
Fibrotic Changes to Schlemm’s Canal Endothelial Cells in Glaucoma

 ,
,
Abstract
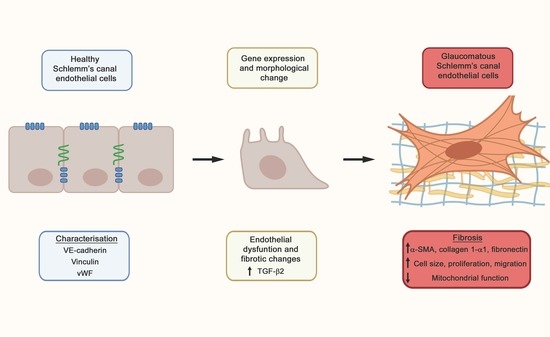
:
1. Introduction
2. Results
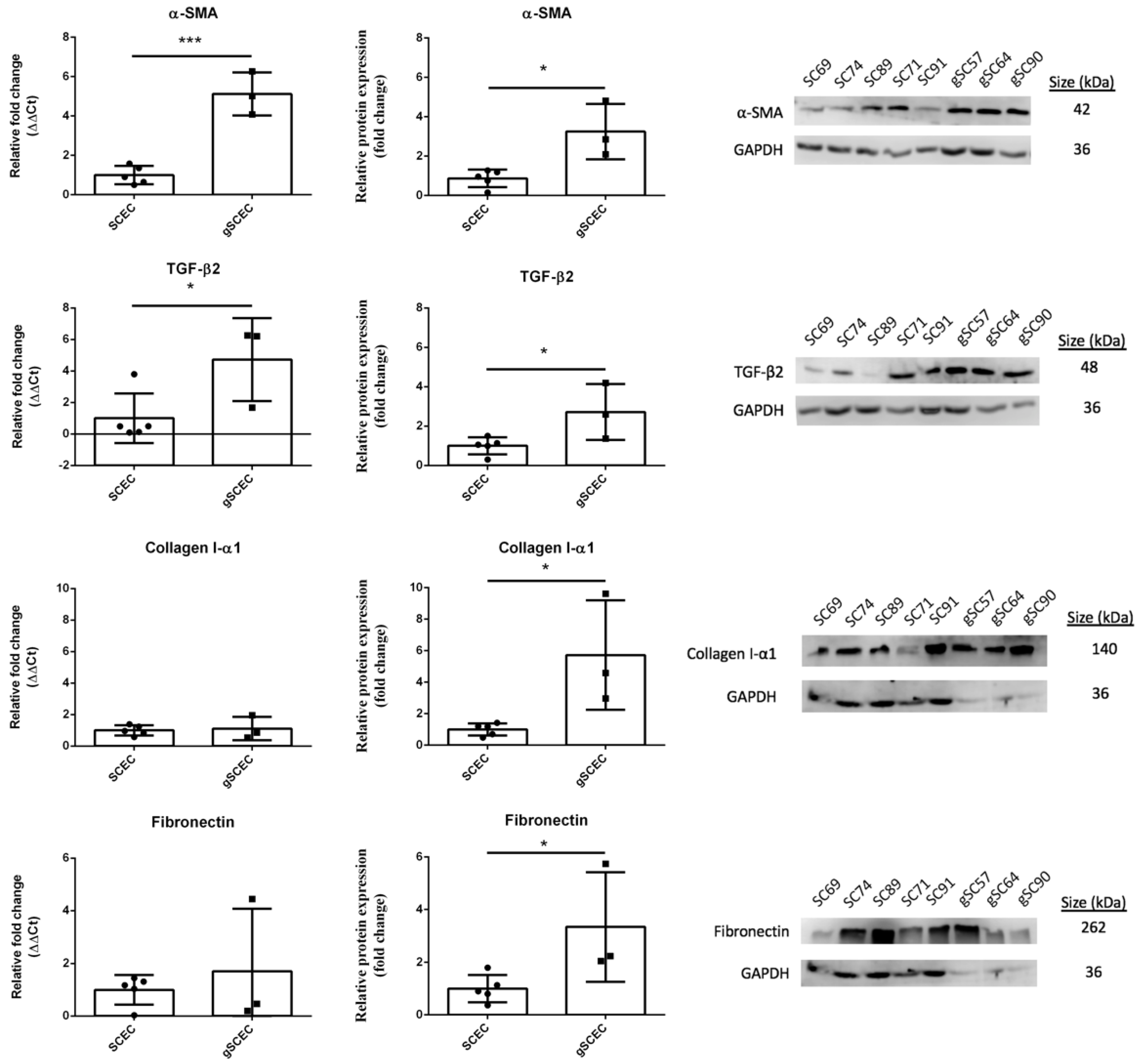
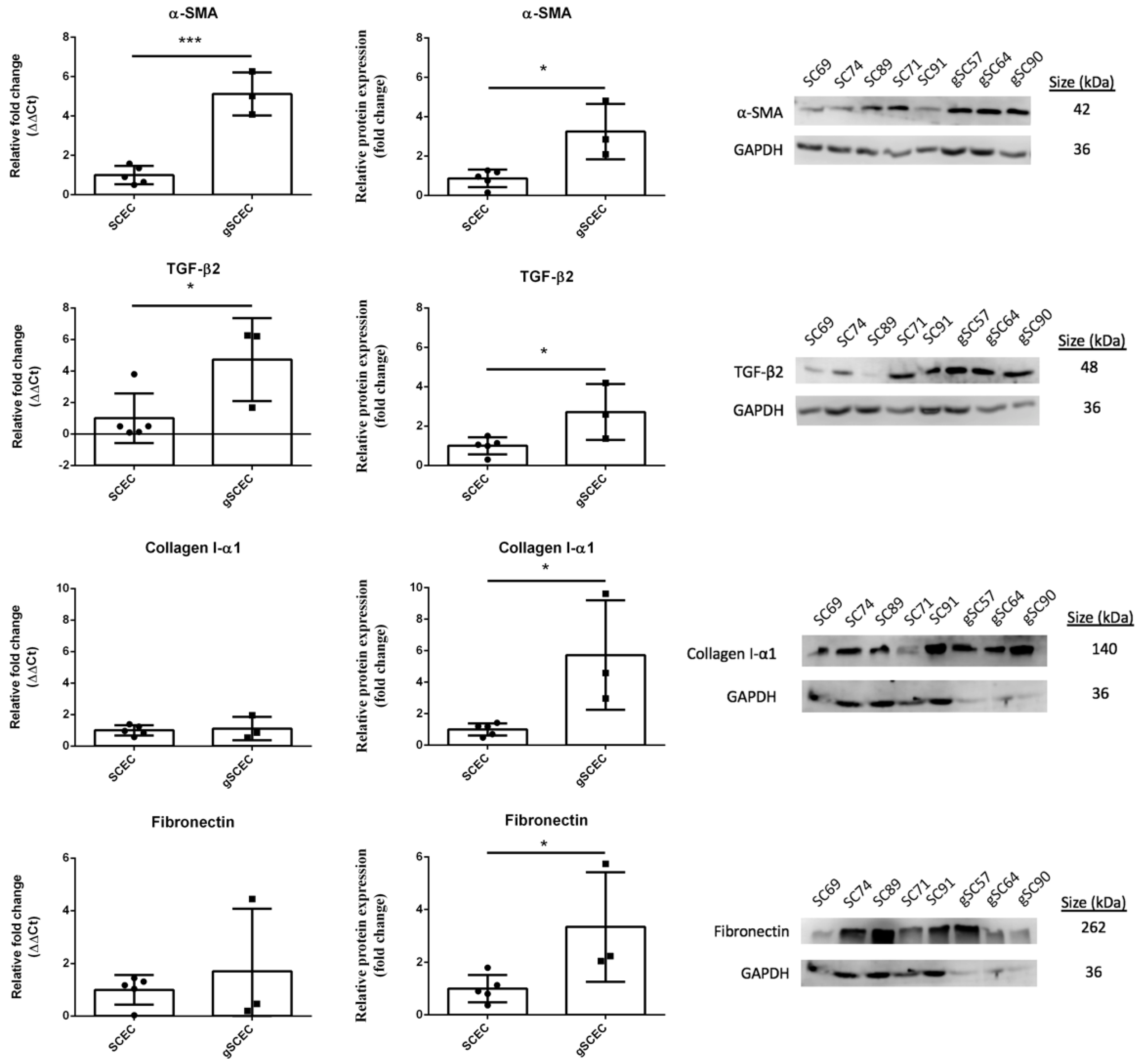
2.1. Fibrotic Marker Expression in Healthy versus Glaucomatous SCECs
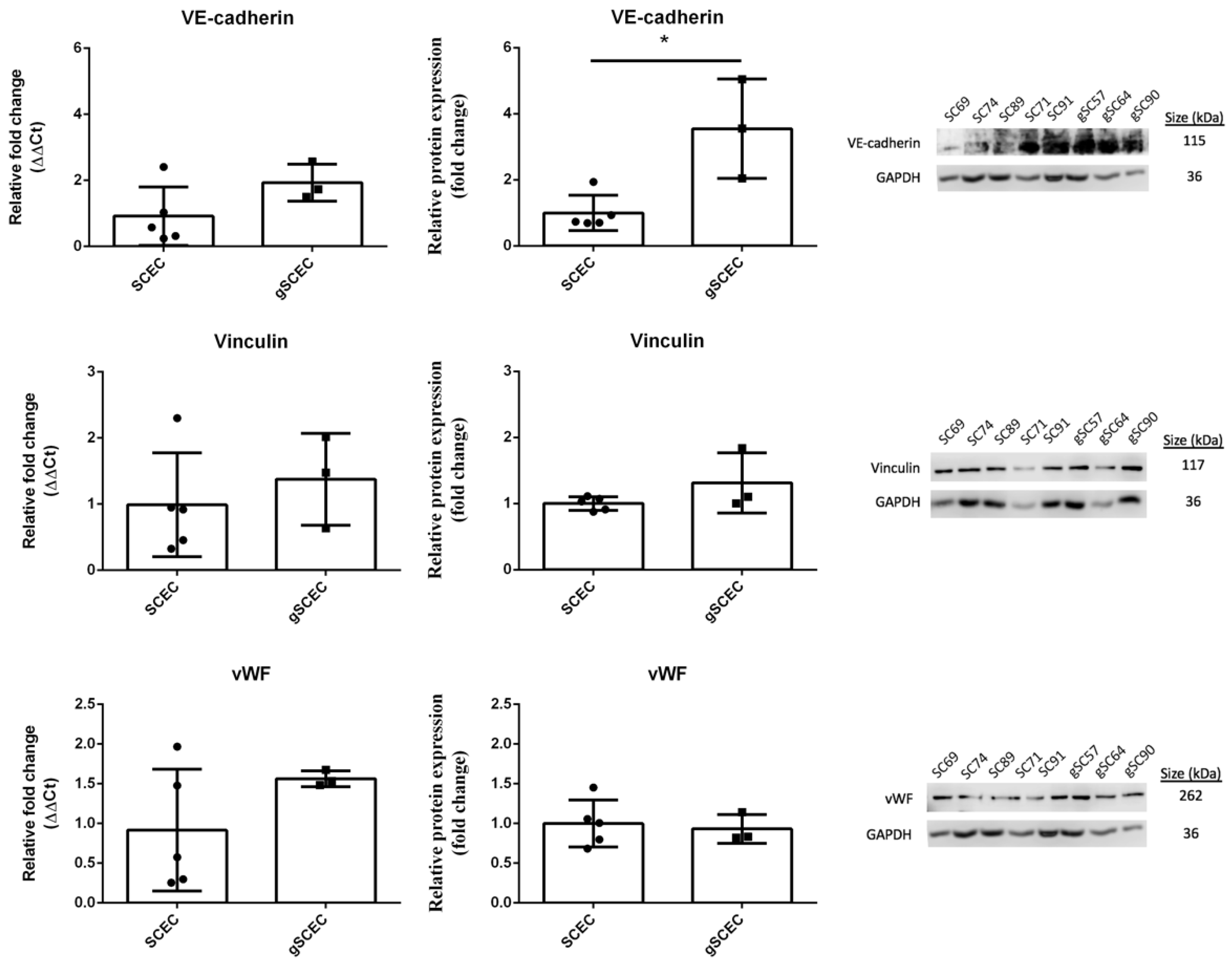
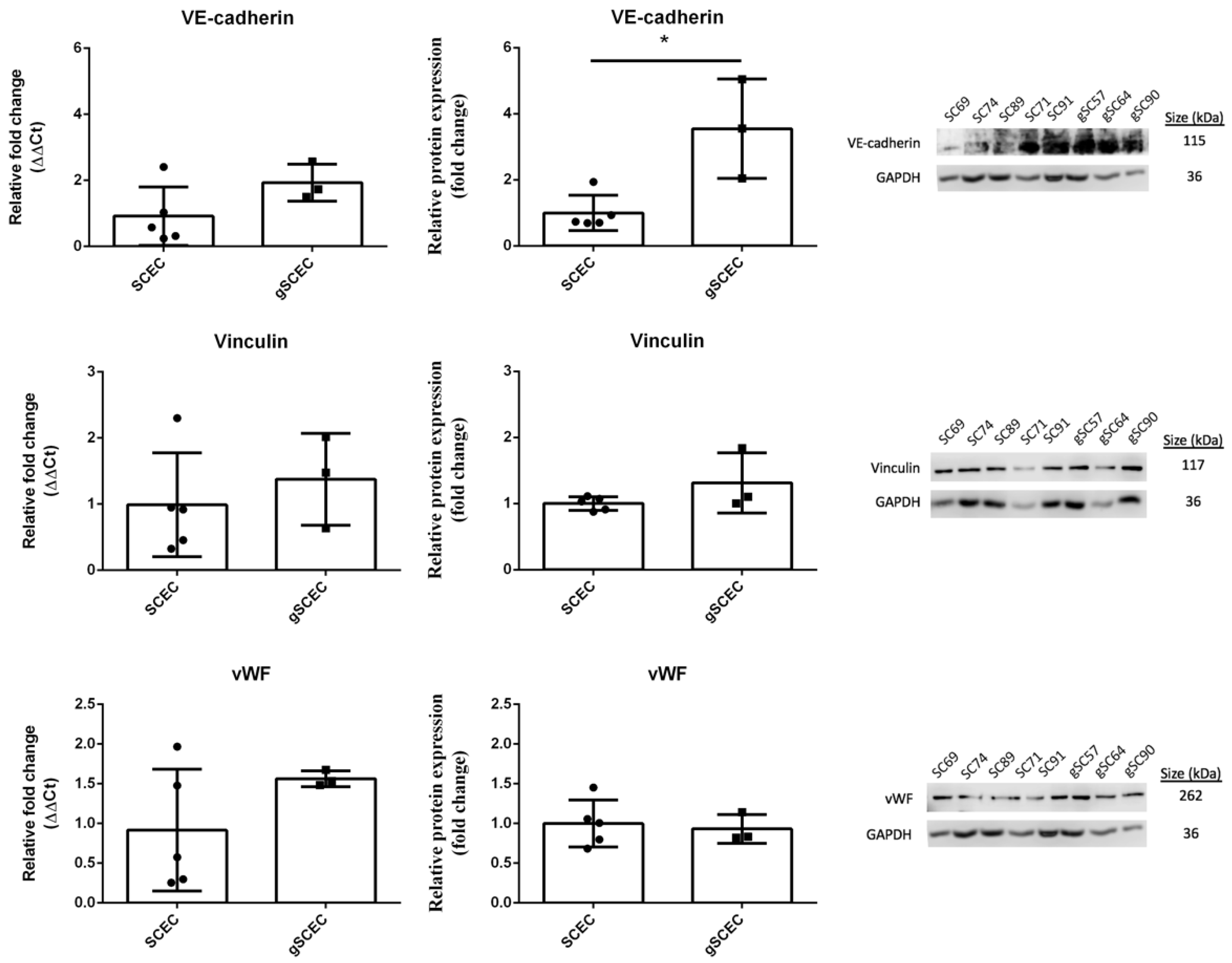
2.2. Comparison of Endothelial Cell Marker Expression in Healthy versus Glaucomatous SCECs
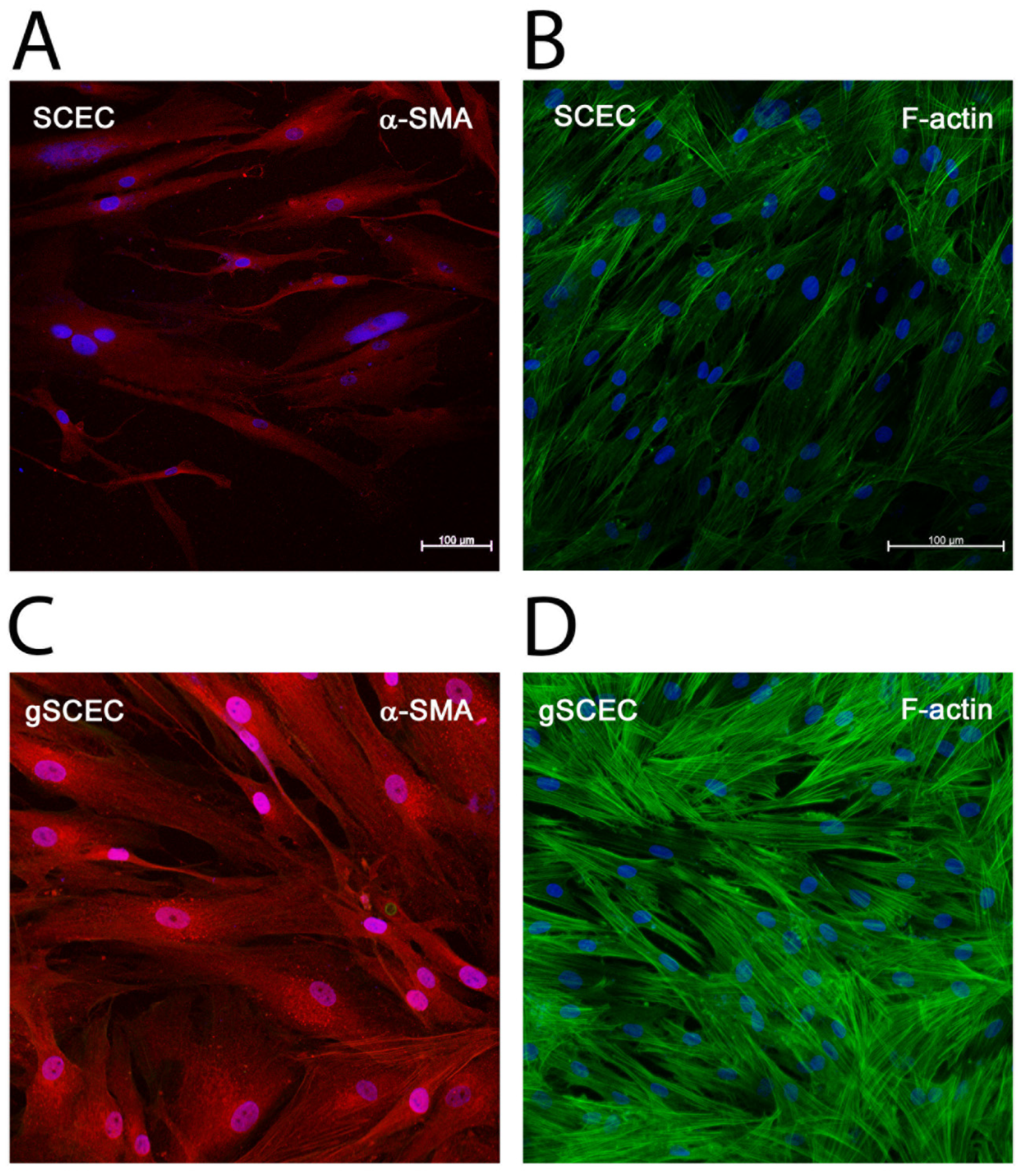
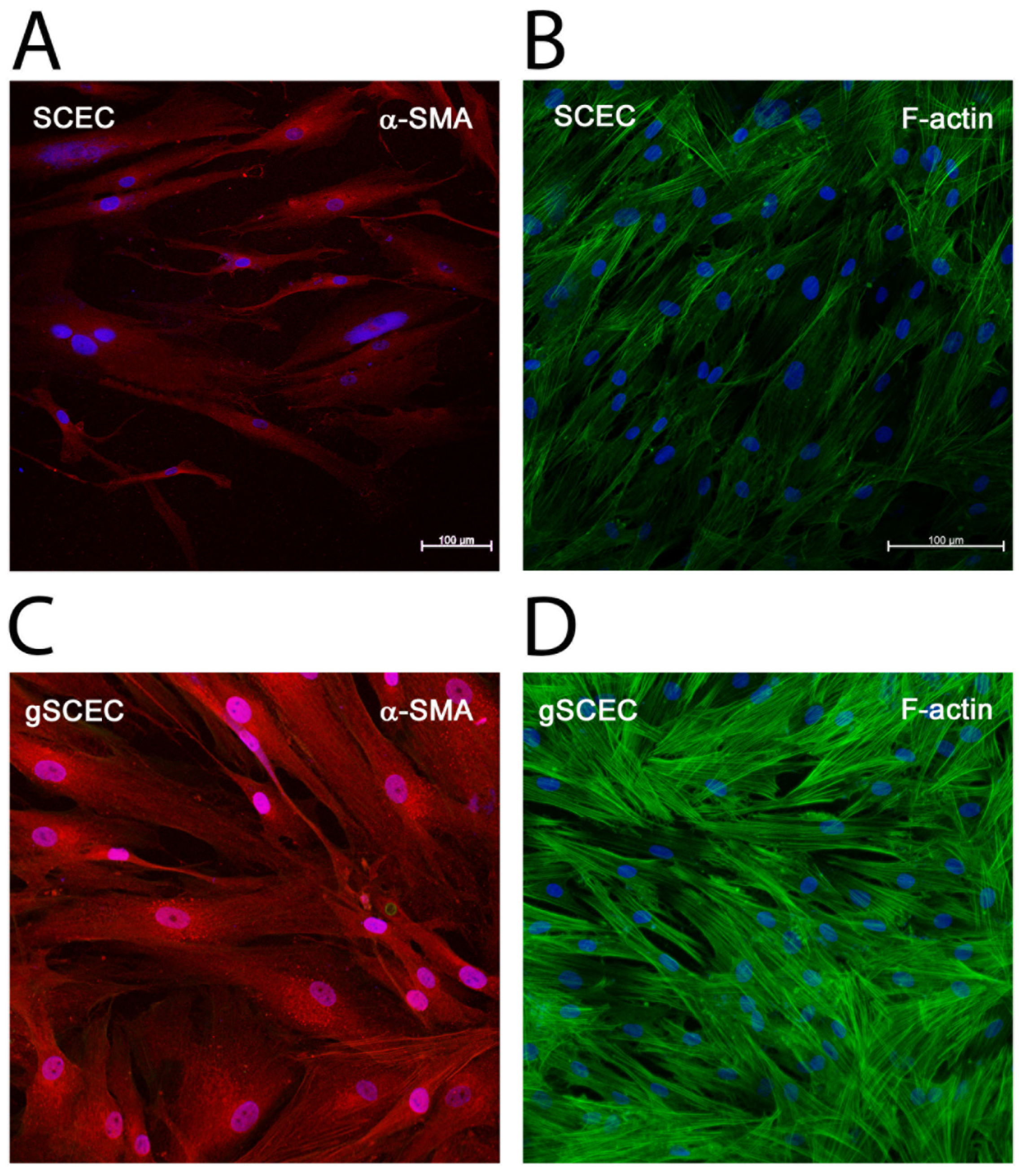
2.3. Elevated Levels of α-SMA and F-Actin Protein in gSCECs, Compared to Healthy SCECs
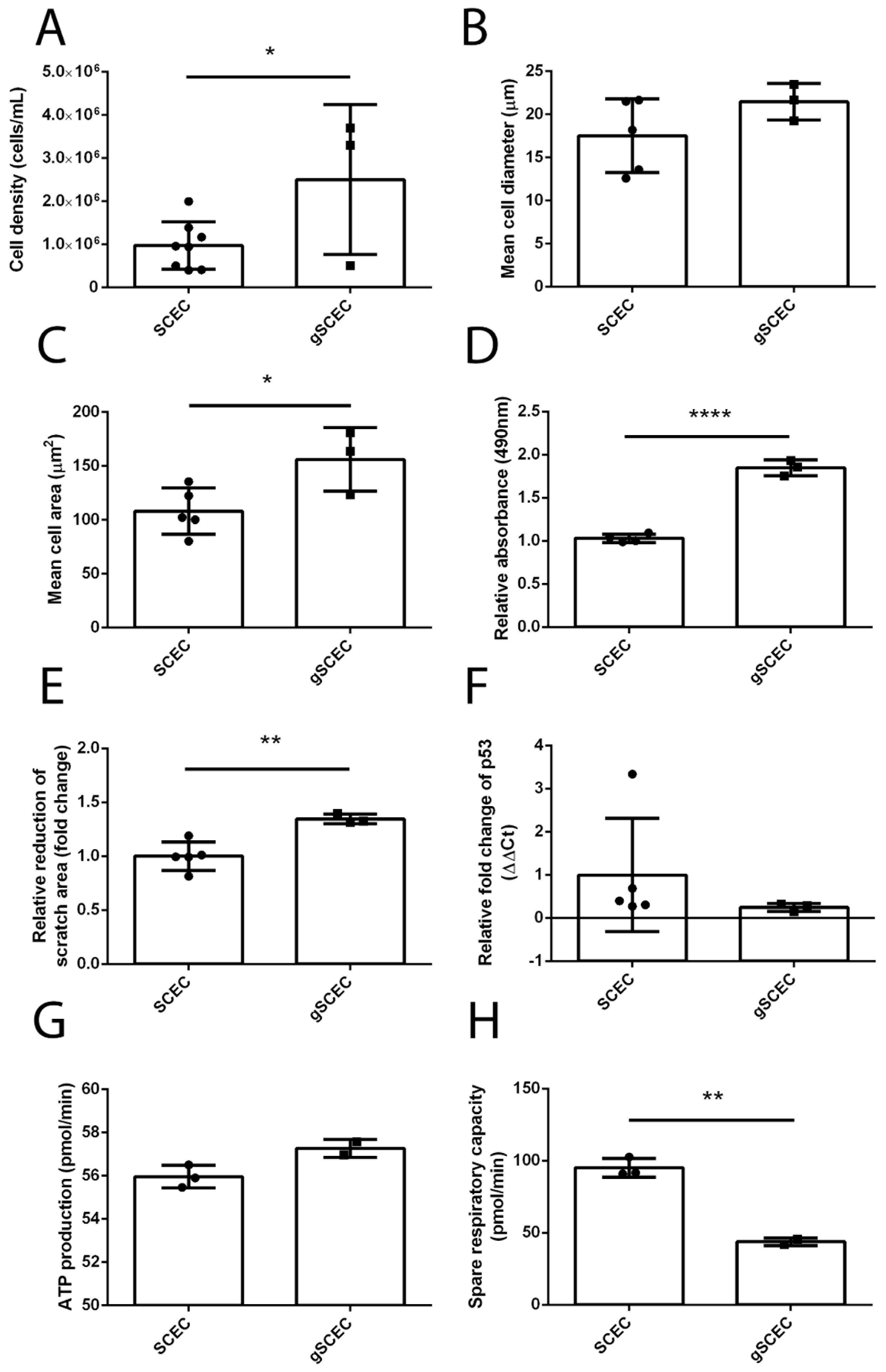
2.4. Increased Cell Size, Proliferation, Migration and Reduced Mitochondrial Activity in Glaucomatous SCECs, Compared to Healthy SCECs
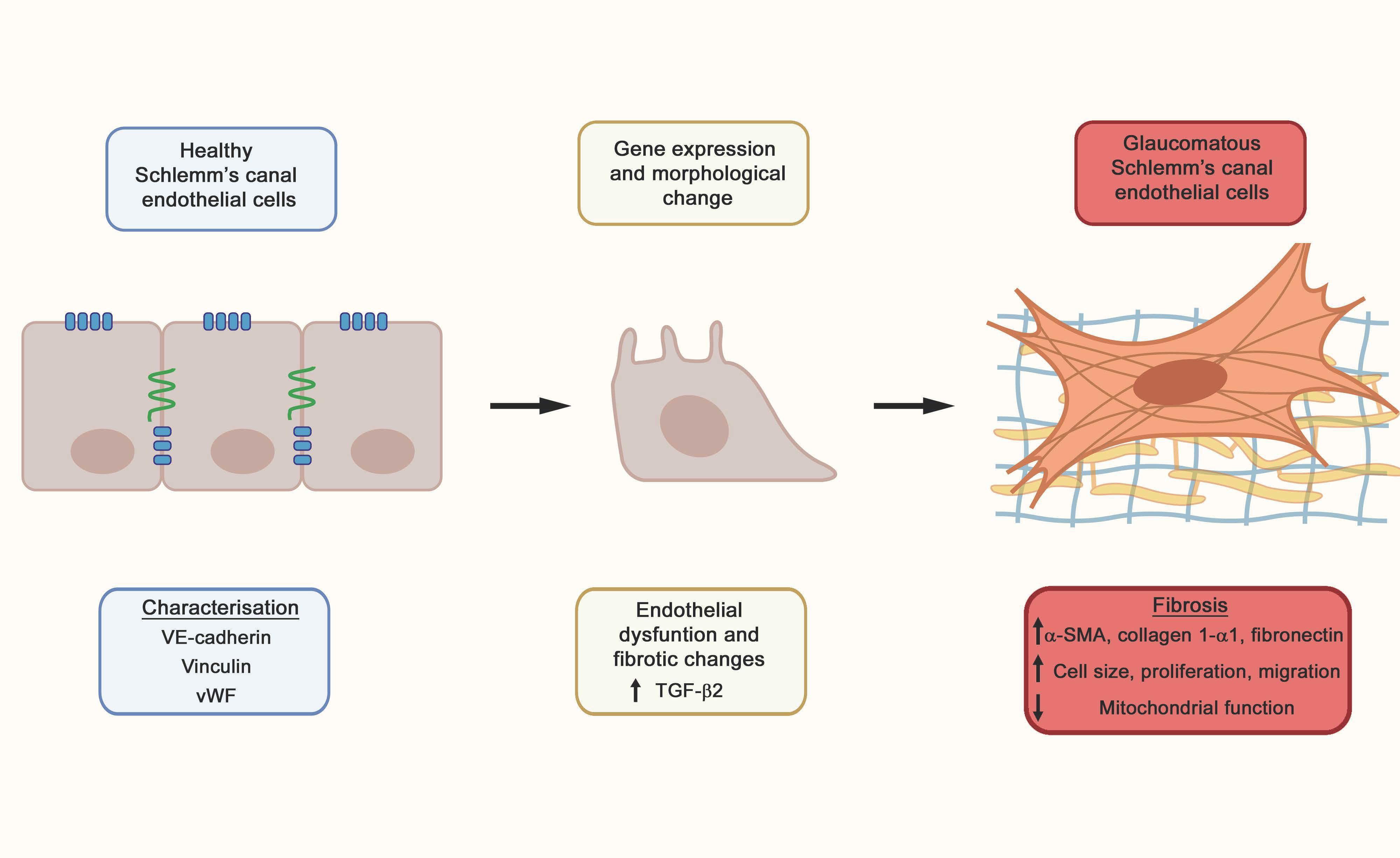
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Quantitative Real-Time PCR
4.3. Western Blot
4.4. Immunocytochemistry
4.5. Cell Morphology
4.6. Cell Proliferation (MTS) Assay
4.7. Migration Scratch Assay
4.8. Seahorse Assay
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Kass, M.A.; Heuer, D.K.; Higginbotham, E.J.; Johnson, C.A.; Keltner, J.L.; Miller, J.P.; Parrish, R.K., 2nd; Wilson, M.R.; Gordon, M.O. The Ocular Hypertension Treatment Study: A randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch. Ophthalmol. 2002, 120, 701–713. [Google Scholar] [CrossRef]
- Goel, M.; Picciani, R.G.; Lee, R.K.; Bhattacharya, S.K. Aqueous humor dynamics: A review. Open Ophthalmol. J. 2010, 4, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Stamer, W.D. The Cell and Molecular Biology of Glaucoma: Mechanisms in the Conventional Outflow Pathway. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2470–2472. [Google Scholar] [CrossRef] [Green Version]
- Vahabikashi, A.; Gelman, A.; Dong, B.; Gong, L.; Cha, E.D.K.; Schimmel, M.; Tamm, E.R.; Perkumas, K.; Stamer, W.D.; Sun, C.; et al. Increased stiffness and flow resistance of the inner wall of Schlemm’s canal in glaucomatous human eyes. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Overby, D.R.; Zhou, E.H.; Vargas-Pinto, R.; Pedrigi, R.M.; Fuchshofer, R.; Braakman, S.T.; Gupta, R.; Perkumas, K.M.; Sherwood, J.M.; Vahabikashi, A.; et al. Altered mechanobiology of Schlemm’s canal endothelial cells in glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 13876–13881. [Google Scholar] [CrossRef] [Green Version]
- Allingham, R.R.; de Kater, A.W.; Ethier, C.R.; Anderson, P.J.; Hertzmark, E.; Epstein, D.L. The relationship between pore density and outflow facility in human eyes. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1661–1669. [Google Scholar]
- Braakman, S.T.; Read, A.T.; Chan, D.W.H.; Ethier, C.R.; Overby, D.R. Colocalization of outflow segmentation and pores along the inner wall of Schlemm’s canal. Exp. Eye Res. 2015, 130, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Chan, D.; Read, A.T.; Christensen, C.; Sit, A.; Ethier, C.R. The pore density in the inner wall endothelium of Schlemm’s canal of glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2950–2955. [Google Scholar]
- Cai, J.; Perkumas, K.M.; Qin, X.; Hauser, M.A.; Stamer, W.D.; Liu, Y. Expression Profiling of Human Schlemm’s Canal Endothelial Cells From Eyes With and Without Glaucoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6747–6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, R.C.; Li, J.; Chan, W.F.; Tripathi, B.J. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp. Eye Res. 1994, 59, 723–727. [Google Scholar] [CrossRef]
- Agarwal, P.; Daher, A.M.; Agarwal, R. Aqueous humor TGF-β2 levels in patients with open-angle glaucoma: A meta-analysis. Mol. Vis. 2015, 21, 612–620. [Google Scholar] [PubMed]
- Min, S.H.; Lee, T.-I.; Chung, Y.S.; Kim, H.K. Transforming growth factor-beta levels in human aqueous humor of glaucomatous, diabetic and uveitic eyes. Korean J. Ophthalmol. 2006, 20, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, Y.; Ochiai, H. Higher concentration of transforming growth factor-beta in aqueous humor of glaucomatous eyes and diabetic eyes. Jpn. J. Ophthalmol. 2002, 46, 249–253. [Google Scholar] [CrossRef]
- Inatani, M.; Tanihara, H.; Katsuta, H.; Honjo, M.; Kido, N.; Honda, Y. Transforming growth factor-beta 2 levels in aqueous humor of glaucomatous eyes. Graefes Arch. Clin. Exp. Ophthalmol. 2001, 239, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, R.F. Cellular and biochemical aspects of normal and abnormal wound healing: An overview. J. Urol. 1997, 157, 298–302. [Google Scholar] [CrossRef]
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586. [Google Scholar] [CrossRef]
- Takahashi, E.; Inoue, T.; Fujimoto, T.; Kojima, S.; Tanihara, H. Epithelial mesenchymal transition-like phenomenon in trabecular meshwork cells. Exp. Eye Res. 2014, 118, 72–79. [Google Scholar] [CrossRef]
- Tamm, E.R.; Siegner, A.; Baur, A.; Lutjen-Drecoll, E. Transforming growth factor-beta 1 induces alpha-smooth muscle-actin expression in cultured human and monkey trabecular meshwork. Exp. Eye Res. 1996, 62, 389–397. [Google Scholar] [CrossRef]
- Fleenor, D.L.; Shepard, A.R.; Hellberg, P.E.; Jacobson, N.; Pang, I.H.; Clark, A.F. TGFbeta2-induced changes in human trabecular meshwork: Implications for intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2006, 47, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.Y. The extracellular matrix and its modulation in the trabecular meshwork. Surv. Ophthalmol. 1996, 40, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Acott, T.S.; Kelley, M.J. Extracellular matrix in the trabecular meshwork. Exp. Eye Res. 2008, 86, 543–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchshofer, R.; Tamm, E.R. The role of TGF-beta in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012, 347, 279–290. [Google Scholar] [CrossRef]
- Tovar-Vidales, T.; Clark, A.F.; Wordinger, R.J. Transforming growth factor-beta2 utilizes the canonical Smad-signaling pathway to regulate tissue transglutaminase expression in human trabecular meshwork cells. Exp. Eye Res. 2011, 93, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Ramsey, K.E.; Stephan, D.A.; Russell, P. Gene and protein expression changes in human trabecular meshwork cells treated with transforming growth factor-beta. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4023–4034. [Google Scholar] [CrossRef]
- Gottanka, J.; Chan, D.; Eichhorn, M.; Lütjen-Drecoll, E.; Ethier, C.R. Effects of TGF-beta2 in perfused human eyes. Investig. Ophthalmol. Vis. Sci. 2004, 45, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Junglas, B.; Kuespert, S.; Seleem, A.A.; Struller, T.; Ullmann, S.; Bosl, M.; Bosserhoff, A.; Kostler, J.; Wagner, R.; Tamm, E.R.; et al. Connective tissue growth factor causes glaucoma by modifying the actin cytoskeleton of the trabecular meshwork. Am. J. Pathol. 2012, 180, 2386–2403. [Google Scholar] [CrossRef]
- Zhu, Y.-T.; Chen, H.-C.; Chen, S.-Y.; Tseng, S.C.G. Nuclear p120 catenin unlocks mitotic block of contact-inhibited human corneal endothelial monolayers without disrupting adherent junctions. J. Cell Sci. 2012, 125, 3636–3648. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.-S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunger, B.M.; Fuchshofer, R.; Tamm, E.R. The aqueous humor outflow pathways in glaucoma: A unifying concept of disease mechanisms and causative treatment. Eur. J. Pharm. Biopharm. 2015, 95, 173–181. [Google Scholar] [CrossRef]
- Pattabiraman, P.P.; Maddala, R.; Rao, P.V. Regulation of plasticity and fibrogenic activity of trabecular meshwork cells by Rho GTPase signaling. J. Cell Physiol. 2014, 229, 927–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabiraman, P.P.; Rao, P.V. Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. Am. J. Physiol. Cell Physiol. 2010, 298, C749–C763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Ma, Y.; Liu, Y.; Wan, Q. Measurement of mitochondrial respiration in adherent cells by Seahorse XF96 Cell Mito Stress Test. Star Protoc. 2021, 2, 100245. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhang, J.; Gan, T.Y.; Xu, G.J.; Tang, B.P. Advanced glycation end products induce endothelial-to-mesenchymal transition via downregulating Sirt 1 and upregulating TGF-β in human endothelial cells. Biomed. Res. Int. 2015, 2015, 684242. [Google Scholar] [CrossRef] [Green Version]
- Zhavoronkov, A.; Izumchenko, E.; Kanherkar, R.R.; Teka, M.; Cantor, C.; Manaye, K.; Sidransky, D.; West, M.D.; Makarev, E.; Csoka, A.B. Pro-fibrotic pathway activation in trabecular meshwork and lamina cribrosa is the main driving force of glaucoma. Cell Cycle 2016, 15, 1643–1652. [Google Scholar] [CrossRef]
- McDonnell, F.; O’Brien, C.; Wallace, D. The role of epigenetics in the fibrotic processes associated with glaucoma. J. Ophthalmol. 2014, 2014, 750459. [Google Scholar] [CrossRef] [Green Version]
- Wary, A.L.; Sreedhar, S. The Structure and Function of VE-cadherin in Endothelial Cells. FASEB J. 2017, 31, lb97. [Google Scholar] [CrossRef]
- Perkumas, K.M.; Stamer, W.D. Protein markers and differentiation in culture for Schlemm’s canal endothelial cells. Exp. Eye Res. 2012, 96, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Zhou, E.H.; Krishnan, R.; Stamer, W.D.; Perkumas, K.M.; Rajendran, K.; Nabhan, J.F.; Lu, Q.; Fredberg, J.J.; Johnson, M. Mechanical responsiveness of the endothelial cell of Schlemm’s canal: Scope, variability and its potential role in controlling aqueous humour outflow. J. R. Soc. Interface 2012, 9, 1144–1155. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef]
- Yao, L.; Romero, M.J.; Toque, H.A.; Yang, G.; Caldwell, R.B.; Caldwell, R.W. The role of RhoA/Rho kinase pathway in endothelial dysfunction. J. Cardiovasc. Dis. Res. 2010, 1, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanna, A.P.; Johnson, M. Rho Kinase Inhibitors as a Novel Treatment for Glaucoma and Ocular Hypertension. J. Ophthalmol. 2018, 125, 1741–1756. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Lee, C.; Read, A.T.; Wang, K.; Ha, J.; Kuhn, M.; Navarro, I.; Cui, J.; Young, K.; Gorijavolu, R.; et al. Anti-fibrotic activity of a rho-kinase inhibitor restores outflow function and intraocular pressure homeostasis. Elife 2021, 10. [Google Scholar] [CrossRef]
- Bylsma, S.S.; Samples, J.R.; Acott, T.S.; Van Buskirk, E.M. Trabecular cell division after argon laser trabeculoplasty. Arch. Ophthalmol. 1988, 106, 544–547. [Google Scholar] [CrossRef]
- Napoli, C.; Benincasa, G.; Loscalzo, J. Epigenetic Inheritance Underlying Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 653–664. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef] [PubMed]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Kamel, K.; O’Brien, C.J.; Zhdanov, A.V.; Papkovsky, D.B.; Clark, A.F.; Stamer, W.D.; Irnaten, M. Reduced Oxidative Phosphorylation and Increased Glycolysis in Human Glaucoma Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4. [Google Scholar] [CrossRef]
- O’Brien, C.J.; McNally, S.; Kirwan, R.; Clark, A.F.; Wallace, D. Reduced Expression of the Cell Cycle Inhibitors p27 and p57 and Increased Proliferation in Glaucoma Lamina Cribrosa Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3675. [Google Scholar]
- Stamer, W.D.; Roberts, B.C.; Howell, D.N.; Epstein, D.L. Isolation, culture, and characterization of endothelial cells from Schlemm’s canal. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1804–1812. [Google Scholar]
- Heimark, R.L.; Kaochar, S.; Stamer, W.D. Human Schlemm’s canal cells express the endothelial adherens proteins, VE-cadherin and PECAM-1. Curr. Eye Res. 2002, 25, 299–308. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Underwood, E.; Redell, J.B.; Zhao, J.; Moore, A.N.; Dash, P.K. A method for assessing tissue respiration in anatomically defined brain regions. Sci. Rep. 2020, 10, 13179. [Google Scholar] [CrossRef]
- Leung, D.T.H.; Chu, S. Measurement of Oxidative Stress: Mitochondrial Function Using the Seahorse System. Methods Mol. Biol. 2018, 1710, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Repp, B.; Biagosch, C.; Terrile, C.; Prokisch, H. Assessing Mitochondrial Bioenergetics in Isolated Mitochondria from Various Mouse Tissues Using Seahorse XF96 Analyzer. Methods Mol. Biol. 2017, 1567, 217–230. [Google Scholar] [CrossRef] [PubMed]
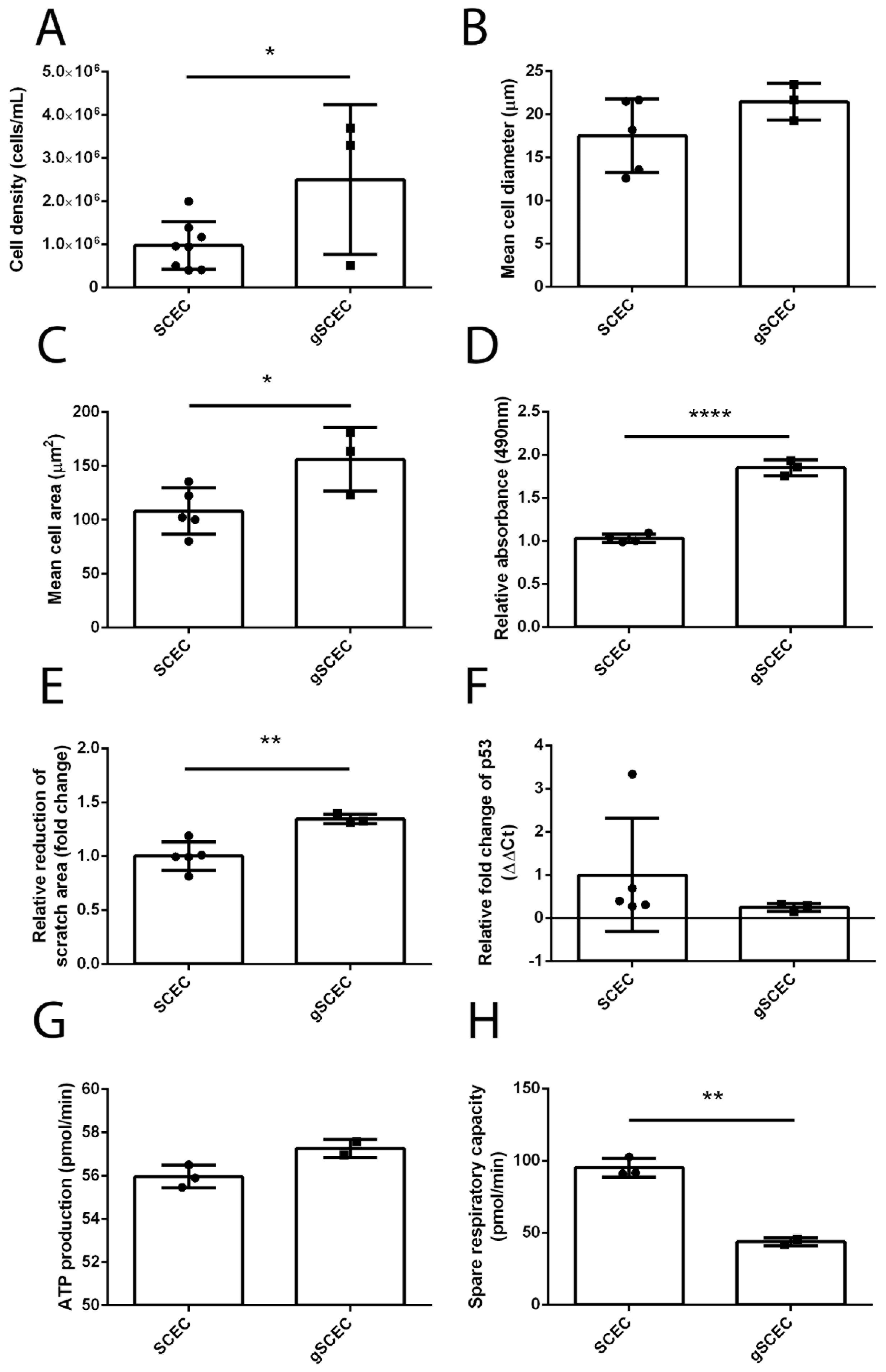

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Significant Changes |
|---|---|
| α-SMA transcript expression | ↑ *** |
| α-SMA protein expression | ↑ * |
| TGF-β2 transcript expression | ↑ * |
| TGF-β2 protein expression | ↑ * |
| Collagen I-α1 protein expression | ↑ * |
| Fibronectin protein expression | ↑ * |
| VE-cadherin protein expression | ↑ * |
| Cell density | ↑ * |
| Cell area (ICC) | ↑ * |
| Cell migration | ↑ ** |
| Cell proliferation | ↑ **** |
| Spare respiratory capacity | ↓ ** |
| Group | Cell Strain No. | Age (Years) | Sex |
|---|---|---|---|
| Healthy | SCEC 71 | 44 | Male |
| SCEC 91a | 74 | Female | |
| SCEC 89 | 68 | Male | |
| SCEC 69 | 45 | Male | |
| SCEC 74 | 0.7 | Male | |
| SCEC 68 | 30 | Unknown | |
| SCEC 81 | 75 | Unknown | |
| SCEC 86 | 62 | Male | |
| Glaucoma | gSCEC 57 | 78 | Male |
| gSCEC 64 | 78 | Male | |
| gSCEC 90 | 71 | Female |
| Primer Pair | Forward Sequence (5’ to 3’) | Reverse Sequence (5’ to 3’) |
|---|---|---|
| α-SMA | CCGACGAATGCAGAAGGA | ACAGAGTATTTGCGCTCCGAA |
| TGF-β2 | ACGGATTGAGCTATATCAGATTCTCA | TGCAGCAGGGACAGTGTAAG |
| Collagen Iα-1 | TTTGGATGGTGCCAAGGGAG | CACCATCATTTCCACGAGCA |
| Fibronectin | AGCGGACCTACCTAGGCAAT | GGTTTGCGATGGTACAGCTT |
| VE-cadherin | GCACCAGTTTGGCCAATATA | GGGTTTTTGCATAATAAGCAGG |
| Vinculin | TGGACGAAGCCATTGATACCA | AGCTCTTTTGCAGTCCAGGG |
| vWF | GTTCGTCCTGGAAGGATCGG | CACTGACACCTGAGTGAGAC |
| β-actin | GGGAAATCGTGCGTGACAT | GTGATGACCTGGCCGTAG |
| GAPDH | TGTAGTTGAGGTCAATGAAGGG | ACATCGCTCAGACACCATG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly, R.A.; Perkumas, K.M.; Campbell, M.; Farrar, G.J.; Stamer, W.D.; Humphries, P.; O’Callaghan, J.; O’Brien, C.J. Fibrotic Changes to Schlemm’s Canal Endothelial Cells in Glaucoma. Int. J. Mol. Sci. 2021, 22, 9446. https://doi.org/10.3390/ijms22179446
Kelly RA, Perkumas KM, Campbell M, Farrar GJ, Stamer WD, Humphries P, O’Callaghan J, O’Brien CJ. Fibrotic Changes to Schlemm’s Canal Endothelial Cells in Glaucoma. International Journal of Molecular Sciences. 2021; 22(17):9446. https://doi.org/10.3390/ijms22179446
Chicago/Turabian StyleKelly, Ruth A., Kristin M. Perkumas, Matthew Campbell, G. Jane Farrar, W. Daniel Stamer, Pete Humphries, Jeffrey O’Callaghan, and Colm J. O’Brien. 2021. "Fibrotic Changes to Schlemm’s Canal Endothelial Cells in Glaucoma" International Journal of Molecular Sciences 22, no. 17: 9446. https://doi.org/10.3390/ijms22179446
APA StyleKelly, R. A., Perkumas, K. M., Campbell, M., Farrar, G. J., Stamer, W. D., Humphries, P., O’Callaghan, J., & O’Brien, C. J. (2021). Fibrotic Changes to Schlemm’s Canal Endothelial Cells in Glaucoma. International Journal of Molecular Sciences, 22(17), 9446. https://doi.org/10.3390/ijms22179446