
One-Pot Process: Microwave-Assisted Keratin Extraction and Direct Electrospinning to Obtain Keratin-Based Bioplastic

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract
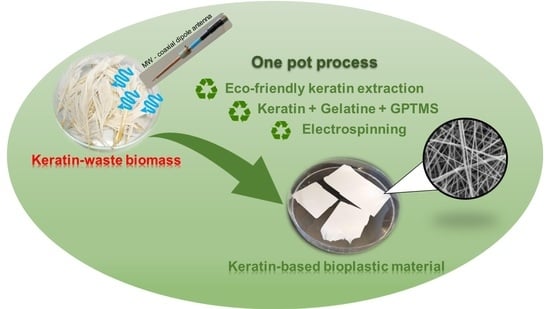
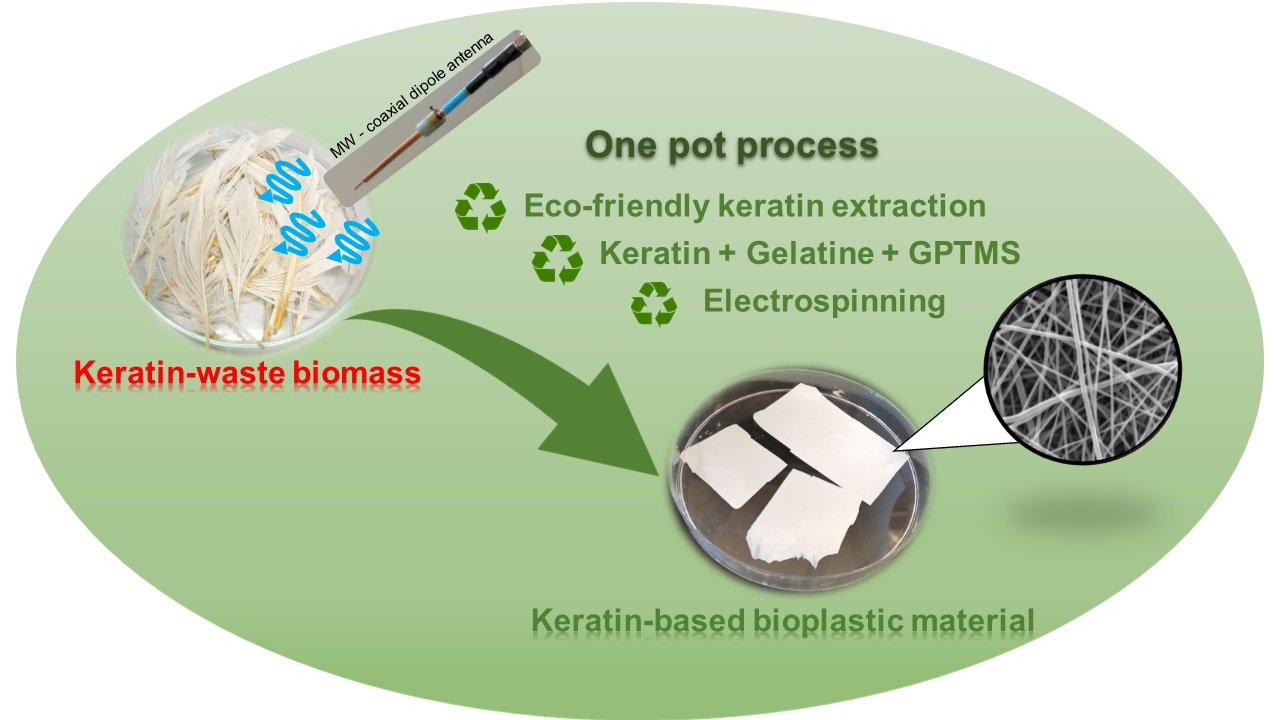
:
1. Introduction
2. Results
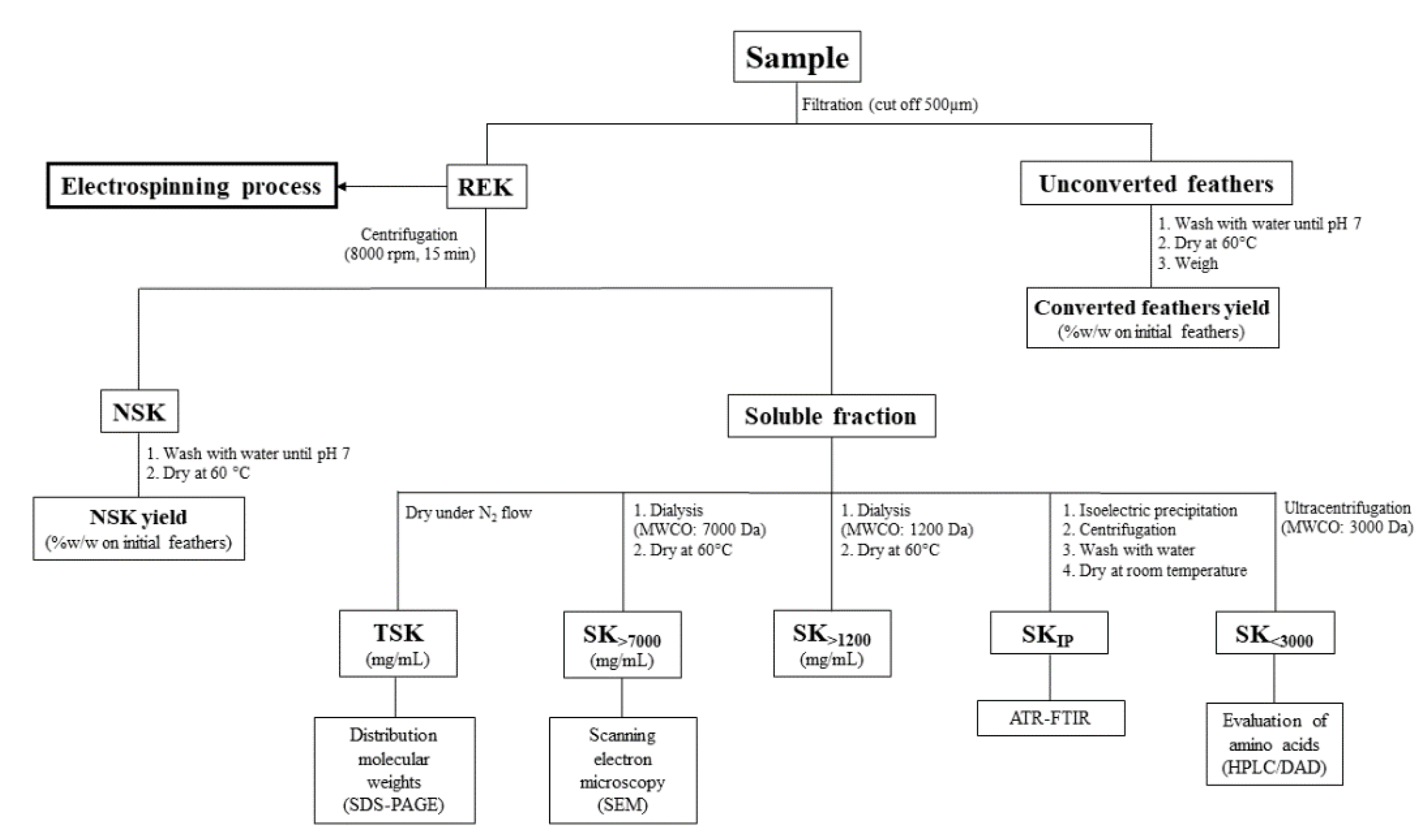
2.1. Keratin Extraction and Characterization
2.2. Mechanical, Barrier and Morphology Properties of Keratin-Based Materials
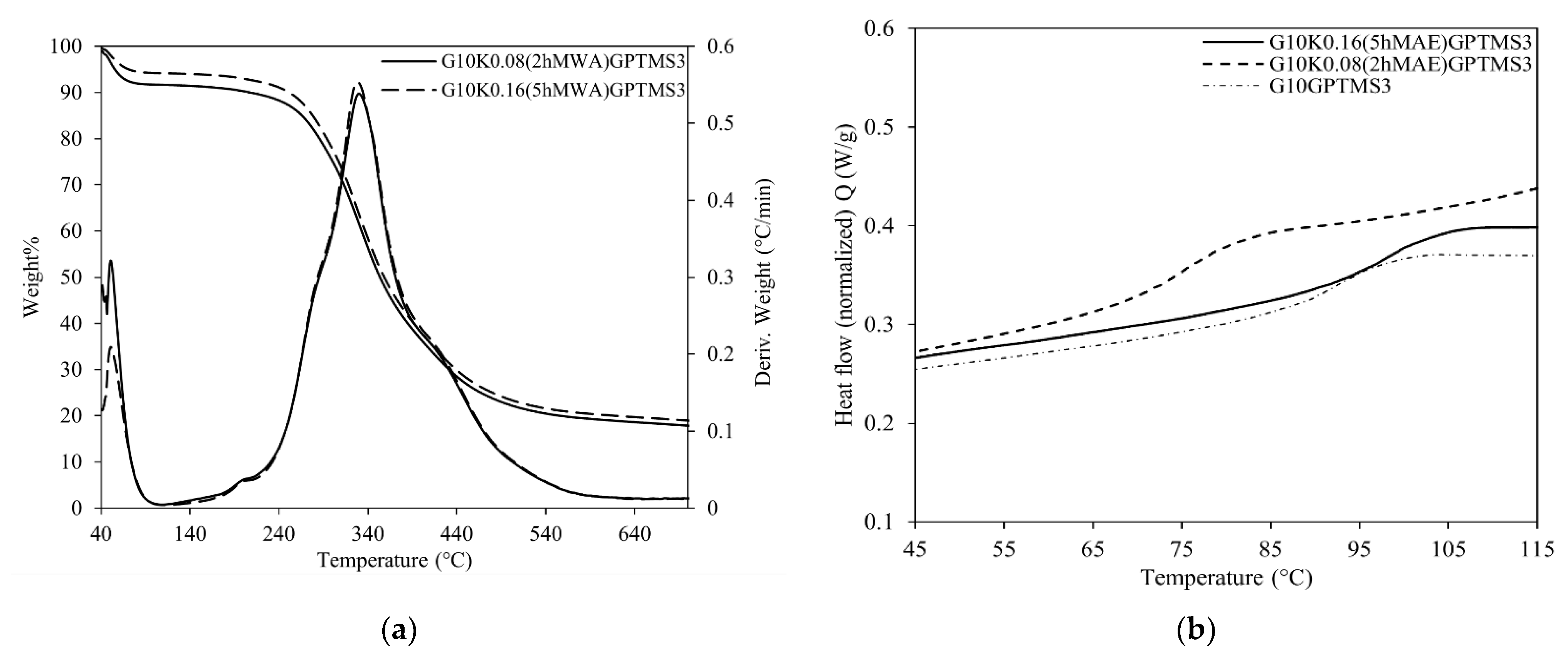
2.3. Physical-Chemical Properties of Keratin-Based Materials
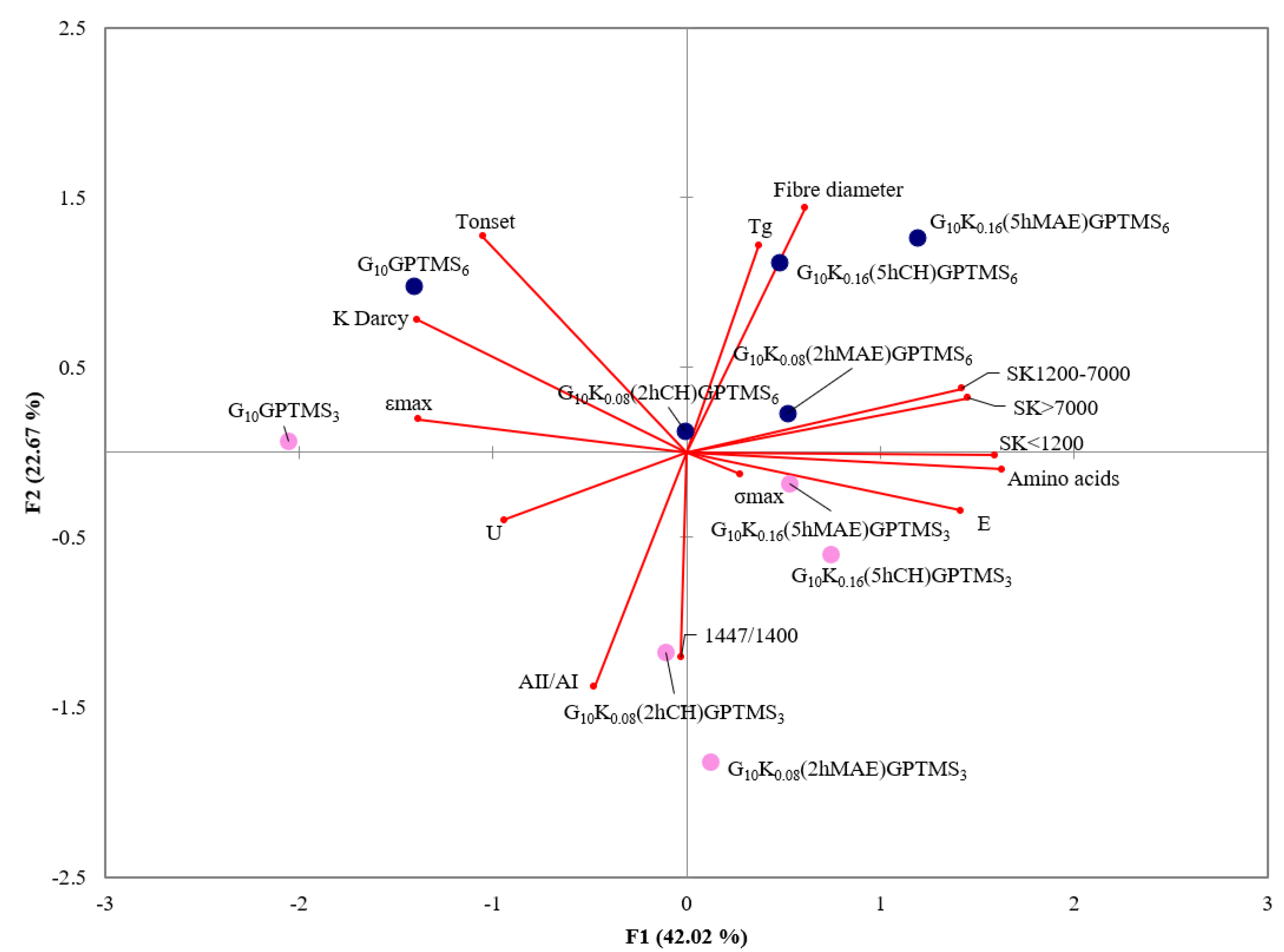
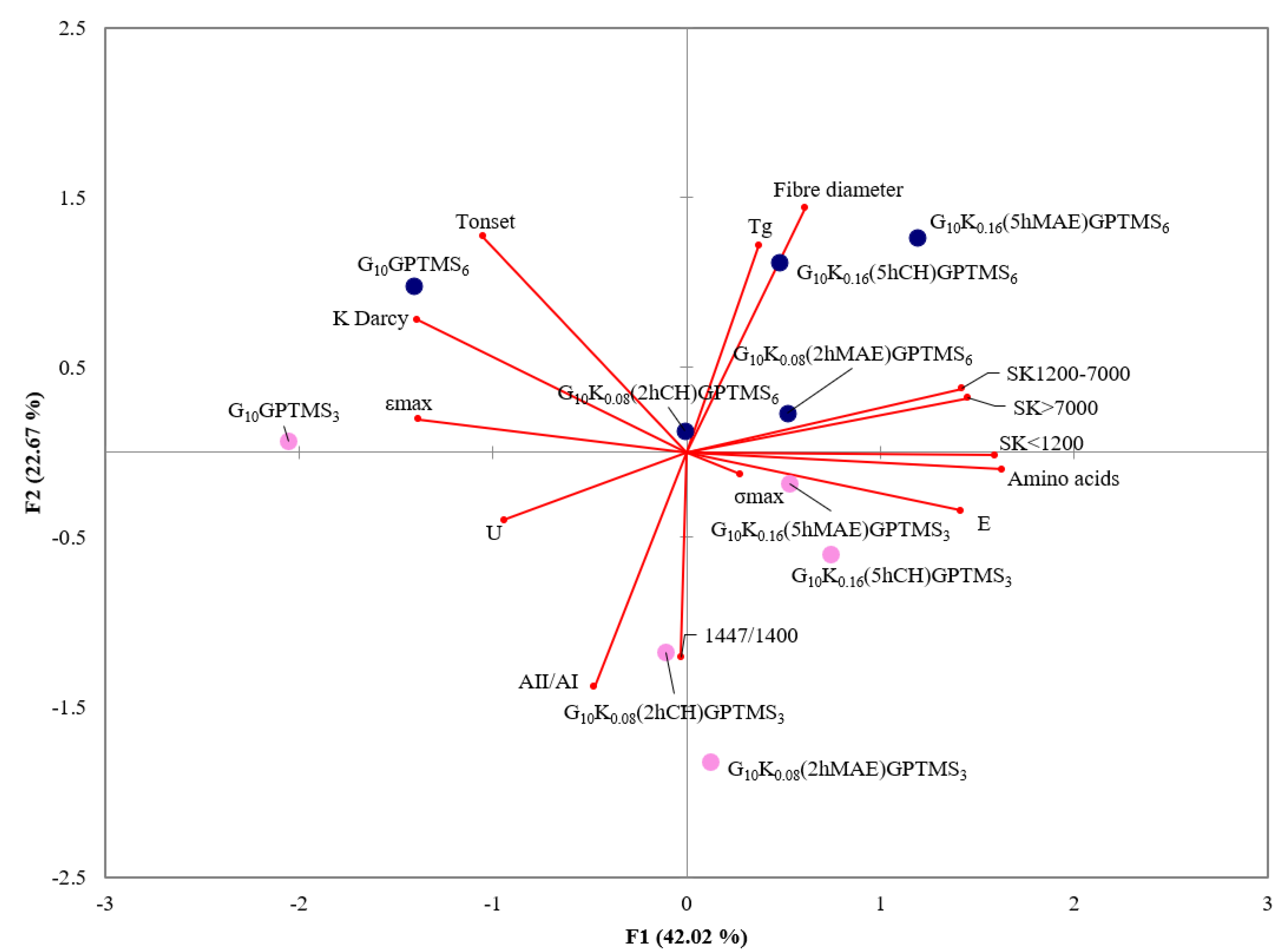
2.4. Statistical Analysis
3. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
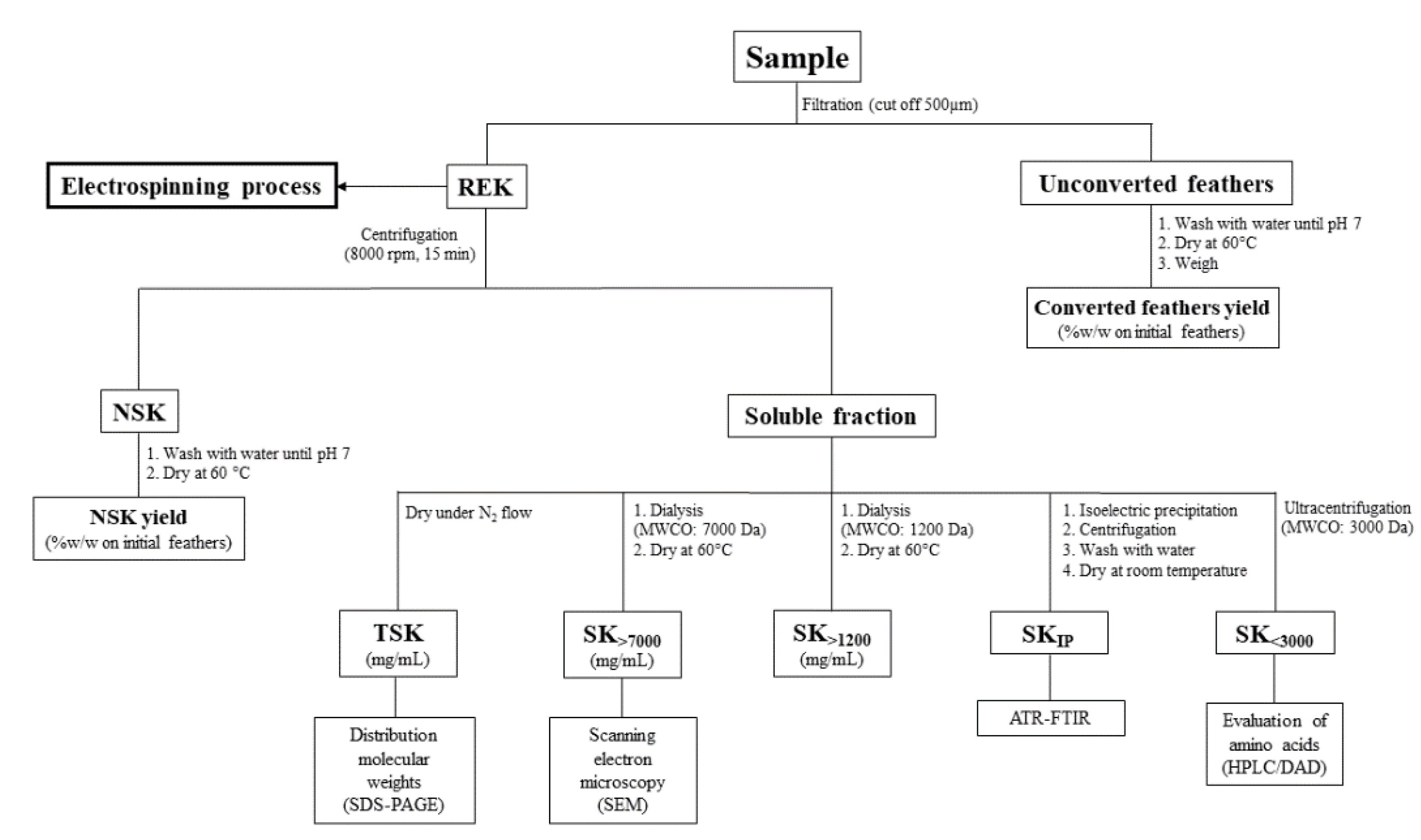{kind=link}
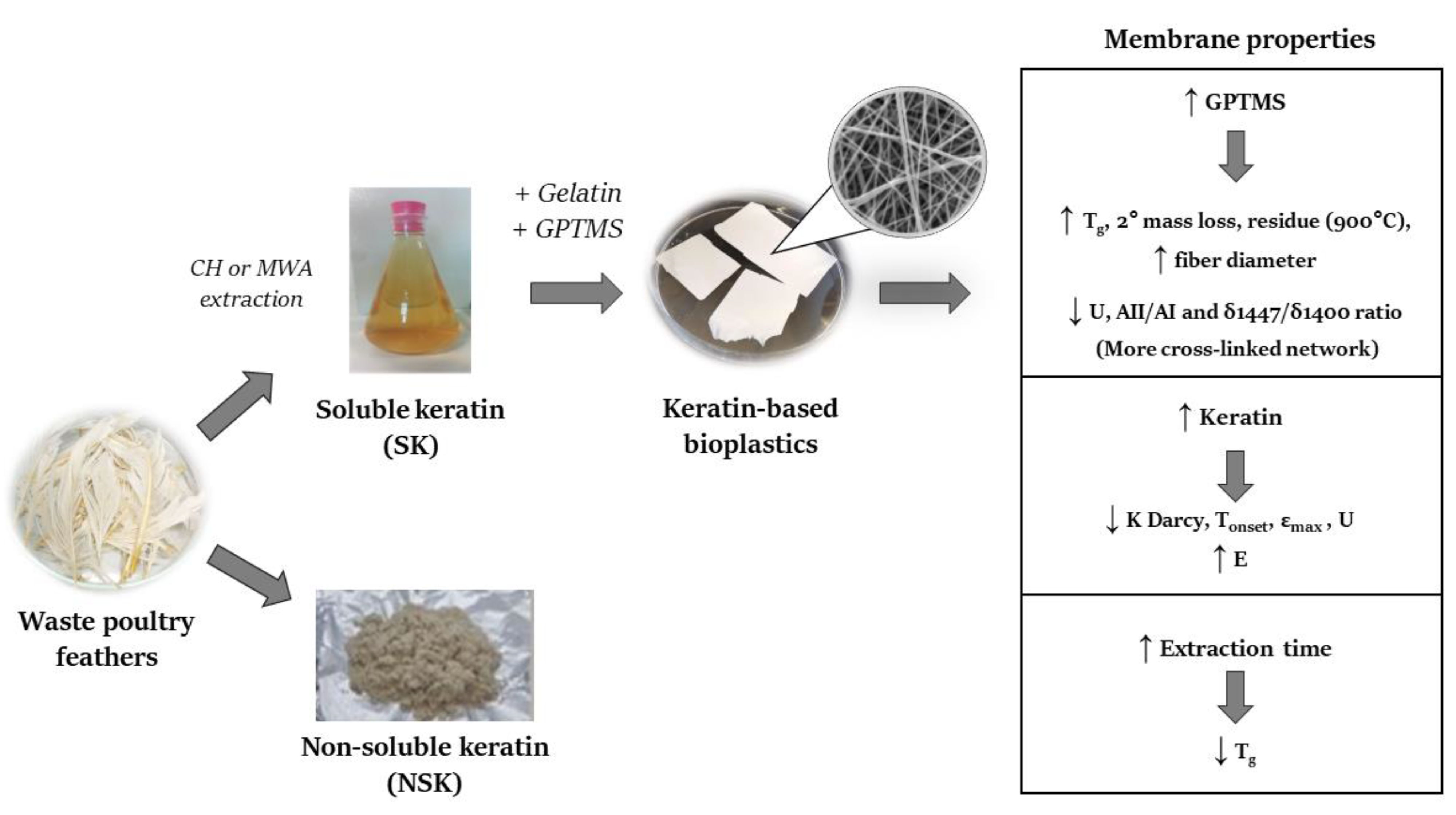{kind=link}
| Extraction Method | Extraction Conditions | Yields |
|---|---|---|
| Alkaline/MAE [44] | NaOH 0.5M 800W Solid: liquid (w/w) = 1:50 Temperature (°C) = n.r Time = 10 min | Thiol = 24.72 mM Protein (Folin Ciocalteau) = 26.74 mg/mL Amino Acid (Ninhydrin Assay) = 69.4 mg/g Molecular Weight = n.r |
| Reductive [14] | Na2S (500mM) Solid: liquid (w/w) = 1:40 Temperature (°C) = 50 Time = 6 h | Keratin yield % = Extracted keratin/feathers (w/w) = 80% Protein (Bradford assay) = 1.6 mg/mL Molecular Weight = n.r |
| Sulphitolysis [28] | Urea 8M; Na2S2O5 0.2M; SDS/feathers 0.6; NaOH up to pH 6.5 Solid: liquid (w/w) = 1:36 Temperature (°C) = 65 Time = 5 h | Keratin yield % = (Feathers-unconverted feathers)/(feathers) = 87.6% Molecular Weight (SDS-PAGE) = 11–20, 32, 37, 50, 75 kDa |
| Enzymatic and microbial [17] | Keratinases-Bacillus subtilitis pH 8 Temperature (°C) = 28 Time = 5 days | Protein (Lowry method) = 95% Molecular Weight (MALDI-TOF) = 0.8–1.1 kDa |
| Ionic liquid [19] | [Bmim][Cl]+10%Na2SO3 Solid: liquid (w/w) = 1:20 Temperature (°C) = 90 Time = 1 h | Keratin yield % = Extracted keratin/feathers (w/w) = 75.1% Protein (hydrolysis HCl-sum amino acids) = 72% Molecular Weight (GPC) = 8.83–9.74 kDa |
| Acid [45] | H2O-H2SO4 1:1 Temperature: 50 °C pH: 4–5 Time: 12 h Recrystallization in EtOH/H2O) with microwaves or ultrasounds | Not reported |
| Acid/MAE (This work) | Acetic acid 70% v/v 90 W Solid: liquid (w/w) = 1:75 Temperature (°C) = 104.6 Time = 5 h | Converted feathers % = (Feathers-unconverted feathers)/Feathers = 38 ± 5% NSK = 12 ± 2% - TSK = 26 ± 1% Amino acids = 0.94 ± 0.02 mg/mL, SK<1200 = 0.80 ± 0.03 mg/mL SK1200–7000 = 0.87 ± 0.02 mg/mL, SK>7000 = 0.49 ± 0.04 mg/mL Molecular Weight (SDS-PAGE) = 5–10 kDa |
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sharma, S.; Gupta, A. Sustainable Management of Keratin Waste Biomass: Applications and Future Perspectives. Braz. Arch. Biol. Technol. 2016, 59. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-D. Purification and Characterization of a Keratinase from a Feather-Degrading Fungus, Aspergillus flavus Strain K-03. Mycobiology 2007, 35, 219. [Google Scholar] [CrossRef] [Green Version]
- Korniłłowicz-Kowalska, T.; Bohacz, J. Biodegradation of keratin waste: Theory and practical aspects. Waste Manag. 2011, 31, 1689–1701. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Omary, M.B. ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 2002, 14, 110–122. [Google Scholar] [CrossRef]
- McKittrick, J.; Chen, P.-Y.; Bodde, S.G.; Yang, W.; Novitskaya, E.E.; Meyers, M.A. The Structure, Functions, and Mechanical Properties of Keratin. JOM 2012, 64, 449–468. [Google Scholar] [CrossRef]
- Sun, P.; Liu, Z.-T.; Liu, Z.-W. Particles from bird feather: A novel application of an ionic liquid and waste resource. J. Hazard. Mater. 2009, 170, 786–790. [Google Scholar] [CrossRef]
- Sharma, S.; Gupta, A.; Kumar, A. Keratin: An Introduction; Springer: Cham, Switzerland, 2019; pp. 1–18. [Google Scholar]
- Vineis, C.; Varesano, A.; Varchi, G.; Aluigi, A. Extraction and Characterization of Keratin from Different Biomasses; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Secchi, G. Role of protein in cosmetics. Clin. Dermatol. 2008, 26, 321–325. [Google Scholar] [CrossRef]
- Rouse, J.G.; Van Dyke, M.E. A Review of Keratin-Based Biomaterials for Biomedical Applications. Materials 2010, 3, 999–1014. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Cai, S.; Xu, L.; Yang, Y. Water-Stable Three-Dimensional Ultrafine Fibrous Scaffolds from Keratin for Cartilage Tissue Engineering. Langmuir 2014, 30, 8461–8470. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, N.; Sharma, S.; Gupta, A.; Alashwal, B.Y. Keratin based bioplastic film from chicken feathers and its characterization. Int. J. Biol. Macromol. 2018, 111, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Schrooyen, P.M.M.; Dijkstra, P.J.; Oberthür, R.C.; Bantjes, A.; Feijen, J. Partially Carboxymethylated Feather Keratins. 1. Properties in Aqueous Systems. J. Agric. Food Chem. 2000, 48, 4326–4334. [Google Scholar] [CrossRef]
- Sharma, S.; Gupta, A.; Kumar, A.; Kee, C.G.; Kamyab, H.; Saufi, S.M. An efficient conversion of waste feather keratin into ecofriendly bioplastic film. Clean Technol. Environ. Policy 2018, 20, 2157–2167. [Google Scholar] [CrossRef]
- Sinkiewicz, I.; Śliwińska, A.; Staroszczyk, H.; Kołodziejska, I. Alternative Methods of Preparation of Soluble Keratin from Chicken Feathers. Waste Biomass Valorization 2017, 8, 1043–1048. [Google Scholar] [CrossRef]
- Esparza, Y.; Ullah, A.; Boluk, Y.; Wu, J. Preparation and characterization of thermally crosslinked poly(vinyl alcohol)/feather keratin nanofiber scaffolds. Mater. Des. 2017, 133, 1–9. [Google Scholar] [CrossRef]
- Villa, A.L.V.; Aragão, M.R.S.; dos Santos, E.P.; Mazotto, A.M.; Zingali, R.B.; de Souza, E.P.; Vermelho, A.B. Feather keratin hydrolysates obtained from microbial keratinases: Effect on hair fiber. BMC Biotechnol. 2013, 13, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahni, N.; Phutela, U.G. Bacterial keratinases and their prospective applications: A review. Int. J. Curr. Microbiol. App. Sci. 2015, 4, 768–783. [Google Scholar]
- Ji, Y.; Chen, J.; Lv, J.; Li, Z.; Xing, L.; Ding, S. Extraction of keratin with ionic liquids from poultry feather. Sep. Purif. Technol. 2014, 132, 577–583. [Google Scholar] [CrossRef]
- Yin, J.; Rastogi, S.; Terry, A.E.; Popescu, C. Self-organization of Oligopeptides Obtained on Dissolution of Feather Keratins in Superheated Water. Biomacromolecules 2007, 8, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Longo, I.; Simone Ricci, A. Chemical Activation Using an Open-End Coaxial Applicator. J. Microw. Power Electromagn. Energy 2006, 41, 4–19. [Google Scholar] [CrossRef] [PubMed]
- González-Rivera, J.; Duce, C.; Falconieri, D.; Ferrari, C.; Ghezzi, L.; Piras, A.; Tine, M.R. Coaxial microwave assisted hydrodistillation of essential oils from five different herbs (lavender, rosemary, sage, fennel seeds and clove buds): Chemical composition and thermal analysis. Innov. Food Sci. Emerg. Technol. 2016, 33, 308–318. [Google Scholar] [CrossRef]
- González-Rivera, J.; Spepi, A.; Ferrari, C.; Duce, C.; Longo, I.; Falconieri, D.; Piras, A.; Tinè, M.R. Novel configurations for a citrus waste based biorefinery: From solventless to simultaneous ultrasound and microwave assisted extraction. Green Chem. 2016, 18, 6482–6492. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rivera, J.; Duce, C.; Campanella, B.; Bernazzani, L.; Ferrari, C.; Tanzini, E.; Onor, M.; Longo, I.; Ruiz, J.C.; Tinè, M.R.; et al. In situ microwave assisted extraction of clove buds to isolate essential oil, polyphenols, and lignocellulosic compounds. Ind. Crops Prod. 2021, 161, 113203. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Fujihara, K.; Teo, W.E.; Lim, T.C.; Ma, Z. An Introduction to Electrospinning and Nanofibers; World Scientific Publishing Co: Singapore, 2005; ISBN 9789812567611. [Google Scholar]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and Electrospun Nanofibers: Methods, Materials, and Applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.M.D.; Siqueira, N.M.; Prabhakaram, M.P.; Ramakrishna, S. Electrospinning and electrospray of bio-based and natural polymers for biomaterials development. Mater. Sci. Eng. C 2018, 92, 969–982. [Google Scholar] [CrossRef]
- Isarankura, N.A.S.; Tanpichai, S.; Wootthikanokkhan, J. Keratin Extracted from Chicken Feather Waste: Extraction, Preparation, and Structural Characterization of the Keratin and Keratin/Biopolymer Films and Electrospuns. J. Polym. Environ. 2015, 23, 506–516. [Google Scholar] [CrossRef]
- Aluigi, A.; Varesano, A.; Montarsolo, A.; Vineis, C.; Ferrero, F.; Mazzuchetti, G.; Tonin, C. Electrospinning of keratin/poly(ethylene oxide)blend nanofibers. J. Appl. Polym. Sci. 2007, 104, 863–870. [Google Scholar] [CrossRef]
- Chae, W.P.; Kang, I.K.; Kim, S.Y. Keratin Nanofibers as a Biomaterial. In Proceedings of the 2010 International Conference on Nanotechnology and Biosensors, Hong Kong, China, 28–30 December 2010; Volume 2, pp. 120–124. [Google Scholar]
- Yuan, J.; Xing, Z.C.; Park, S.W.; Geng, J.; Kang, I.K.; Shen, J.; Meng, W.; Shim, K.J.; Han, I.S.; Kim, J.C. Fabrication of PHBV/keratin composite nanofibrous mats for biomedical applications. Macromol. Res. 2009, 17, 850–855. [Google Scholar] [CrossRef]
- Tonin, C.; Aluigi, A.; Vineis, C.; Varesano, A.; Montarsolo, A.; Ferrero, F. Thermal and structural characterization of poly(ethylene-oxide)/keratin blend films. J. Therm. Anal. Calorim. 2007, 89, 601–608. [Google Scholar] [CrossRef]
- Katoh, K.; Shibayama, M.; Tanabe, T.; Yamauchi, K. Preparation and properties of keratin-poly(vinyl alcohol) blend fiber. J. Appl. Polym. Sci. 2004, 91, 756–762. [Google Scholar] [CrossRef]
- Fortunato, G.M.; Da Ros, F.; Bisconti, S.; De Acutis, A.; Biagini, F.; Lapomarda, A.; Magliaro, C.; De Maria, C.; Montemurro, F.; Bizzotto, D.; et al. Electrospun Structures Made of a Hydrolyzed Keratin-Based Biomaterial for Development of in vitro Tissue Models. Front. Bioeng. Biotechnol. 2019, 7, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasong, S.; Wasan, T. Preparation and Characterization of Hair Keratin/Gelatin Blend Films. Pak. J. Biol. Sci. 2011, 14, 351–356. [Google Scholar] [CrossRef]
- Sow, L.C.; Toh, N.Z.Y.; Wong, C.W.; Yang, H. Combination of sodium alginate with tilapia fish gelatin for improved texture properties and nanostructure modification. Food Hydrocoll. 2019, 94, 459–467. [Google Scholar] [CrossRef]
- Sow, L.C.; Nicole Chong, J.M.; Liao, Q.X.; Yang, H. Effects of κ-carrageenan on the structure and rheological properties of fish gelatin. J. Food Eng. 2018, 239, 92–103. [Google Scholar] [CrossRef]
- Sow, L.C.; Tan, S.J.; Yang, H. Rheological properties and structure modification in liquid and gel of tilapia skin gelatin by the addition of low acyl gellan. Food Hydrocoll. 2019, 90, 9–18. [Google Scholar] [CrossRef]
- Yang, D.; Gao, S.; Yang, H. Effects of sucrose addition on the rheology and structure of iota-carrageenan. Food Hydrocoll. 2020, 99, 105317. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, H.; Yang, H. Effects of sucrose addition on the rheology and microstructure of κ-carrageenan gel. Food Hydrocoll. 2018, 75, 164–173. [Google Scholar] [CrossRef]
- Tonda-Turo, C.; Cipriani, E.; Gnavi, S.; Chiono, V.; Mattu, C.; Gentile, P.; Perroteau, I.; Zanetti, M.; Ciardelli, G. Crosslinked gelatin nanofibres: Preparation, characterisation and in vitro studies using glial-like cells. Mater. Sci. Eng. C 2013, 33, 2723–2735. [Google Scholar] [CrossRef]
- Said, M.I.; Yuliati, F.N.; Sukma, M. The effects of acidic and alkaline hydrolysis process on some physical and chemical properties of broiler chicken feathers. Iran. J. Appl. Anim. Sci. 2019, 9, 529–540. [Google Scholar]
- Said, M.I.; Abustam, E.; Pakiding, W.; Mide, M.Z.; Sukma, M. Synthesis of Feather Concentrate from Broiler Feather Waste using Different Chemical Hydrolysis Process and Effect on Its Properties. Online J. Biol. Sci. 2018, 18, 270–276. [Google Scholar] [CrossRef]
- Lee, Y.S.; Phang, L.-Y.; Ahmad, S.A.; Ooi, P.T. Microwave-Alkali Treatment of Chicken Feathers for Protein Hydrolysate Production. Waste Biomass Valorization 2016, 7, 1147–1157. [Google Scholar] [CrossRef]
- Rodríguez-Clavel, I.S.; Paredes-Carrera, S.P.; Flores-Valle, S.O.; Paz-García, E.J.; Sánchez-Ochoa, J.C.; Pérez-Gutiérrez, R.M. Effect of Microwave or Ultrasound Irradiation in the Extraction from Feather Keratin. J. Chem. 2019, 2019, 1326063. [Google Scholar] [CrossRef]
- Aluigi, A.; Corbellini, A.; Rombaldoni, F.; Zoccola, M.; Canetti, M. Morphological and structural investigation of wool-derived keratin nanofibres crosslinked by thermal treatment. Int. J. Biol. Macromol. 2013, 57, 30–37. [Google Scholar] [CrossRef]
- He, M.; Zhang, B.; Dou, Y.; Yin, G.; Cui, Y.; Chen, X. Fabrication and characterization of electrospun feather keratin/poly(vinyl alcohol) composite nanofibers. RSC Adv. 2017, 7, 9854–9861. [Google Scholar] [CrossRef] [Green Version]
- Koombhongse, S.; Liu, W.; Reneker, D.H. Flat polymer ribbons and other shapes by electrospinning. J. Polym. Sci. Part. B Polym. Phys. 2001, 39, 2598–2606. [Google Scholar] [CrossRef]
- Parker, F.S. Applications of Infrared Spectroscopy in Biochemistry, Biology, and Medicine; Springer: Berlin/Heidelberg, Germany, 1971. [Google Scholar]
- Bandekar, J. Amide modes and protein conformation. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1992, 1120, 123–143. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramanti, E.; Lenci, F.; Sgarbossa, A. Effects of hypericin on the structure and aggregation properties of β-amyloid peptides. Eur. Biophys. J. 2010, 39, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Maiti, T.K.; Ghosh, K.S.; Dasgupta, S. Interaction of (−)-epigallocatechin-3-gallate with human serum albumin: Fluorescence, fourier transform infrared, circular dichroism, and docking studies. Proteins Struct. Funct. Bioinforma. 2006, 64, 355–362. [Google Scholar] [CrossRef]
- Shi, W.; Dumont, M.-J. Review: Bio-based films from zein, keratin, pea, and rapeseed protein feedstocks. J. Mater. Sci. 2014, 49, 1915–1930. [Google Scholar] [CrossRef]
- Biagini, F.; Calvigioni, M.; Lapomarda, A.; Vecchione, A.; Magliaro, C.; De Maria, C.; Montemurro, F.; Celandroni, F.; Mazzantini, D.; Mattioli-Belmonte, M.; et al. A novel 3D in vitro model of the human gut microbiota. Sci. Rep. 2020, 10, 21499. [Google Scholar] [CrossRef]


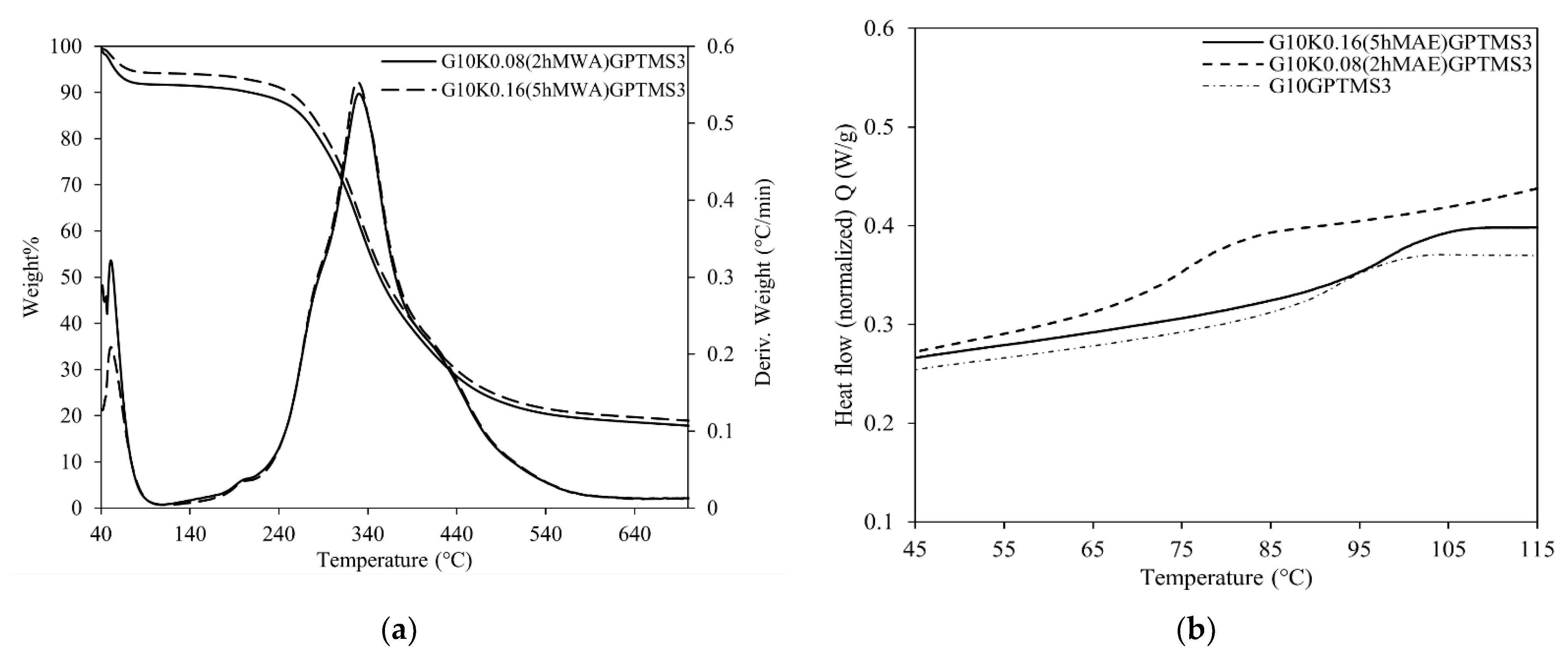

| MAE | CH | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Solvent/Feathers | Converted Feathers (% w/w) | NSK (% w/w) | TSK (% w/w) | Time (h) | Solvent/Feathers | Converted Feathers (% w/w) | NSK (% w/w) | TSK (% w/w) |
| 2 | 75:1 | 30 ± 4 | 18 ± 3 | 13 ± 2 | 2 | 75:1 | 29 ± 2 | 16 ± 1 | 12 ± 2 |
| 5 | 75:1 | 38 ± 5 | 12 ± 2 | 26 ± 1 | 5 | 75:1 | 52 ± 3 | 31 ± 2 | 25 ± 1 |
| 2 | 150:1 | 39 ± 1 | 26 ± 2 | 12 ± 2 | 5 | 150:1 | 50 ± 1 | 27 ± 2 | 26 ± 2 |
| Time (h) | TSK (mg/mL) | Amino Acids (mg/mL) | SK<1200 * (mg/mL) | SK1200–7000 * (mg/mL) | SK>7000 * (mg/mL) |
|---|---|---|---|---|---|
| 2h-MAE | 1.6 ± 0.2 | 0.74 ± 0.09 | 0.45 ± 0.02 | 0.27 ± 0.03 | 0.14 ± 0.02 |
| 5h-MAE | 3.1 ± 0.1 | 0.94 ± 0.02 | 0.80 ± 0.03 | 0.87 ± 0.02 | 0.49 ± 0.04 |
| 2h-CH | 1.4 ± 0.3 | 0.6 ± 0.2 | 0.40 ± 0.05 | 0.20 ± 0.04 | 0.20 ± 0.05 |
| 5h-CH | 2.7 ± 0.1 | 1.13 ± 0.03 | 0.48 ± 0.04 | 0.68 ± 0.02 | 0.41 ± 0.01 |
| Keratin/Gelatin Bioplastic | Uniaxial Tensile Test | Water Permeability Test | SEM | |||
|---|---|---|---|---|---|---|
| σmax (MPa) | εmax (%) | E (MPa) | U (kJ·m−3) | K Darcy (10−15·m2) | Fiber Diameter (nm) | |
| G10GPTMS3 | 0.8 ± 0.5 | 6.8 ± 1.6 | 24 ± 3 | 77 ± 58 | 22 ± 2 | 76 ± 26 |
| G10K0.08(2hMAE)GPTMS3 | 1.9 ± 0.4 | 2.5 ± 0.5 | 160 ± 7 | 46 ± 22 | 2.0 ± 0.4 | 85 ± 33 |
| G10K0.16(5hMAE)GPTMS3 | 1.2 ± 0.5 | 4.5 ± 1.1 | 116 ± 43 | 60 ± 31 | 7.5 ± 1.0 | 86 ± 25 |
| G10K0.08(2hCH)GPTMS3 | 0.5 ± 0.1 | 1.9 ± 1.2 | 106 ± 4 | 7 ± 5 | 3.1 ± 0.1 | 72 ± 26 |
| G10K0.16(5hCH)GPTMS3 | 1.4 ± 0.3 | 2.8 ± 0.8 | 176 ± 37 | 26 ± 9 | 3.4 ± 0.3 | 79 ± 24 |
| G10GPTMS6 | 1.7 ± 0.6 | 4.8 ± 1.4 | 34 ± 5 | 32 ± 14 | 17 ± 6 | 125 ± 53 |
| G10K0.08(2hMAE)GPTMS6 | 2.0 ± 0.5 | 1.3 ± 0.3 | 230 ± 74 | 16 ± 5 | 0.65 ± 0.03 | 126 ± 59 |
| G10K0.16(5hMAE)GPTMS6 | 1.3 ± 0.8 | 1.5 ± 0.8 | 176 ± 72 | 17 ± 13 | 8.8 ± 1.3 | 161 ± 69 |
| G10K0.08(2hCH)GPTMS6 | 1.0 ± 0.4 | 2.4 ± 0.6 | 146 ± 54 | 13 ± 10 | 6.4 ± 0.7 | 114 ± 40 |
| G10K0.16(5hCH)GPTMS6 | 0.7 ± 0.1 | 3.0 ± 0.8 | 89 ± 5 | 6 ± 2 | 7.0 ± 0.5 | 149 ± 59 |
| Gelatin/Keratin Bioplastic | TGA | DSC | IR | |||||
|---|---|---|---|---|---|---|---|---|
| Tonset (°C) | Degradation Step Temperature (°C)/ Weight Loss % w/w | Residue (900 °C) | Tg (°C) | AII/AI | δ1447/δÄ1400 | |||
| 1 | 2 | 3 | ||||||
| G10GPTMS3 | 278 | 44 | 331 | 439 | 19% | 89.6 | 0.642 ± 0.013 | 1.652 ± 0.083 |
| (11%) | (48%) | (22%) | ||||||
| G10K0.08(2hMAE)GPTMS3 | 264 | 51 | 330 | 422 | 16% | 74.3 | 0.638 ± 0.013 | 1.736 ± 0.087 |
| (8%) | (51%) | (25%) | ||||||
| G10K0.16(5hMAE)GPTMS3 | 262 | 51 | 328 | 423 | 17% | 95.4 | 0.628 ± 0.013 | 1.678 ± 0.084 |
| (6%) | (53%) | (24%) | ||||||
| G10K0.08(2hCH)GPTMS3 | 260 | 44 | 327 | 425 | 16% | 73.3 | 0.651 ± 0.013 | 1.650 ± 0.082 |
| (8%) | (51%) | (23%) | ||||||
| G10K0.16(5hCH)GPTMS3 | 266 | 53 | 329 | 429 | 16% | 88.1 | 0.630 ± 0.013 | 1.667 ± 0.083 |
| (8%) | (50%) | (25%) | ||||||
| G10GPTMS6 | 281 | 39 | 330 | 406 | 23% | 87.9 | 0.613 ± 0.012 | 1.661 ± 0.083 |
| (7%) | (41%) | (29%) | ||||||
| G10K0.08(2hMAE)GPTMS6 | 269 | 44 | 329 | 416 | 20% | 86.8 | 0.622 ± 0.012 | 1.632 ± 0.082 |
| (4%) | (47%) | (28%) | ||||||
| G10K0.16(5hMAE)GPTMS6 | 276 | 51 | 328 | 423 | 20% | 103.7 | 0.615 ± 0.012 | 1.651 ± 0.083 |
| (5%) | (47%) | (28%) | ||||||
| G10K0.08(2hCH)GPTMS6 | 273 | 50 | 331 | 427 | 20% | 83.9 | 0.620 ± 0.012 | 1.672 ± 0.084 |
| (4%) | (48%) | (28%) | ||||||
| G10K0.16(5hCH)GPTMS6 | 274 | 50 | 328 | 424 | 20% | 84.5 | 0.619 ± 0.012 | 1.620 ± 0.081 |
| Sample 1 | Gelatin (% w/v) | Keratin (% w/v) 2 | Extraction Time (h) | Heating Mode | GPTMS (% v/v) | Acetic Acid (% v/v) |
|---|---|---|---|---|---|---|
| G10GPTMS3 | 10 | 0 | / | / | 3 | 90 |
| G10K0.08(2hMAE)GPTMS3 | 10 | 0.08 | 2 | MAE | 3 | 80 |
| G10K0.16(5hMAE)GPTMS3 | 10 | 0.16 | 5 | MAE | 3 | 80 |
| G10K0.08(2hCH)GPTMS3 | 10 | 0.08 | 2 | CH | 3 | 80 |
| G10K0.16(5hCH)GPTMS3 | 10 | 0.16 | 5 | CH | 3 | 80 |
| G10GPTMS6 | 10 | 0 | / | / | 6 | 90 |
| G10K0.08(2hMAE)GPTMS6 | 10 | 0.08 | 2 | MAE | 6 | 80 |
| G10K0.16(5hMAE)GPTMS6 | 10 | 0.16 | 5 | MAE | 6 | 80 |
| G10K0.08(2hCH)GPTMS6 | 10 | 0.08 | 2 | CH | 6 | 80 |
| G10K0.16(5hCH)GPTMS6 | 10 | 0.16 | 5 | CH | 6 | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulidori, E.; Micalizzi, S.; Bramanti, E.; Bernazzani, L.; Duce, C.; De Maria, C.; Montemurro, F.; Pelosi, C.; De Acutis, A.; Vozzi, G.; et al. One-Pot Process: Microwave-Assisted Keratin Extraction and Direct Electrospinning to Obtain Keratin-Based Bioplastic. Int. J. Mol. Sci. 2021, 22, 9597. https://doi.org/10.3390/ijms22179597
Pulidori E, Micalizzi S, Bramanti E, Bernazzani L, Duce C, De Maria C, Montemurro F, Pelosi C, De Acutis A, Vozzi G, et al. One-Pot Process: Microwave-Assisted Keratin Extraction and Direct Electrospinning to Obtain Keratin-Based Bioplastic. International Journal of Molecular Sciences. 2021; 22(17):9597. https://doi.org/10.3390/ijms22179597
Chicago/Turabian StylePulidori, Elena, Simone Micalizzi, Emilia Bramanti, Luca Bernazzani, Celia Duce, Carmelo De Maria, Francesca Montemurro, Chiara Pelosi, Aurora De Acutis, Giovanni Vozzi, and et al. 2021. "One-Pot Process: Microwave-Assisted Keratin Extraction and Direct Electrospinning to Obtain Keratin-Based Bioplastic" International Journal of Molecular Sciences 22, no. 17: 9597. https://doi.org/10.3390/ijms22179597
APA StylePulidori, E., Micalizzi, S., Bramanti, E., Bernazzani, L., Duce, C., De Maria, C., Montemurro, F., Pelosi, C., De Acutis, A., Vozzi, G., & Tinè, M. R. (2021). One-Pot Process: Microwave-Assisted Keratin Extraction and Direct Electrospinning to Obtain Keratin-Based Bioplastic. International Journal of Molecular Sciences, 22(17), 9597. https://doi.org/10.3390/ijms22179597