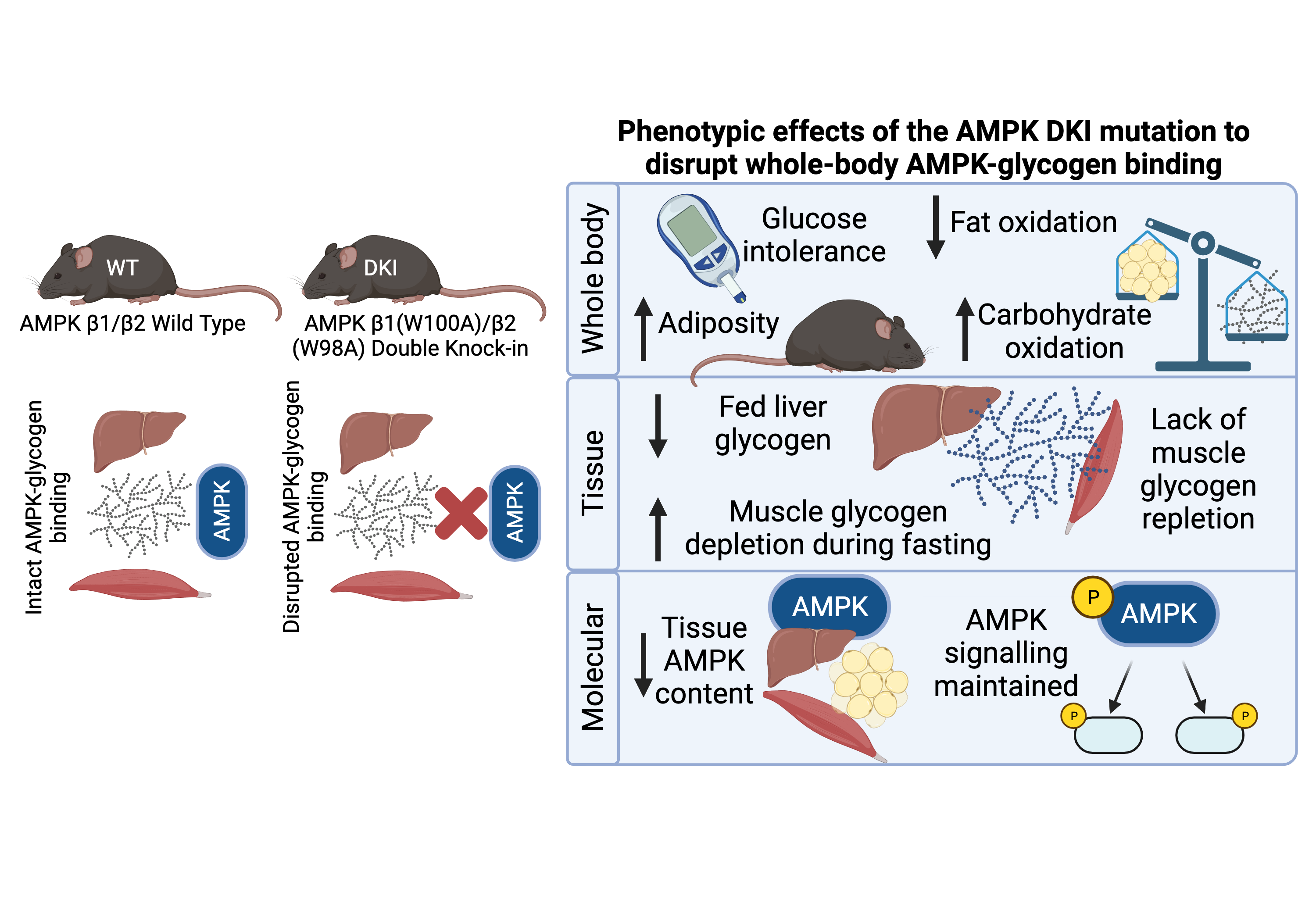
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Whole-Body Composition Measurements
4.3. Metabolic Caging
4.4. Intraperitoneal Glucose Tolerance and Insulin Tolerance Testing
4.5. Fasting and Refeeding Protocol
4.6. Serum Analysis
4.7. Assessment of Tissue Glycogen Content
4.8. Immunoblotting
4.9. RNA Extraction and Gene Expression Analysis
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Iseli, T.J.; Walter, M.; van Denderen, B.J.W.; Katsis, F.; Witters, L.A.; Kemp, B.E.; Michell, B.J.; Stapleton, D. AMP-activated Protein Kinase β Subunit Tethers α and γ Subunits via Its C-terminal Sequence (186–270). J. Biol. Chem. 2005, 280, 13395–13400. [Google Scholar] [CrossRef] [Green Version]
- Janzen, N.R.; Whitfield, J.; Hoffman, N.J. Interactive Roles for AMPK and Glycogen from Cellular Energy Sensing to Exercise Metabolism. Int. J. Mol. Sci. 2018, 19, 3344. [Google Scholar] [CrossRef] [Green Version]
- Hudson, E.R.; Pan, D.A.; James, J.; Lucocq, J.M.; Hawley, S.A.; Green, K.A.; Baba, O.; Terashima, T.; Hardie, D.G. A Novel Domain in AMP-Activated Protein Kinase Causes Glycogen Storage Bodies Similar to Those Seen in Hereditary Cardiac Arrhythmias. Curr. Biol. 2003, 13, 861–866. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.; Ghilagaber, S.; Nikolaev, A.; Hardie, D.G. The Glycogen-Binding Domain on the AMPK β Subunit Allows the Kinase to Act as a Glycogen Sensor. Cell Metab. 2009, 9, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Polekhina, G.; Gupta, A.; Michell, B.J.; van Denderen, B.; Murthy, S.; Feil, S.C.; Jennings, I.G.; Campbell, D.J.; Witters, L.A.; Parker, M.W.; et al. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr. Biol. 2003, 13, 867–871. [Google Scholar] [CrossRef] [Green Version]
- Polekhina, G.; Gupta, A.; van Denderen, B.J.; Feil, S.C.; Kemp, B.E.; Stapleton, D.; Parker, M.W. Structural Basis for Glycogen Recognition by AMP-Activated Protein Kinase. Structure 2005, 13, 1453–1462. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszewski, J.F.; Jørgensen, S.B.; Hellsten, Y.; Hardie, D.G.; Richter, E.A. Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes 2002, 51, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Watt, M.J.; McGee, S.L.; Chan, S.; Hargreaves, M.; Febbraio, M.A.; Stapleton, D.; Kemp, B.E. Reduced glycogen availability is associated with increased AMPKα2 activity, nuclear AMPKα2 protein abundance, and GLUT4 mRNA expression in contracting human skeletal muscle. Appl. Physiol. Nutr. Metab. 2006, 31, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Watt, M.J.; Steinberg, G.R.; Chan, S.; Garnham, A.; Kemp, B.E.; Febbraio, M.A. β-adrenergic stimulation of skeletal muscle HSL can be overridden by AMPK signaling. FASEB J. 2004, 18, 1445–1446. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszewski, J.F.P.; Macdonald, C.; Nielsen, J.N.; Hellsten, Y.; Hardie, D.G.; Kemp, B.E.; Kiens, B.; Richter, E.A. Regulation of 5′AMP-activated protein kinase activity and substrate utilization in exercising human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E813–E822. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.J.; Whitfield, J.; Janzen, N.R.; Belhaj, M.R.; Galic, S.; Murray-Segal, L.; Smiles, W.J.; Ling, N.X.Y.; Dite, T.A.; Scott, J.W.; et al. Genetic loss of AMPK-glycogen binding destabilises AMPK and disrupts metabolism. Mol. Metab. 2020, 41, 101048. [Google Scholar] [CrossRef]
- López-Soldado, I.; Bertini, A.; Adrover, A.; Duran, J.; Guinovart, J.J. Maintenance of liver glycogen during long-term fasting preserves energy state in mice. FEBS Lett. 2020, 594, 1698–1710. [Google Scholar] [CrossRef]
- Wijngaarden, M.A.; Van Der Zon, G.C.; van Dijk, K.W.; Pijl, H.; Guigas, B. Effects of prolonged fasting on AMPK signaling, gene expression, and mitochondrial respiratory chain content in skeletal muscle from lean and obese individuals. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1012–E1021. [Google Scholar] [CrossRef] [Green Version]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.-B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferre, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Crosson, S.M.; Khan, A.; Printen, J.; Pessin, J.E.; Saltiel, A.R. PTG gene deletion causes impaired glycogen synthesis and developmental insulin resistance. J. Clin. Investig. 2003, 111, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.M.; Dobrzyn, A.; Lee, S.-H.; Dobrzyn, P.; Miyazaki, M.; Ntambi, J.M. Stearoyl-CoA desaturase 1 deficiency increases insulin signaling and glycogen accumulation in brown adipose tissue. Am. J. Physiol. Endocrionol. Metab. 2005, 288, E381–E387. [Google Scholar] [CrossRef] [Green Version]
- Ceperuelo-Mallafré, V.; Ejarque, M.; Serena, C.; Duran, X.; Montori-Grau, M.; Rodríguez, M.A.; Yanes, O.; Núñez-Roa, C.; Roche, K.; Puthanveetil, P.; et al. Adipose tissue glycogen accumulation is associated with obesity-linked inflammation in humans. Mol. Metab. 2016, 5, 5–18. [Google Scholar] [CrossRef]
- Flores-Opazo, M.; Trieu, J.; Naim, T.; Valladares-Ide, D.; Zbinden-Foncea, H.; Stapleton, D. Defective fasting-induced PKA activation impairs adipose tissue glycogen degradation in obese Zucker rats. Int. J. Obes. 2020, 44, 500–509. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, L.; Li, B.; Jiang, H.; Duan, Y.; Xie, Z.; Shuai, L.; Li, J.; Li, J. AMP-Activated Protein Kinase (AMPK) Regulates Energy Metabolism through Modulating Thermogenesis in Adipose Tissue. Front. Physiol. 2018, 9, 122. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [Green Version]
- Hughey, C.C.; James, F.D.; Bracy, D.P.; Donahue, E.P.; Young, J.D.; Viollet, B.; Foretz, M.; Wasserman, D.H. Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice. J. Biol. Chem. 2017, 292, 20125–20140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Soldado, I.; Zafra, D.; Duran, J.; Adrover, A.; Calbó, J.; Guinovart, J.J. Liver Glycogen Reduces Food Intake and Attenuates Obesity in a High-Fat Diet–Fed Mouse Model. Diabetes 2015, 64, 796–807. [Google Scholar] [CrossRef]
- Saad, M.F.; Knowler, W.C.; Pettitt, D.J.; Nelson, R.G.; Mott, D.M.; Bennett, P.H. Sequential changes in serum insulin concentration during development of non-insulin-dependent diabetes. Lancet 1989, 1, 1356–1359. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamshoum, H.; Medak, K.D.; Townsend, L.K.; Ashworth, K.E.; Bush, N.D.; Halm, M.K.; Kemp, B.E.; Wright, D.C. AMPK β1 activation suppresses antipsychotic-induced hyperglycemia in mice. FASEB J. 2019, 33, 14010–14021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; O’Neill, H.M.; Dzamko, N.L.; Galic, S.; Naim, T.; Koopman, R.; Jørgensen, S.B.; Honeyman, J.; Hewitt, K.; Chen, Z.-P.; et al. Whole Body Deletion of AMP-activated Protein Kinase β2 Reduces Muscle AMPK Activity and Exercise Capacity. J. Biol. Chem. 2010, 285, 37198–37209. [Google Scholar] [CrossRef] [Green Version]
- Conlee, R.K.; Lawler, R.M.; Ross, P.E. Effects of Glucose or Fructose Feeding on Glycogen Repletion in Muscle and Liver after Exercise or Fasting. Ann. Nutr. Metab. 1987, 31, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Tantiwong, P.; Stuenaes, J.T.; Molina-Carrion, M.; DeFronzo, R.A.; Sakamoto, K.; Musi, N. Effect of acute exercise on glycogen synthase in muscle from obese and diabetic subjects. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E82–E89. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.; Sakamoto, K.; McManus, E.J.; Alessi, D.R. Role of the PDK1-PKB-GSK3 pathway in regulating glycogen synthase and glucose uptake in the heart. FEBS Lett. 2005, 579, 3632–3638. [Google Scholar] [CrossRef] [Green Version]
- Prats, C.; Helge, J.W.; Nordby, P.; Qvortrup, K.; Ploug, T.; Dela, F.; Wojtaszewski, J.F.P. Dual Regulation of Muscle Glycogen Synthase during Exercise by Activation and Compartmentalization. J. Biol. Chem. 2009, 284, 15692–15700. [Google Scholar] [CrossRef] [Green Version]
- Skurat, A.V.; Roach, P.J. Phosphorylation of Sites 3a and 3b (Ser640 and Ser644) in the Control of Rabbit Muscle Glycogen Synthase. J. Biol. Chem. 1995, 270, 12491–12497. [Google Scholar] [CrossRef] [Green Version]
- Roach, P.J.; DePaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [Green Version]
- Irimia, J.M.; Rovira, J.; Nielsen, J.N.; Guerrero, M.; Wojtaszewski, J.F.P.; Cussó, R. Hexokinase 2, Glycogen Synthase and Phosphorylase Play a Key Role in Muscle Glycogen Supercompensation. PLoS ONE 2012, 7, e42453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irimia, J.M.; Tagliabracci, V.S.; Meyer, C.M.; Segvich, D.M.; DePaoli-Roach, A.A.; Roach, P.J. Muscle Glycogen Remodeling and Glycogen Phosphate Metabolism following Exhaustive Exercise of Wild Type and Laforin Knockout Mice. J. Biol. Chem. 2015, 290, 22686–22698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, L.M.; van Loon, L.J.C.; Hawley, J.A. Postexercise muscle glycogen resynthesis in humans. J. Appl. Physiol. 2017, 122, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and Skeletal Muscle Glucose Uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingst, J.R.; Bruhn, L.; Hansen, M.B.; Rosschou, M.F.; Birk, J.B.; Fentz, J.; Foretz, M.; Viollet, B.; Sakamoto, K.; Færgeman, N.J.; et al. Exercise-induced molecular mechanisms promoting glycogen supercompensation in human skeletal muscle. Mol. Metab. 2018, 16, 24–34. [Google Scholar] [CrossRef]
- Yeo, W.K.; McGee, S.L.; Carey, A.; Paton, C.D.; Garnham, A.P.; Hargreaves, M.; Hawley, J.A. Acute signalling responses to intense endurance training commenced with low or normal muscle glycogen. Exp. Physiol. 2010, 95, 351–358. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jorgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) 1 2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, B.; Ju, J.S.; Sasaki, Y.; Liu, X.; Jung, S.-R.; Higashida, K.; Lindquist, D.; Milbrandt, J. The AMPK β2 Subunit Is Required for Energy Homeostasis during Metabolic Stress. Mol. Cell. Biol. 2012, 32, 2837–2848. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.; van Denderen, B.J.; Hevener, A.L.; Jørgensen, S.B.; Honeyman, J.; Galic, S.; Chen, Z.-P.; Watt, M.J.; Campbell, D.J.; Steinberg, G.R.; et al. AMPK β1 Deletion Reduces Appetite, Preventing Obesity and Hepatic Insulin Resistance. J. Biol. Chem. 2010, 285, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-Y.; Irwin, P.M.; Nikolov, Z.L. Effects of mutations in the starch-binding domain of Bacillus macerans cyclodextrin glycosyltransferase. J. Biotechnol. 1998, 65, 191–202. [Google Scholar] [CrossRef]
- Péronnet, F.; Massicotte, D. Table of nonprotein respiratory quotient: An update. Can. J. Sport Sci. 1991, 16, 23–29. [Google Scholar]
- Bergmeyer, H.U. Methods of Enzymatic Analysis, 2nd ed.; Verlag Chemie: Weinheim, Germany; Academic Press: New York, NY, USA, 1974. [Google Scholar]
- Tachtsis, B.; Whitfield, J.; Hawley, J.A.; Hoffman, N.J. Omega-3 Polyunsaturated Fatty Acids Mitigate Palmitate-Induced Impairments in Skeletal Muscle Cell Viability and Differentiation. Front. Physiol. 2020, 11, 563. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | WT Tissue Mass (mg) | DKI Tissue Mass (mg) | p-Value |
|---|---|---|---|
| Pancreas | 120 ± 12.0 | 167 ± 16.5 * | 0.029 |
| Heart | 159 ± 10.3 | 176 ± 11.6 | 0.27 |
| Gastrocnemius | 306 ± 12.9 | 328 ± 17.1 | 0.11 |
| Soleus | 16 ± 1.8 | 14 ± 1.8 | 0.35 |
| Target | Primary Antibody | Incubation | Secondary Antibody |
|---|---|---|---|
| AMPK α p-T172 | CST 2531 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| AMPK α | CST 2532 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| AMPK β1/β2 | CST 4150 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| ACC p-S79 | CST 11818 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| ACC | CST 3662 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| Akt p-S473 | CST 9271 | 1.5 h | GAR CST 7074 |
| 1:1000 | Room temperature | 1:5000 | |
| Akt | CST 4691 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| AS160 p-T642 | CST 4288 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| AS160 | Abcam ab24469 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| GLUT4 | CST 2213 | Overnight | GAM CST 7076 |
| 1:1000 | 4 °C | 1:2000 | |
| GS p-S641 | CST 47043 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| GS | CST 3886 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:5000 | |
| Glycogenin1 | Abcam ab11171 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:2000 | |
| GBE1 | Abcam ab180596 | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:2000 | |
| GP | Antibody generated by B.E.K. laboratory | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:2000 | |
| GDE | Antibody generated by B.E.K. laboratory | Overnight | GAR CST 7074 |
| 1:1000 | 4 °C | 1:2000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janzen, N.R.; Whitfield, J.; Murray-Segal, L.; Kemp, B.E.; Hawley, J.A.; Hoffman, N.J. Mice with Whole-Body Disruption of AMPK-Glycogen Binding Have Increased Adiposity, Reduced Fat Oxidation and Altered Tissue Glycogen Dynamics. Int. J. Mol. Sci. 2021, 22, 9616. https://doi.org/10.3390/ijms22179616
Janzen NR, Whitfield J, Murray-Segal L, Kemp BE, Hawley JA, Hoffman NJ. Mice with Whole-Body Disruption of AMPK-Glycogen Binding Have Increased Adiposity, Reduced Fat Oxidation and Altered Tissue Glycogen Dynamics. International Journal of Molecular Sciences. 2021; 22(17):9616. https://doi.org/10.3390/ijms22179616
Chicago/Turabian StyleJanzen, Natalie R., Jamie Whitfield, Lisa Murray-Segal, Bruce E. Kemp, John A. Hawley, and Nolan J. Hoffman. 2021. "Mice with Whole-Body Disruption of AMPK-Glycogen Binding Have Increased Adiposity, Reduced Fat Oxidation and Altered Tissue Glycogen Dynamics" International Journal of Molecular Sciences 22, no. 17: 9616. https://doi.org/10.3390/ijms22179616
APA StyleJanzen, N. R., Whitfield, J., Murray-Segal, L., Kemp, B. E., Hawley, J. A., & Hoffman, N. J. (2021). Mice with Whole-Body Disruption of AMPK-Glycogen Binding Have Increased Adiposity, Reduced Fat Oxidation and Altered Tissue Glycogen Dynamics. International Journal of Molecular Sciences, 22(17), 9616. https://doi.org/10.3390/ijms22179616