
The Metabolic Flux Probe (MFP)—Secreted Protein as a Non-Disruptive Information Carrier for 13C-Based Metabolic Flux Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Cultivation Workflow
2.3. Continuous Cultivations for 13C-Based Metabolic Flux Ratio Analysis (METAFoR) and Metabolic Flux Analysis (MFA)
2.4. Batch Cultivation for 13C-Based Metabolic Flux Ratio Analysis (METAFoR) and Metabolic Flux Analysis (MFA)
2.5. HPLC Analysis of Cultivation Supernatants
2.6. Protein Purification
2.7. Protein Quantification According to Bradford
2.8. SDS-PAGE Analysis
2.9. Protein and Biomass Processing for GC-MS Analysis
2.10. Stable 13C-Isotope-Based Metabolic Flux Ratio Analysis (METAFoR)
3. Results
3.1. Development of a Sample Processing Workflow for Isotope Mapping with Secreted Protein
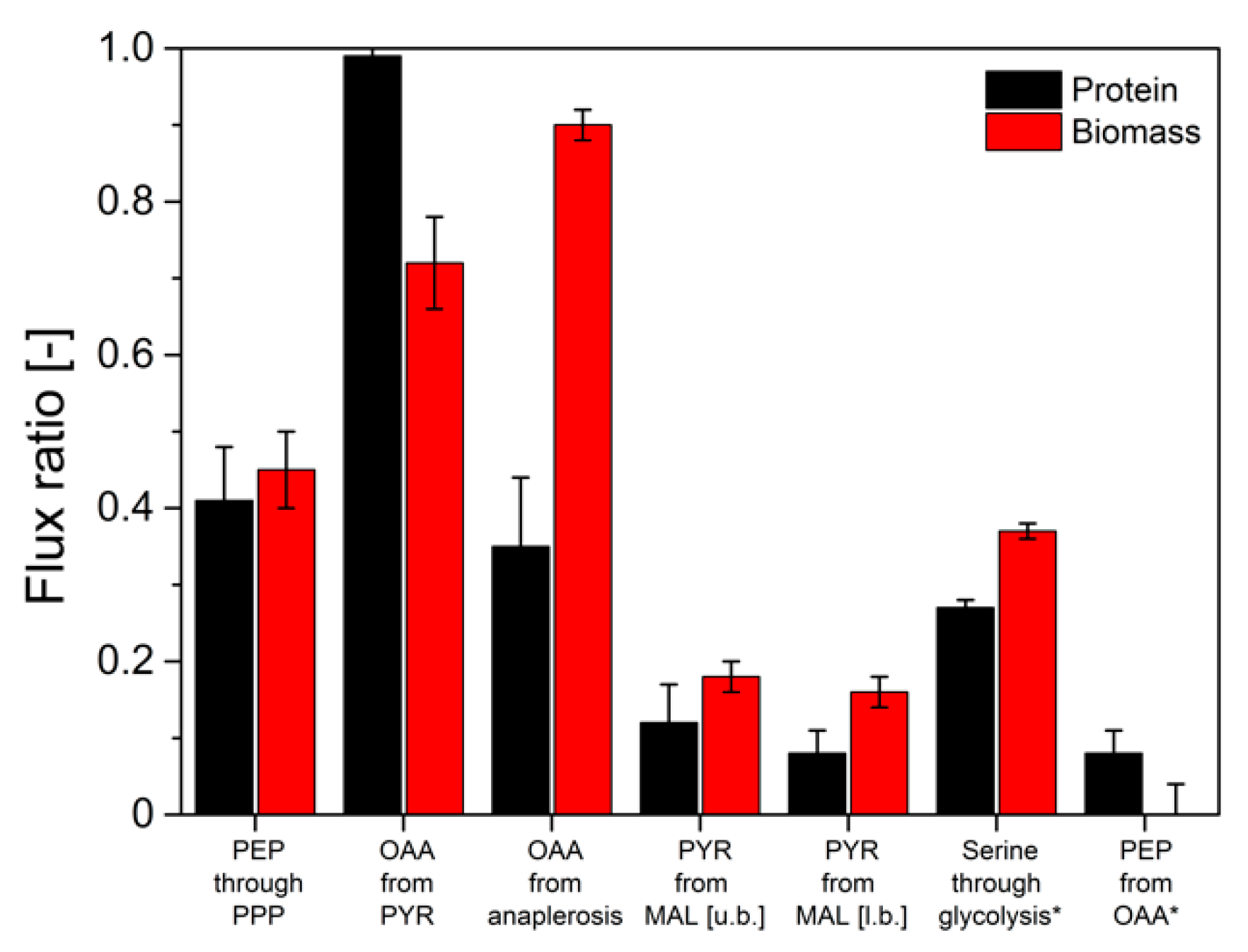
3.2. 13C-Isotope Mapping from Secreted Protein and Biomass in Aerobic Batch Cultivations (Metabolic and Isotopic Steady-State)
3.3. 13C-Isotope Mapping from Secreted Protein and Biomass in Continuous Glucose-Limited Chemostat Cultivations (Metabolic and Isotopic Steady-State)
3.4. Dynamic 13C-Labeling Experiments in Metabolic Steady-State and Isotopic Nonstationary State
4. Discussion
The Single-Cell Flux Probe: Opportunities and Challenges
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Heyland, J.; Fu, J.; Blank, L.M. Correlation between TCA cycle flux and glucose uptake rate during respiro-fermentative growth of Saccharomyces cerevisiae. Microbiology 2009, 155, 3827–3837. [Google Scholar] [CrossRef]
- Blank, L.M.; Kuepfer, L.; Sauer, U. Large-scale 13C-flux analysis reveals mechanistic principles of metabolic network robustness to null mutations in yeast. Genome Biol. 2005, 6, R49. [Google Scholar] [CrossRef] [PubMed]
- Blank, L.M.; Kuepfer, L. Metabolic flux distributions: Genetic information, computational predictions, and experimental validation. Appl. Microbiol. Biotechnol. 2010, 86, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Lange, S.; Wilhelm, N.; Bureik, M.; Yang, T.H.; Heinzle, E.; Schneider, K. Overcoming the metabolic burden of protein secretion in Schizosaccharomyces pombe—A quantitative approach using 13C-based metabolic flux analysis. Metab. Eng. 2014, 21, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.; Fritzsch, F.S.; Zhang, X.; Wendisch, V.F.; Blank, L.M.; Buhler, B.; Schmid, A. Subtoxic product levels limit the epoxidation capacity of recombinant E. coli by increasing microbial energy demands. J. Biotechnol. 2013, 163, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Blank, L.M.; Ebert, B.E.; Buhler, B.; Schmid, A. Metabolic capacity estimation of Escherichia coli as a platform for redox biocatalysis: Constraint-based modeling and experimental verification. Biotechnol. Bioeng. 2008, 100, 1050–1065. [Google Scholar] [CrossRef]
- Sauer, U.; Zamboni, N.; Fendt, S.M.; Ruhl, M. (13)C-based metabolic flux analysis. Nat. Protoc. 2009, 4, 878–892. [Google Scholar] [CrossRef]
- Wittmann, C.; Hans, M.; Heinzle, E. In vivo analysis of intracellular amino acid labelings by GC/MS. Anal. Biochem. 2002, 307, 379–382. [Google Scholar] [CrossRef]
- Zamboni, N.; Fischer, E.; Sauer, U. FiatFlux—A software for metabolic flux analysis from 13C-glucose experiments. BMC Bioinform. 2005, 6, 209. [Google Scholar] [CrossRef]
- Weitzel, M.; Noh, K.; Dalman, T.; Niedenfuhr, S.; Stute, B.; Wiechert, W. 13CFLUX2—High-performance software suite for (13)C-metabolic flux analysis. Bioinformatics 2013, 29, 143–145. [Google Scholar] [CrossRef]
- Quek, L.E.; Wittmann, C.; Nielsen, L.K.; Kromer, J.O. OpenFLUX: Efficient modelling software for 13C-based metabolic flux analysis. Microb. Cell Fact. 2009, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2006, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U.; Zamboni, N. Novel biological insights through metabolomics and (13)C-flux analysis. Curr. Opin. Microbiol. 2009, 12, 553–558. [Google Scholar] [CrossRef]
- Fischer, E.; Sauer, U. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 2003, 270, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U.; Ruhl, M.; Hardt, W.D. Subpopulation-Specific Metabolic pathway usage in mixed cultures as revealed by reporter protein-based (13)C analysis. Appl. Environ. Microbiol. 2011, 77, 1816–1821. [Google Scholar] [CrossRef]
- Tsioris, K.; Torres, A.J.; Douce, T.B.; Love, J.C. A new toolbox for assessing single cells. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, M.; Zenobi, R. Single cell metabolomics. Curr. Opin. Biotechnol. 2011, 22, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Rubakhin, S.S.; Lanni, E.J.; Sweedler, J.V. Progress toward single cell metabolomics. Curr. Opin. Biotechnol. 2013, 24, 95–104. [Google Scholar] [CrossRef]
- Urban, P.L.; Schmidt, A.M.; Fagerer, S.R.; Amantonico, A.; Ibanez, A.; Jefimovs, K.; Heinemann, M.; Zenobi, R. Carbon-13 labelling strategy for studying the ATP metabolism in individual yeast cells by micro-arrays for mass spectrometry. Mol. Biosyst. 2011, 7, 2837–2840. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Carnicer, M.; Baumann, K.; Toplitz, I.; Sanchez-Ferrando, F.; Mattanovich, D.; Ferrer, P.; Albiol, J. Macromolecular and elemental composition analysis and extracellular metabolite balances of Pichia pastoris growing at different oxygen levels. Microb. Cell Fact. 2009, 8, 65. [Google Scholar] [CrossRef]
- Van Winden, W.A.; van Dam, J.C.; Ras, C.; Kleijn, R.J.; Vinke, J.L.; van Gulik, W.M.; Heijnen, J.J. Metabolic-flux analysis of Saccharomyces cerevisiae CEN.PK113-7D based on mass isotopomer measurements of (13)C-labeled primary metabolites. FEMS Yeast Res. 2005, 5, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Zamboni, N.; Sauer, U. High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal. Biochem. 2004, 325, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Kostrewa, D.; Wyss, M.; Brugger, R.; D’Arcy, A.; Pasamontes, L.; van Loon, A.P. From DNA sequence to improved functionality: Using protein sequence comparisons to rapidly design a thermostable consensus phytase. Protein Eng. 2000, 13, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Gellissen, G.; Veenhuis, M. The methylotrophic yeast Hansenula polymorpha: Its use in fundamental research and as a cell factory. Yeast 2001, 18, i–iii. [Google Scholar] [CrossRef]
- Mayer, A.F.; Hellmuth, K.; Schlieker, H.; Lopez-Ulibarri, R.; Oertel, S.; Dahlems, U.; Strasser, A.W.M.; van Loon, A.P.G.M. An expression system matures: A highly efficient and cost-effective process for phytase production by recombinant strains of Hansenula polymorpha. Biotechnol. Bioeng. 1999, 63, 373–381. [Google Scholar] [CrossRef]
- Nanchen, A.; Fuhrer, T.; Sauer, U. Determination of metabolic flux ratios from 13C-experiments and gas chromatography-mass spectrometry data: Protocol and principles. Methods Mol. Biol. 2007, 358, 177–197. [Google Scholar] [CrossRef]
- Mandelstam, J. The intracellular turnover of protein and nucleic acids and its role in biochemical differentiation. Bacteriol. Rev. 1960, 24, 289–308. [Google Scholar] [CrossRef] [PubMed]
- Halvorson, H. Intracellular protein and nucleic acid turnover in resting yeast cells. Biochim. Biophys. Acta 1958, 27, 255–266. [Google Scholar] [CrossRef]
- Shilo, B.; Riddle, V.G.; Pardee, A.B. Protein turnover and cell-cycle initiation in yeast. Exp. Cell Res. 1979, 123, 221–227. [Google Scholar] [CrossRef]
- Li, Q. Advances in protein turnover analysis at the global level and biological insights. Mass Spectrom. Rev. 2010, 29, 717–736. [Google Scholar] [CrossRef]
- Geva-Zatorsky, N.; Issaeva, I.; Mayo, A.; Cohen, A.; Dekel, E.; Danon, T.; Cohen, L.; Liron, Y.; Alon, U.; Eden, E. Using bleach-chase to measure protein half-lives in living cells. Nat. Protoc. 2012, 7, 801–811. [Google Scholar] [CrossRef]
- Cazzador, L. Characterization of cell population growth by cell cycle parameters. J. Biotechnol. 1999, 71, 245–249. [Google Scholar] [CrossRef]
- Dano, S.; Hynne, F.; De Monte, S.; d’Ovidio, F.; Sorensen, P.G.; Westerhoff, H. Synchronization of glycolytic oscillations in a yeast cell population. Faraday Discuss. 2001, 120, 261–276; discussion 325–351. [Google Scholar] [CrossRef] [PubMed]
- Madsen, M.F.; Dano, S.; Sorensen, P.G. On the mechanisms of glycolytic oscillations in yeast. FEBS J. 2005, 272, 2648–2660. [Google Scholar] [CrossRef]
- Hans, M.A.; Heinzle, E.; Wittmann, C. Free intracellular amino acid pools during autonomous oscillations in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2003, 82, 143–151. [Google Scholar] [CrossRef]
- O’Shea, E.K.; Belle, A.; Tanay, A.; Bitincka, L.; Shamir, R. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. USA 2006, 103, 13004–13009. [Google Scholar] [CrossRef]
- Doerr, A. A day in the half-life of a protein. Nat. Methods 2011, 8, 201. [Google Scholar] [CrossRef]
- Noh, K.; Noack, S.; Moch, M.; Oldiges, M.; Wiechert, W. Stationary versus nonstationary (13)C-MFA: A comparison using a consistent dataset. J. Biotechnol. 2011, 154, 179–190. [Google Scholar] [CrossRef]
- Wahl, S.; Noh, K.; Wiechert, W. 13C labeling experiments at metabolic nonstationary conditions: An exploratory study. BMC Bioinform. 2008, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, J.; Sutherland, A.; Wei, W.; Shin, Y.S.; Xue, M.; Heath, J.R. Microfluidics-based single-cell functional proteomics for fundamental and applied biomedical applications. Annu. Rev. Anal. Chem. 2014, 7, 275–295. [Google Scholar] [CrossRef] [PubMed]
- Zenobi, R.; Amantonico, A.; Urban, P.L.; Fagerer, S.R.; Balabin, R.M. Single-Cell MALDI-MS as an analytical tool for studying intrapopulation metabolic heterogeneity of unicellular organisms. Anal. Chem. 2010, 82, 7394–7400. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dusny, C.; Schmid, A. The Metabolic Flux Probe (MFP)—Secreted Protein as a Non-Disruptive Information Carrier for 13C-Based Metabolic Flux Analysis. Int. J. Mol. Sci. 2021, 22, 9438. https://doi.org/10.3390/ijms22179438
Dusny C, Schmid A. The Metabolic Flux Probe (MFP)—Secreted Protein as a Non-Disruptive Information Carrier for 13C-Based Metabolic Flux Analysis. International Journal of Molecular Sciences. 2021; 22(17):9438. https://doi.org/10.3390/ijms22179438
Chicago/Turabian StyleDusny, Christian, and Andreas Schmid. 2021. "The Metabolic Flux Probe (MFP)—Secreted Protein as a Non-Disruptive Information Carrier for 13C-Based Metabolic Flux Analysis" International Journal of Molecular Sciences 22, no. 17: 9438. https://doi.org/10.3390/ijms22179438
APA StyleDusny, C., & Schmid, A. (2021). The Metabolic Flux Probe (MFP)—Secreted Protein as a Non-Disruptive Information Carrier for 13C-Based Metabolic Flux Analysis. International Journal of Molecular Sciences, 22(17), 9438. https://doi.org/10.3390/ijms22179438