
MECP2-Related Disorders in Males

Abstract
1. MECP2 Gene
1.1. General Characteristics of MECP2
1.2. One Gene, Multiple Phenotypes
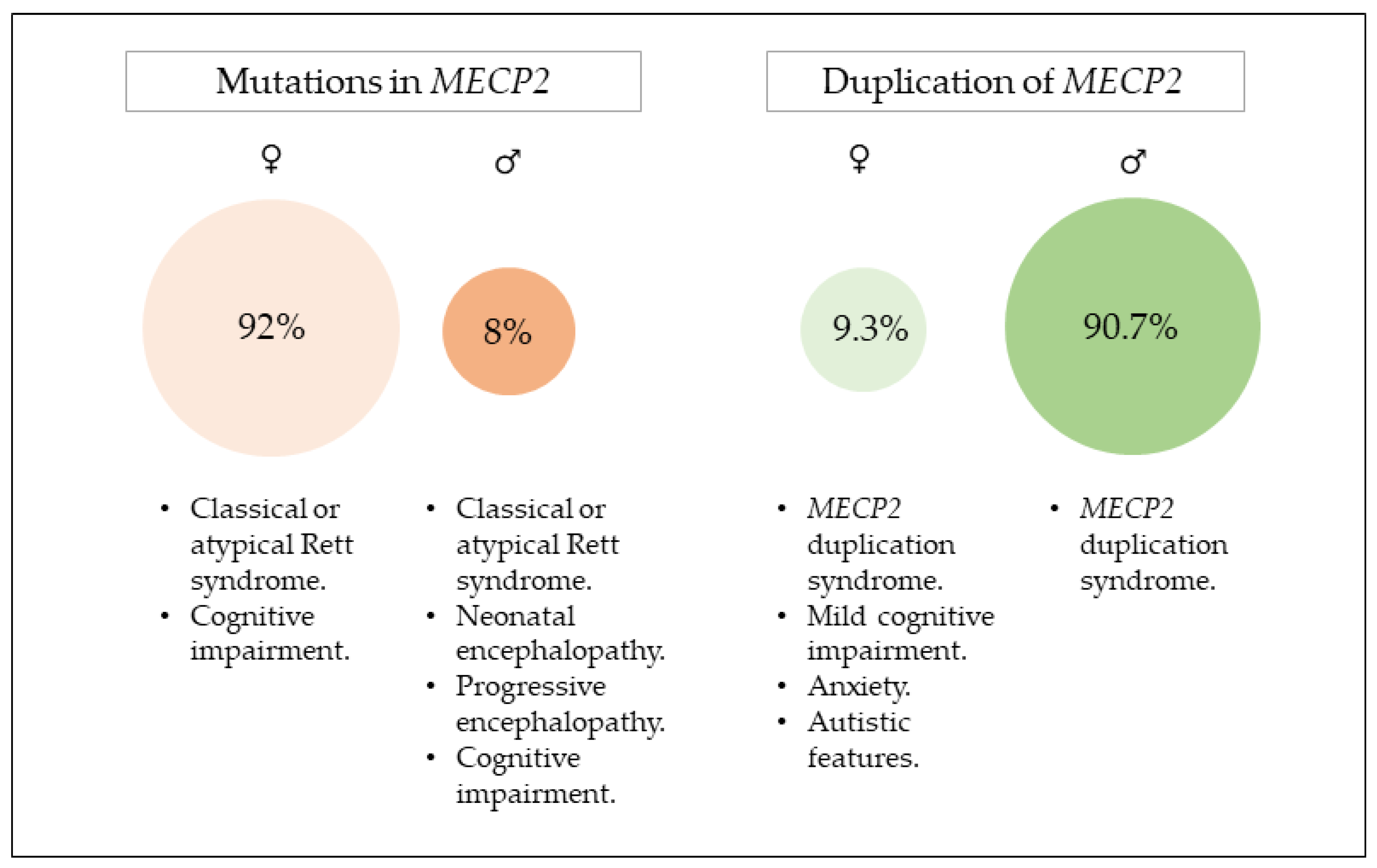
2. Mutations in MECP2
2.1. Clinical Presentation
2.2. Genotype–Phenotype Correlation
3. Duplication of MECP2
3.1. Clinical Presentations
3.1.1. Neurological Aspects
3.1.2. Immunological Aspects
3.1.3. Gastrointestinal Aspects
3.1.4. Dysmorphological Aspects
3.1.5. Evolution of the Syndrome with Age
3.2. Characteristics of the Duplication
3.3. Genotype–Phenotype Correlation
4. Modeling RTT and MDS for Future Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Xinhua, B.; Shengling, J.; Fuying, S.; Hong, P.; Meirong, L.; Wu, X.R. X chromosome inactivation in rett syndrome and Its correlations with mecp2 mutations and phenotype. J. Child Neurol. 2008, 23, 22–25. [Google Scholar] [CrossRef]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.M.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004, 36, 339–341. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Bardai, F.H.; Ma, C.; Price, V.; Rawat, V.; Verma, P.; Narayanan, V.; D’Mello, S.R. Isoform-specific toxicity of MeCP2 in postmitotic neurons: Suppression of neurotoxicity by FoxG1. J. Neurosci. 2012, 32, 2846–2855. [Google Scholar] [CrossRef]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.B.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef]
- Martínez De Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; De Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef]
- Sharma, K.; Singh, J.; Frost, E.E.; Pillai, P.P. MeCP2 in central nervous system glial cells: Current updates. Acta Neurobiol. Exp. 2018, 78, 30–40. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Brandt, V.L.; Zoghbi, H.Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: Prospects for future therapies. Lancet Neurol. 2020, 19, 689–698. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The molecular basis of MeCP2 function in the brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, F.; Rossi, R.L.; Galli, F.; Cobolli Gigli, C.; Gandaglia, A.; Kilstrup-Nielsen, C.; Landsberger, N. Rett syndrome and the urge of novel approaches to study MeCP2 functions and mechanisms of action. Neurosci. Biobehav. Rev. 2014, 46, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The genetic driver of Rett syndrome epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue-and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Sung, S.; Lee, J.; Zhang, X.; Houwink-Manville, I.; Song, H.-R.; Amir, R.E.; Budden, S.; Naidu, S.; Luiz, J.; et al. Rett Syndrome and beyond: Recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am. J. Hum. Genet 1999, 65, 1520–1529. [Google Scholar] [CrossRef]
- Armstrong, J.; Póo, P.; Pineda, M.; Aibar, E.; Geán, E.; Català, V.; Nia Monrós, E. Classic Rett syndrome in a Boy as a Result of Somatic Mosaicism for a MECP2 Mutation; Wiley-Liss: Hoboken, NJ, USA, 2001. [Google Scholar]
- Maiwald, R.; Bönte, A.; Jung, H.; Bitter, P.; Storm, Z.; Laccone, F.; Herkenrath, P. De novo MECP2 mutation in a 46, XX male patient with Rett syndrome. Neurogenetics 2002, 4, 107–108. [Google Scholar] [CrossRef]
- Lundvall, M.; Samuelsson, L.; Kyllerman, M. Male Rett phenotypes in T158M and R294X MeCP2-mutations. Neuropediatrics 2006, 37, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Psoni, S.; Sofocleous, C.; Traeger-Synodinos, J.; Kitsiou-Tzeli, S.; Kanavakis, E.; Fryssira-Kanioura, H. Phenotypic and genotypic variability in four males with MECP2 gene sequence aberrations including a novel deletion. Pediatr. Res. 2010, 67, 551–556. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef]
- Neul, J.L.; Lane, J.B.; Lee, H.S.; Geerts, S.; Barrish, J.O.; Annese, F.; Baggett, L.M.N.; Barnes, K.; Skinner, S.A.; Motil, K.J.; et al. Developmental delay in Rett syndrome: Data from the natural history study. J. Neurodev. Disord. 2014, 6, 20. [Google Scholar] [CrossRef]
- Couvert, P.; Bienvenu, T.; Aquaviva, C.; Poirier, K.; Moraine, C.; Gendrot, C.; Verloes, A.; Andrès, C.; Le Fevre, A.C.; Souville, I.; et al. MECP2 is highly mutated in X-linked mental retardation. Hum. Mol. Genet. 2001, 10, 941–946. [Google Scholar] [CrossRef]
- Yntema, H.G.; Kleesfstra, T.; Oudakker, A.R.; Romein, T.; de Vries, B.B.A.; Nillesen, W.; Sistermans, E.A.; Brunner, H.G.; Hamel, B.C.J.; van Bokhoven, H. Low frequency of MECP2 mutations in mentally retarded males. Eur. J. Hum. Genet. 2002, 10, 487–490. [Google Scholar] [CrossRef]
- Bourdon, V.; Philippe, C.; Martin, D.; Verlò, A.; Grandemenge, A.; Jonveaux, P. MECP2 mutations or polymorphisms in mentally retarded boys diagnostic implications. Mol Diagn 2003, 7, 3–7. [Google Scholar] [PubMed]
- Moog, U.; Van Roozendaal, K.; Smeets, E.; Tserpelis, D.; Devriendt, K.; Van Buggenhout, G.; Frijns, J.P.; Schrander-Stumpel, C. MECP2 mutations are an infrequent cause of mental retardation associated with neurological problems in male patients. Brain Dev. 2006, 28, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Ravn, K.; Nielsen, J.B.; Uldall, P.; Hansen, F.J. No correlation between phenotype and genotype in boys with a truncating MECP2 mutation. J Med Genet 2003, 40, e5. [Google Scholar] [CrossRef]
- Yntema, H.G.; Oudakker, A.R.; Kleefstra, T.; Hamel, B.C.J.; van Bokhoven, H.; Chelly, J.; Kalscheuer, V.M.; Fryns, J.P.; Raynaud, M.; Moizard, M.P.; et al. In-frame deletion in MECP2 causes mild nonspecific mental retardation. Am. J. Med. Genet. 2002, 107, 81–83. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. International Statistical Classification of Disease and Related Health Problems, 10th ed. World Health Organization: Geneva, Switzerland, 1992.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; Barber, J.P., Connolly, M.B., Crits-Christoph, P., Gladis, L., Siqueland, L., Eds.; American Psychiatric Pub: Washington, DC, USA, 1994; Volume 68, pp. 1027–1032. [Google Scholar]
- Hagberg, B.; Hanefeld, F.; Percy, A.; Skjeldal, O. An update on clinically applicable diagnostic criteria in Rett syndrome: Comments to Rett syndrome clinical criteria consensus panel satellite to European Paediatric Neurology Society Meeting Baden Baden, Germany, 11 September 2001. Eur. J. Paediatr. Neurol. 2002, 6, 293–297. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Monrós, E.; Armstrong, J.; Aibar, E.; Poo, P.; Canós, I.; Pineda, M. Rett syndrome in Spain: Mutation analysis and clinical correlations. Brain Dev. 2001, 23, S251–S253. [Google Scholar] [CrossRef]
- Kerr, A.M.; Nomura, Y.; Armstrong, D.; Anvret, M.; Belichenko, P.V.; Budden, S.; Cass, H.; Christodoulou, J.; Clarke, A.; Ellaway, C.; et al. Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev. 2001, 23, 208–211. [Google Scholar] [CrossRef]
- Colvin, L.; Fyfe, S.; Leonard, S.; Schiavello, T.; Ellaway, C.; De Klerk, N.; Christodoulou, J.; Msall, M. Describing the phenotype in Rett syndrome using a population database. Arch. Dis. Child. 2003, 88, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Huppke, P.; Gärtner, J. Molecular diagnosis of Rett Syndrome. J. Child Neurol. 2005, 20, 732–736. [Google Scholar] [CrossRef]
- Moretti, P.; Zoghbi, H.Y. MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev. 2006, 16, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Villard, L. MECP2 mutations in males. J. Med. Genet. 2007, 44, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L. The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin. Neurosci. 2012, 14, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, T.; Matsuo, M.; Jing, J.J.; Tabara, Y.; Kitsuki, K.; Yamagata, H.; Kan, Y.; Miki, T.; Ishii, K.; Kondo, I. Classic Rett syndrome in a boy with R133C mutation of MECP2. Brain Dev. 2005, 27, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Benke, T.A.; Marsh, E.D.; Skinner, S.A.; Merritt, J.; Lieberman, D.N.; Standridge, S.; Feyma, T.; Heydemann, P.; Peters, S.; et al. The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2019, 180, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Buyse, I.M.; Fang, P.; Hoon, K.T.; Amir, R.E.; Zoghbi, H.Y.; Roa, B.B. Diagnostic testing for Rett Syndrome by DHPLC and direct sequencing analysis of the MECP2 Gene: Identification of several novel mutations and polymorphisms. Am. J. Hum. Genet 2000, 67, 1428–1436. [Google Scholar] [CrossRef]
- Bianciardi, L.; Fichera, M.; Failla, P.; Di Marco, C.; Grozeva, D.; Mencarelli, M.A.; Spiga, O.; Mari, F.; Meloni, I.; Raymond, L.; et al. MECP2 missense mutations outside the canonical MBD and TRD domains in males with intellectual disability. J. Hum. Genet. 2016, 61, 95–101. [Google Scholar] [CrossRef][Green Version]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Parker, M.J.; Archer, H.; Firth, H.V.; Park, S.M.; Canham, N.; Holder, S.E.; Wilson, M.; et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 2014, 94, 618–624. [Google Scholar] [CrossRef]
- Vidal, S.; Brandi, N.; Pacheco, P.; Maynou, J.; Fernandez, G.; Xiol, C.; Pascual-Alonso, A.; Pineda, M.; O’Callaghan, M.; Garcia-Cazorla, À.; et al. The most recurrent monogenic disorders that overlap with the phenotype of Rett syndrome. Eur. J. Paediatr. Neurol. 2019, 23, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Schönewolf-Greulich, B.; Bisgaard, A.M.; Dunø, M.; Jespersgaard, C.; Rokkjær, M.; Hansen, L.K.; Tsoutsou, E.; Sofokleous, C.; Topcu, M.; Kaur, S.; et al. Mosaic MECP2 variants in males with classical Rett syndrome features, including stereotypical hand movements. Clin. Genet. 2019, 95, 403–408. [Google Scholar] [CrossRef]
- Gomot, M.; Gendrot, C.; Verloes, A.; Raynaud, M.; David, A.; Yntema, H.G.; Dessay, S.; Kalscheuer, V.; Frints, S.; Couvert, P.; et al. MECP2 gene mutations in non-syndromic X-linked mental retardation: Phenotype-genotype correlation. Am. J. Med. Genet. 2003, 123, 129–139. [Google Scholar] [CrossRef]
- Sanlaville, D.; Prieur, M.; de Blois, M.C.; Genevieve, D.; Lapierre, J.M.; Ozilou, C.; Picq, M.; Gosset, P.; Morichon-Delvallez, N.; Munnich, A.; et al. Functional disomy of the Xq28 chromosome region. Eur. J. Hum. Genet. 2005, 13, 579–585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lubs, H.; Abidi, F.; Blaymore Bier, J.-A.; Abuelo, D.; Ouzts, L.; Voeller, K.; Fennell, E.; Stevenson, R.E.; Schwartz, C.E.; Arena, F. XLMR syndrome characterized by multiple respiratory infections, hypertelorism, severe CNS deterioration and early death localizes to distal Xq28. J. Med. Genet 1999, 85, 243–248. [Google Scholar] [CrossRef]
- Friez, M.J.; Jones, J.R.; Clarkson, K.; Lubs, H.; Abuelo, D.; Bier, J.A.B.; Pai, S.; Simensen, R.; Williams, C.; Giampietro, P.F.; et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics 2006, 118, e1687–e1695. [Google Scholar] [CrossRef] [PubMed]
- Meins, M.; Lehmann, J.; Gerresheim, F.; Herchenbach, J.; Hagedorn, M.; Hameister, K.; Epplen, J.T. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J. Med. Genet. 2005, 42, e12. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gecz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef]
- Shao, L.; Shaw, C.A.; Lu, X.Y.; Sahoo, T.; Bacino, C.A.; Lalani, S.R.; Stankiewicz, P.; Yatsenko, S.A.; Li, Y.; Neill, S.; et al. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: A study of 5380 cases. Am. J. Med. Genet. Part A 2008, 146A, 2242–2251. [Google Scholar] [CrossRef]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 duplication syndrome. Am. J. Med. Genet. Part A 2010, 152A, 1079–1088. [Google Scholar] [CrossRef]
- de Brouwer, A.P.M.; Yntema, H.G.; Kleefstra, T.; Lugtenberg, D.; Oudakker, A.R.; de Vries, B.B.A.; van Bokhoven, H.; Van Esch, H.; Frints, S.G.M.; Froyen, G.; et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum. Mutat. 2007, 28, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, D.; Kleefstra, T.; Oudakker, A.R.; Nillesen, W.M.; Yntema, H.G.; Tzschach, A.; Raynaud, M.; Rating, D.; Journel, H.; Chelly, J.; et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur. J. Hum. Genet. 2009, 17, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Hayashi, S.; Nakane, T.; Imoto, I.; Kurosawa, K.; Mizuno, S.; Okamoto, N.; Kato, M.; Yoshihashi, H.; Kubota, T.; et al. The incidence of hypoplasia of the corpus callosum in patients with dup (X)(q28) involving MECP2 is associated with the location of distal breakpoints. Am. J. Med. Genet. Part A 2012, 158A, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Del Gaudio, D.; Fang, P.; Scaglia, F.; Ward, P.A.; Craigen, W.J.; Glaze, D.G.; Neul, J.L.; Patel, A.; Lee, J.A.; Irons, M.; et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 2006, 8, 784–792. [Google Scholar] [CrossRef]
- Campos, M.; Churchman, S.M.; Santos-Rebouças, C.B.; Ponchel, F.; Pimentel, M.M.G. High frequency of nonrecurrent MECP2 duplications among Brazilian males with mental retardation. J. Mol. Neurosci. 2010, 41, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, D.; De Brouwer, A.P.M.; Kleefstra, T.; Oudakker, A.R.; Frints, S.G.M.; Schrander-Stumpel, C.T.R.M.; Fryns, J.P.; Jensen, L.R.; Chelly, J.; Moraine, C.; et al. Chromosomal copy number changes in patients with non-syndromic X linked mental retardation detected by array CGH. J. Med. Genet. 2006, 43, 362–370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosenberg, C.; Knijnenburg, J.; Bakker, E.; Vianna-Morgante, A.M.; Sloos, W.; Otto, P.A.; Kriek, M.; Hansson, K.; Krepischi-Santos, A.C.V.; Fiegler, H.; et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: Clinical significance of imbalances present both in affected children and normal parents. J. Med. Genet. 2006, 43, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, I.; Rodríguez-Revenga, L.; Armengol, L.; González, E.; Rodriguez, B.; Badenas, C.; Sánchez, A.; Martínez, F.; Guitart, M.; Fernández, I.; et al. X-chromosome tiling path array detection of copy number variants in patients with chromosome X-linked mental retardation. BMC Genom. 2007, 8, 443. [Google Scholar] [CrossRef] [PubMed]
- Bauters, M.; Van Esch, H.; Friez, M.J.; Boespflug-Tanguy, O.; Zenker, M.; Vianna-Morgante, A.M.; Rosenberg, C.; Ignatius, J.; Raynaud, M.; Hollanders, K.; et al. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res. 2008, 18, 847–858. [Google Scholar] [CrossRef]
- Smyk, M.; Obersztyn, E.; Nowakowska, B.; Nawara, M.; Cheung, S.W.; Mazurczak, T.; Stankiewicz, P.; Bocian, E. Different-sized duplications of Xq28, including MECP2, in three males with mental retardation, absent or delayed speech, and recurrent infections. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147B, 799–806. [Google Scholar] [CrossRef]
- Velinov, M.; Novelli, A.; Gu, H.; Fenko, M.; Dolzhanskaya, N.; Bernardini, L.; Capalbo, A.; Dallapiccola, B.; Jenkins, E.C.; Brown, W.T. De-novo 2.15 Mb terminal Xq duplication involving MECP2 but not L1CAM gene in a male patient with mental retardation. Clin. Dysmorphol. 2009, 18, 9–12. [Google Scholar] [CrossRef]
- Kirk, E.P.; Malaty-Brevaud, V.; Martini, N.; Lacoste, C.; Levy, N.; Maclean, K.; Davies, L.; Philip, N.; Badens, C. The clinical variability of the MECP2 duplication syndrome: Description of two families with duplications excluding L1CAM and FLNA. Clin. Genet. 2009, 75, 301–303. [Google Scholar] [CrossRef]
- Clayton-Smith, J.; Walters, S.; Hobson, E.; Burkitt-Wright, E.; Smith, R.; Toutain, A.; Amiel, J.; Lyonnet, S.; Mansour, S.; Fitzpatrick, D.; et al. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur. J. Hum. Genet. 2009, 17, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Prescott, T.E.; Rødningen, O.K.; Bjørnstad, A.; Stray-Pedersen, A. Two brothers with a microduplication including the MECP2 gene: Rapid head growth in infancy and resolution of susceptibility to infection. Clin. Dysmorphol. 2009, 18, 78–82. [Google Scholar] [CrossRef]
- Carvalho, C.M.B.B.; Zhang, F.; Liu, P.; Patel, A.; Sahoo, T.; Bacino, C.A.; Shaw, C.; Peacock, S.; Pursley, A.; Tavyev, J.Y.; et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009, 18, 2188–2203. [Google Scholar] [CrossRef] [PubMed]
- Echenne, B.; Roubertie, A.; Lugtenberg, D.; Kleefstra, T.; Hamel, B.C.J.; Van Bokhoven, H.; Lacombe, D.; Philippe, C.; Jonveaux, P.; de Brouwer, A.P.M. Neurologic aspects of MECP2 gene duplication in male patients. Pediatr. Neurol. 2009, 41, 187–191. [Google Scholar] [CrossRef]
- Ramocki, M.B.; Peters, S.U.; Tavyev, Y.J.; Zhang, F.; Carvalho, C.M.B.; Schaaf, C.P.; Richman, R.; Fang, P.; Glaze, D.G.; Lupski, J.R.; et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MECP2 duplication syndrome. Ann. Neurol. 2009, 66, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Belligni, E.F.; Palmer, R.W.; Hennekam, R.C.M. MECP2 duplication in a patient with congenital central hypoventilation. Am. J. Med. Genet. Part A 2010, 152, 1591–1593. [Google Scholar] [CrossRef]
- Bartsch, O.; Gebauer, K.; Lechno, S.; Van Esch, H.; Froyen, G.; Bonin, M.; Seidel, J.; Thamm-Mücke, B.; Horn, D.; Klopocki, E.; et al. Four unrelated patients with Lubs X-linked mental retardation syndrome and different Xq28 duplications. Am. J. Med. Genet. Part A 2010, 152, 305–312. [Google Scholar] [CrossRef]
- Reardon, W.; Donoghue, V.; Murphy, A.M.; King, M.D.; Mayne, P.D.; Horn, N.; Birk Møller, L. Progressive cerebellar degenerative changes in the severe mental retardation syndrome caused by duplication of MECP2 and adjacent loci on Xq28. Eur. J. Pediatr. 2010, 169, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Makrythanasis, P.; Moix, I.; Gimelli, S.; Fluss, J.; Aliferis, K.; Antonarakis, S.E.; Morris, M.A.; Béna, F.; Bottani, A. De novo duplication of MECP2 in a girl with mental retardation and no obvious dysmorphic features. Clin. Genet. 2010, 78, 175–180. [Google Scholar] [CrossRef]
- Honda, S.; Hayashi, S.; Imoto, I.; Toyama, J.; Okazawa, H.; Nakagawa, E.; Goto, Y.I.; Inazawa, J. Copy-number variations on the X chromosome in Japanese patients with mental retardation detected by array-based comparative genomic hybridization analysis. J. Hum. Genet. 2010, 55, 590–599. [Google Scholar] [CrossRef]
- Fernández, R.M.; Núñez-Torres, R.; González-Meneses, A.; Antiñolo, G.; Borrego, S. Novel association of severe neonatal encephalopathy and Hirschsprung disease in a male with a duplication at the Xq28 region. BMC Med. Genet. 2010, 11, 137. [Google Scholar] [CrossRef]
- Jezela-Stanek, A.; Ciara, E.; Juszczak, M.; Pelc, M.; Materna-Kiryluk, A.; Krajewska-Walasek, M.; Cryptic, X. Autosome translocation in a boy—Delineation of the phenotype. Pediatr. Neurol. 2011, 44, 221–224. [Google Scholar] [CrossRef]
- Breman, A.M.; Ramocki, M.B.; Kang, S.H.L.; Williams, M.; Freedenberg, D.; Patel, A.; Bader, P.I.; Cheung, S.W. MECP2 duplications in six patients with complex sex chromosome rearrangements. Eur. J. Hum. Genet. 2011, 19, 409–415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grasshoff, U.; Bonin, M.; Goehring, I.; Ekici, A.; Dufke, A.; Cremer, K.; Wagner, N.; Rossier, E.; Jauch, A.; Walter, M.; et al. De novo MECP2 duplication in two females with random X-inactivation and moderate mental retardation. Eur. J. Hum. Genet. 2011, 19, 507–512. [Google Scholar] [CrossRef]
- Budisteanu, M.; Papuc, S.M.; Tutulan-Cunita, A.; Budisteanu, B.; Arghir, A. Novel clinical finding in MECP2 duplication syndrome. Eur. Child Adolesc. Psychiatry 2011, 20, 373–375. [Google Scholar] [CrossRef]
- Mayo, S.; Monfort, S.; Roselló, M.; Orellana, C.; Oltra, S.; Armstrong, J.; Català, V.; Martínez, F. De novo interstitial triplication of MECP2 in a girl with neurodevelopmental disorder and random X chromosome inactivation. Cytogenet. Genome Res. 2011, 135, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Ramocki, M.B.; Pehlivan, D.; Franco, L.M.; Gonzaga-Jauregui, C.; Fang, P.; McCall, A.; Pivnick, E.K.; Hines-Dowell, S.; Seaver, L.H.; et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat. Genet. 2011, 43, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Utine, G.E.; Kiper, P.Ö.; Alanay, Y.; Haliloǧlu, G.; Aktaş, D.; Boduroǧlu, K.; Tunçbilek, E.; Alikaşifoǧlu, M. Searching for copy number changes in nonsyndromic X-linked intellectual disability. Mol. Syndromol. 2012, 2, 64–71. [Google Scholar] [CrossRef]
- Tang, S.S.; Fernandez, D.; Lazarou, L.P.; Singh, R.; Fallon, P. MECP2 triplication in 3 brothers—A rarely described cause of familial neurological regression in boys. Eur. J. Paediatr. Neurol. 2012, 16, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Satomura, S.; Hayashi, S.; Imoto, I.; Nakagawa, E.; Goto, Y.I.; Inazawa, J. Concomitant microduplications of MECP2 and ATRX in male patients with severe mental retardation. J. Hum. Genet. 2012, 57, 73–77. [Google Scholar] [CrossRef]
- Bijlsma, E.K.; Collins, A.; Papa, F.T.; Tejada, M.I.; Wheeler, P.; Peeters, E.A.J.; Gijsbers, A.C.J.; van de Kamp, J.M.; Kriek, M.; Losekoot, M.; et al. Xq28 duplications including MECP2 in five females: Expanding the phenotype to severe mental retardation. Eur. J. Med. Genet. 2012, 55, 404–413. [Google Scholar] [CrossRef]
- Sanmann, J.N.; Bishay, D.L.; Starr, L.J.; Bell, C.A.; Pickering, D.L.; Stevens, J.M.; Kahler, S.G.; Olney, A.H.; Schaefer, G.B.; Sanger, W.G. Characterization of six novel patients with MECP2 duplications due to unbalanced rearrangements of the X chromosome. Am. J. Med. Genet. Part A 2012, 158A, 1285–1291. [Google Scholar] [CrossRef]
- Vignoli, A.; Borgatti, R.; Peron, A.; Zucca, C.; Ballarati, L.; Bonaglia, C.; Bellini, M.; Giordano, L.; Romaniello, R.; Bedeschi, M.F.; et al. Electroclinical pattern in MECP2 duplication syndrome: Eight new reported cases and review of literature. Epilepsia 2012, 53, 1146–1155. [Google Scholar] [CrossRef]
- Xu, X.; Xu, Q.; Zhang, Y.; Zhang, X.; Cheng, T.; Wu, B.; Ding, Y.; Lu, P.; Zheng, J.; Zhang, M.; et al. A case report of Chinese brothers with inherited MECP2-containing duplication: Autism and intellectual disability, but not seizures or respiratory infections. BMC Med. Genet. 2012, 13, 75. [Google Scholar] [CrossRef]
- Yang, T.; Ramocki, M.B.; Neul, J.L.; Lu, W.; Roberts, L.; Knight, J.; Ward, C.S.; Zoghbi, H.Y.; Kheradmand, F.; Corry, D.B. Overexpression of methyl-CpG binding protein 2 impairs TH1 responses. Sci. Transl. Med. 2012, 4, 163ra158. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Okamoto, N.; Ito, M.; Arai, Y.; Momosaki, K.; Togawa, M.; Maegaki, Y.; Sugawara, M.; Shimojima, K.; Osawa, M.; et al. MECP2 duplication syndrome in both genders. Brain Dev. 2013, 35, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Okamoto, N.; Hirasawa, K.; Yoshii, K.; Tani, Y.; Sugawara, M.; Shimojima, K.; Osawa, M.; Yamamoto, T. Clinical manifestations of Xq28 functional disomy involving MECP2 in one female and two male patients. Am. J. Med. Genet. Part A 2013, 161A, 1779–1785. [Google Scholar] [CrossRef]
- Wax, J.R.; Pinette, M.G.; Smith, R.; Chard, R.; Cartin, A. Second-trimester prenasal and prefrontal skin thickening-Association with MECP2 triplication syndrome. J. Clin. Ultrasound 2013, 41, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.U.; Hundley, R.J.; Wilson, A.K.; Carvalho, C.M.B.; Lupski, J.R.; Ramocki, M.B. Brief report: Regression timing and associated features in MECP2 duplication syndrome. J. Autism Dev. Disord. 2013, 43, 2484–2490. [Google Scholar] [CrossRef]
- Scott Schwoerer, J.; Laffin, J.; Haun, J.; Raca, G.; Friez, M.J.; Giampietro, P.F. MECP2 duplication: Possible cause of severe phenotype in females. Am. J. Med. Genet. Part A 2014, 164A, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Novara, F.; Simonati, A.; Sicca, F.; Battini, R.; Fiori, S.; Contaldo, A.; Criscuolo, L.; Zuffardi, O.; Ciccone, R. MECP2 duplication phenotype in symptomatic females: Report of three further cases. Mol. Cytogenet. 2014, 7, 10. [Google Scholar] [CrossRef]
- Fukushi, D.; Yamada, K.; Nomura, N.; Naiki, M.; Kimura, R.; Yamada, Y.; Kumagai, T.; Yamaguchi, K.; Miyake, Y.; Wakamatsu, N. Clinical characterization and identification of duplication breakpoints in a Japanese family with Xq28 duplication syndrome including MECP2. Am. J. Med. Genet. Part A 2014, 164A, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Kölsch, U.; Krüger, R.; Unterwalder, N.; Hameister, K.; Kaiser, F.M.; Vignoli, A.; Rossi, R.; Botella, M.P.; Budisteanu, M.; et al. Infectious and immunologic phenotype of MECP2 Duplication syndrome. J. Clin. Immunol. 2015, 35, 168–181. [Google Scholar] [CrossRef]
- Miyatake, S.; Koshimizu, E.; Fujita, A.; Fukai, R.; Imagawa, E.; Ohba, C.; Kuki, I.; Nukui, M.; Araki, A.; Makita, Y.; et al. Detecting copy-number variations in whole-exome sequencing data using the exome hidden markov model: An “exome-first” approach. J. Hum. Genet. 2015, 60, 175–182. [Google Scholar] [CrossRef]
- Trobaugh-Lotrario, A.; Martin, J.; López-Terrada, D. Hepatoblastoma in a male with MECP2 duplication syndrome. Am. J. Med. Genet. Part A 2016, 170A, 790–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, Y.; Yang, Y.; Bao, X. MECP2 duplication syndrome in a Chinese family. BMC Med. Genet. 2015, 16, 112. [Google Scholar] [CrossRef]
- El Chehadeh, S.; Faivre, L.; Mosca-Boidron, A.L.; Malan, V.; Amiel, J.; Nizon, M.; Touraine, R.; Prieur, F.; Pasquier, L.; Callier, P.; et al. Large national series of patients with Xq28 duplication involving MECP2: Delineation of brain MRI abnormalities in 30 affected patients. Am. J. Med. Genet. Part A 2016, 170A, 116–129. [Google Scholar] [CrossRef]
- Nageshappa, S.; Carromeu, C.; Trujillo, C.A.; Mesci, P.; Espuny-Camacho, I.; Pasciuto, E.; Vanderhaeghen, P.; Verfaillie, C.M.; Raitano, S.; Kumar, A.; et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 2016, 21, 178–188. [Google Scholar] [CrossRef]
- Signorini, C.; De Felice, C.; Leoncini, S.; Møller, R.S.; Zollo, G.; Buoni, S.; Cortelazzo, A.; Guerranti, R.; Durand, T.; Ciccoli, L.; et al. MECP2 duplication syndrome: Evidence of enhanced oxidative stress. A comparison with Rett syndrome. PLoS ONE 2016, 11, e0150101. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Pan, H.; Li, L.; Wu, H.; Wang, S.; Ma, Y.; Qi, Y. Chromosome Xq28 duplication encompassing MECP2: Clinical and molecular analysis of 16 new patients from 10 families in China. Eur. J. Med. Genet. 2016, 59, 347–353. [Google Scholar] [CrossRef] [PubMed]
- San Antonio-Arce, V.; Fenollar-Cortés, M.; Ionescu, R.O.; DeSantos-Moreno, T.; Gallego-Merlo, J.; Cámara, F.J.I.; Pérez, M.C.O. MECP2 Duplications in symptomatic females. Child Neurol. Open 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Tsuji-Hosokawa, A.; Matsuda, N.; Kurosawa, K.; Kashimada, K.; Morio, T. A case of MECP2 duplication syndrome with gonadotropin-dependent precocious puberty. Horm. Res. Paediatr. 2017, 87, 271–276. [Google Scholar] [CrossRef]
- Ha, K.; Shen, Y.; Graves, T.; Kim, C.H.; Kim, H.G. The presence of two rare genomic syndromes, 1q21 deletion and Xq28 duplication, segregating independently in a family with intellectual disability. Mol. Cytogenet. 2016, 9, 74. [Google Scholar] [CrossRef]
- Lim, Z.; Downs, J.; Wong, K.; Ellaway, C.; Leonard, H. Expanding the clinical picture of the MECP2 Duplication syndrome. Clin. Genet. 2017, 91, 557–563. [Google Scholar] [CrossRef]
- El Chehadeh, S.; Touraine, R.; Prieur, F.; Reardon, W.; Bienvenu, T.; Chantot-Bastaraud, S.; Doco-Fenzy, M.; Landais, E.; Philippe, C.; Marle, N.; et al. Xq28 duplication including MECP2 in six unreported affected females: What can we learn for diagnosis and genetic counselling? Clin. Genet. 2017, 91, 576–588. [Google Scholar] [CrossRef]
- Moirangthem, A.; Tuteja Bhatia, M.; Srivastava, P.; Mandal, K.; Rai, A.; Phadke, S.R. Expansion of the phenotypic spectrum in three families of methyl CpG-binding protein 2 duplication syndrome. Clin. Dysmorphol. 2017, 26, 73–77. [Google Scholar] [CrossRef]
- Yon, D.K.; Park, J.E.; Kim, S.J.; Shim, S.H.; Chae, K.Y. A sibship with duplication of Xq28 inherited from the mother; genomic characterization and clinical outcomes. BMC Med. Genet. 2017, 18, 1–9. [Google Scholar] [CrossRef]
- Li, X.; Xie, H.; Chen, Q.; Yu, X.; Yi, Z.; Li, E.; Zhang, T.; Wang, J.; Zhong, J.; Chen, X. Clinical and molecular genetic characterization of familial MECP2 duplication syndrome in a Chinese family. BMC Med. Genet. 2017, 18, 131. [Google Scholar] [CrossRef]
- Deshwar, A.R.; Dupuis, L.; Bergmann, C.; Stavropoulos, J.; Mendoza-Londono, R. Severe rhizomelic shortening in a child with a complex duplication/deletion rearrangement of chromosome X. Am. J. Med. Genet. Part A 2018, 176A, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Krüger, R.; Kölsch, U.; Unterwalder, N.; Meisel, C.; Wahn, V.; Von Bernuth, H. Antibiotic prophylaxis, immunoglobulin substitution and supportive measures prevent infections in MECP2 duplication syndrome. Pediatr. Infect. Dis. J. 2018, 37, 466–468. [Google Scholar] [CrossRef]
- Miguet, M.; Faivre, L.; Amiel, J.; Nizon, M.; Touraine, R.; Prieur, F.; Pasquier, L.; Lefebvre, M.; Thevenon, J.; Dubourg, C.; et al. Further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. J. Med. Genet. 2018, 55, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Pitzianti, M.B.; Palombo, A.S.; Esposito, S.; Pasini, A. Rett syndrome in males: The different clinical course in two brothers with the same microduplication MECP2 Xq28. Int. J. Environ. Res. Public Health 2019, 16, 3075. [Google Scholar] [CrossRef]
- Kanai, S.; Okanishi, T.; Fujimoto, A.; Itamura, S.; Baba, S.; Nishimura, M.; Itomi, K.; Enoki, H. Successful corpus callosotomy for post-encephalopathic refractory epilepsy in a patient with MECP2 duplication syndrome. Brain Dev. 2019, 41, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Marafi, D.; Suter, B.; Schultz, R.; Glaze, D.; Pavlik, V.N.; Goldman, A.M. Spectrum and time course of epilepsy and the associated cognitive decline in MECP2 duplication syndrome. Neurology 2019, 92, E108–E114. [Google Scholar] [CrossRef]
- Lotti, F.; Geronzi, U.; Grosso, S. Electroencephalographic and epilepsy findings in mecp2 duplication syndrome. A family study. Brain Dev. 2019, 41, 456–459. [Google Scholar] [CrossRef]
- Giudice-Nairn, P.; Downs, J.; Wong, K.; Wilson, D.; Ta, D.; Gattas, M.; Amor, D.; Thompson, E.; Kirrali-Borri, C.; Ellaway, C.; et al. The incidence, prevalence and clinical features of MECP2 duplication syndrome in Australian children. J. Paediatr. Child Health 2019, 55, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.U.; Fu, C.; Suter, B.; Marsh, E.; Benke, T.A.; Skinner, S.A.; Lieberman, D.N.; Standridge, S.; Jones, M.; Beisang, A.; et al. Characterizing the phenotypic effect of Xq28 duplication size in MECP2 duplication syndrome. Clin. Genet. 2019, 95, 575–581. [Google Scholar] [CrossRef]
- Pascual-Alonso, A.; Blasco, L.; Vidal, S.; Gean, E.; Rubio, P.; O’Callaghan, M.; Martínez-Monseny, A.F.; Castells, A.A.; Xiol, C.; Català, V.; et al. Molecular characterization of Spanish patients with MECP2 duplication syndrome. Clin. Genet. 2020, 97, 610–620. [Google Scholar] [CrossRef]
- Gutiérrez-Sánchez, A.M.; Marín-Andrés, M.; López-Lafuente, A.; Monge-Galindo, L.; López-Pisón, J.; Peña-Segura, J.L. Síndrome de duplicación MECP2 familiar. Rev. Neurol. 2020, 70, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Cutri-French, C.; Armstrong, D.; Saby, J.; Gorman, C.; Lane, J.; Fu, C.; Peters, S.U.; Percy, A.; Neul, J.L.; Marsh, E.D. Comparison of core features in four developmental encephalopathies in the Rett Natural History Study. Ann. Neurol. 2020, 88, 396–406. [Google Scholar] [CrossRef]
- Choi, Y.L.J.; Wong, T.K.M.; Ng, K.K.D. Anesthetic management for a patient with MECP2 duplication syndrome: A case report. A&A Pract. 2020, 14, e01202. [Google Scholar] [CrossRef]
- van Baelen, A.; Verhoustraeten, L.; Kenis, S.; Meuwissen, M.; Boudewyns, A.; van Hoorenbeeck, K.; Verhulst, S. Sleep-disordered breathing and nocturnal hypoventilation in children with the MECP2 duplication syndrome: A case series and review of the literature. Am. J. Med. Genet. Part A 2020, 182A, 2437–2441. [Google Scholar] [CrossRef] [PubMed]
- Tekendo-Ngongang, C.; Dahoun, S.; Nguefack, S.; Moix, I.; Gimelli, S.; Zambo, H.; Morris, M.A.; Sloan-Béna, F.; Wonkam, A. MECP2 duplication syndrome in a patient from Cameroon. Am. J. Med. Genet. Part A 2020, 182, 619–622. [Google Scholar] [CrossRef]
- Peters, S.U.; Fu, C.; Marsh, E.D.; Benke, T.A.; Suter, B.; Skinner, S.A.; Lieberman, D.N.; Standridge, S.; Jones, M.; Beisang, A.; et al. Phenotypic features in MECP2 duplication syndrome: Effects of age. Am. J. Med. Genet. Part A 2021, 185A, 362–369. [Google Scholar] [CrossRef]
- Takeguchi, R.; Takahashi, S.; Akaba, Y.; Tanaka, R.; Nabatame, S.; Kurosawa, K.; Matsuishi, T.; Itoh, M. Early diagnosis of MECP2 duplication syndrome: Insights from a nationwide survey in Japan. J. Neurol. Sci. 2021, 422, 117321. [Google Scholar] [CrossRef] [PubMed]
- Al Ali, A.; Singh, R.; Filler, G. Abdominal compartment syndrome secondary to chronic constipation in MECP2 duplication syndrome. Clin. Care Med. 2021, 49, 291. [Google Scholar] [CrossRef]
- Van Esch, H. MECP2 duplication syndrome. Mol. Syndromol. 2011, 2, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.U.; Hundley, R.J.; Wilson, A.K.; Warren, Z.; Vehorn, A.; Carvalho, C.M.B.; Lupski, J.R.; Ramocki, M.B. The behavioral phenotype in MECP2 duplication syndrome: A comparison with idiopathic autism. Autism Res. 2013, 6, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.; Kamphausen, S.B.; Zenker, M.; Bird, L.M.; et al. DeepGestalt—Identifying rare genetic syndromes using deep learning. arXiv 2018, arXiv:1801.07637 2018. [Google Scholar]
- Vidal, S.; Pascual-Alonso, A.; Rabaza-Gairí, M.; Gerotina, E.; Brandi, N.; Pacheco, P.; Xiol, C.; Pineda, M.; Armstrong, J. Characterization of large deletions of the MECP2 gene in Rett syndrome patients by gene dosage analysis. Mol. Genet. Genomic Med. 2019, 7, e793. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.; Gärtner, J. Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): Mutations in the L1CAM gene. Hum. Mutat. 2001, 18, 1–12. [Google Scholar] [CrossRef]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Heckman, L.D.; Chahrour, M.H.; Zoghbi, H.Y. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife 2014, 3, e02676. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Sztainberg, Y.; Wang, Q.; Bajikar, S.S.; Trostle, A.J.; Wan, Y.-W.; Jafar-Nejad, P.; Rigo, F.; Liu, Z.; Tang, J.; et al. Antisense oligonucleotide therapy in a humanized mouse model of MECP2 duplication syndrome. Sci. Transl. Med 2021, 13, 7785. [Google Scholar] [CrossRef]
- Wojtal, D.; Kemaladewi, D.U.; Malam, Z.; Abdullah, S.; Wong, T.W.Y.; Hyatt, E.; Baghestani, Z.; Pereira, S.; Stavropoulos, J.; Mouly, V.; et al. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am. J. Hum. Genet. 2016, 98, 90–101. [Google Scholar] [CrossRef]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.N.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef]
- Tang, X.; Drotar, J.; Li, K.; Clairmont, C.D.; Brumm, A.S.; Sullins, A.J.; Wu, H.; Liu, S.; Wang, J.; Gray, N.S.; et al. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med 2019, 11, eaau0164. [Google Scholar] [CrossRef]
- Kim, J.J.; Savas, J.N.; Miller, M.T.; Hu, X.; Carromeu, C.; Lavallée-Adam, M.; Freitas, B.C.G.; Muotri, A.R.; Yates, J.R.; Ghosh, A. Proteomic analyses reveal misregulation of LIN28 expression and delayed timing of glial differentiation in human iPS cells with MECP2 loss-of-function. PLoS ONE 2019, 14, e0212553. [Google Scholar] [CrossRef]
- Gomes, A.R.; Fernandes, T.G.; Cabral, J.M.S.; Diogo, M.M. Modeling rett syndrome with human pluripotent stem cells: Mechanistic outcomes and future clinical perspectives. Int. J. Mol. Sci. 2021, 22, 3751. [Google Scholar] [CrossRef]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau, J.; Muotri, A.R.; et al. Modeling Rett syndrome with human patient-specific forebrain organoids. Front. Cell Dev. Biol. 2020, 8, 610427. [Google Scholar] [CrossRef] [PubMed]
- Ash, R.T.; Park, J.; Suter, B.; Smirnakis, S.M.; Zoghbi, H.Y. Excessive formation and stabilization of dendritic spine clusters in the mecp2-duplication syndrome mouse model of autism. eNeuro 2021, 8, 1–13. [Google Scholar] [CrossRef]
- Ash, R.T.; Buffington, S.A.; Park, J.; Suter, B.; Costa-Mattioli, M.; Zoghbi, H.Y.; Smirnakis, S.M. Inhibition of elevated ras-mapk signaling normalizes enhanced motor learning and excessive clustered dendritic spine stabilization in the mecp2-duplication syndrome mouse model of autism. eNeuro 2021, 8. [Google Scholar] [CrossRef]


{kind=link}
| Classification Groups | Reference |
|---|---|
| 1. Boys with severe neonatal encephalopathy. When having normal chromosomal complement (46, XY) they die at an early age. When being mosaic or having Klinefelter syndrome (47, XXY) they develop classic RTT. The same mutations cause RTT in girls. 2. Boys with non-specific mental retardation. They have normal chromosomal complement. The same mutations do not affect girls or cause mild ID. | Ravn et al., 2003 |
| 1. Boys with RTT. RTT consensus criteria are fulfilled. They are mosaic or have Klinefelter syndrome (47, XXY). The same mutations cause RTT in girls. 2. Boys with severe neonatal encephalopathy and early death. They have a normal chromosomal complement (46, XY). The same mutations cause RTT in girls. 3. Boys with less severe neurological and/or psychiatric manifestations. They have normal chromosomal complement. The same mutations do not affect girls, cause mild ID, or have not been reported in female patients. | Huppke and Gärtner 2005, Moretti et al., 2006, Villard 2007, Neul et al., 2012 |
| 1. Boys with Classical or atypical RTT. Consensus criteria for each category are fulfilled. When classical RTT is diagnosed the term “male RTT encephalopathy” is suggested. 2. Neonatal encephalopathy. Impairment of the clinical traits is noted from birth. 3. Progressive encephalopathy. Impairment of the clinical traits appears over the years. 4. Cognitive impairment. No progressive worsening is detected. | Neul et al., 2019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascual-Alonso, A.; Martínez-Monseny, A.F.; Xiol, C.; Armstrong, J. MECP2-Related Disorders in Males. Int. J. Mol. Sci. 2021, 22, 9610. https://doi.org/10.3390/ijms22179610
Pascual-Alonso A, Martínez-Monseny AF, Xiol C, Armstrong J. MECP2-Related Disorders in Males. International Journal of Molecular Sciences. 2021; 22(17):9610. https://doi.org/10.3390/ijms22179610
Chicago/Turabian StylePascual-Alonso, Ainhoa, Antonio F. Martínez-Monseny, Clara Xiol, and Judith Armstrong. 2021. "MECP2-Related Disorders in Males" International Journal of Molecular Sciences 22, no. 17: 9610. https://doi.org/10.3390/ijms22179610
APA StylePascual-Alonso, A., Martínez-Monseny, A. F., Xiol, C., & Armstrong, J. (2021). MECP2-Related Disorders in Males. International Journal of Molecular Sciences, 22(17), 9610. https://doi.org/10.3390/ijms22179610