
Synthetic NAC 71-82 Peptides Designed to Produce Fibrils with Different Protofilament Interface Contacts

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
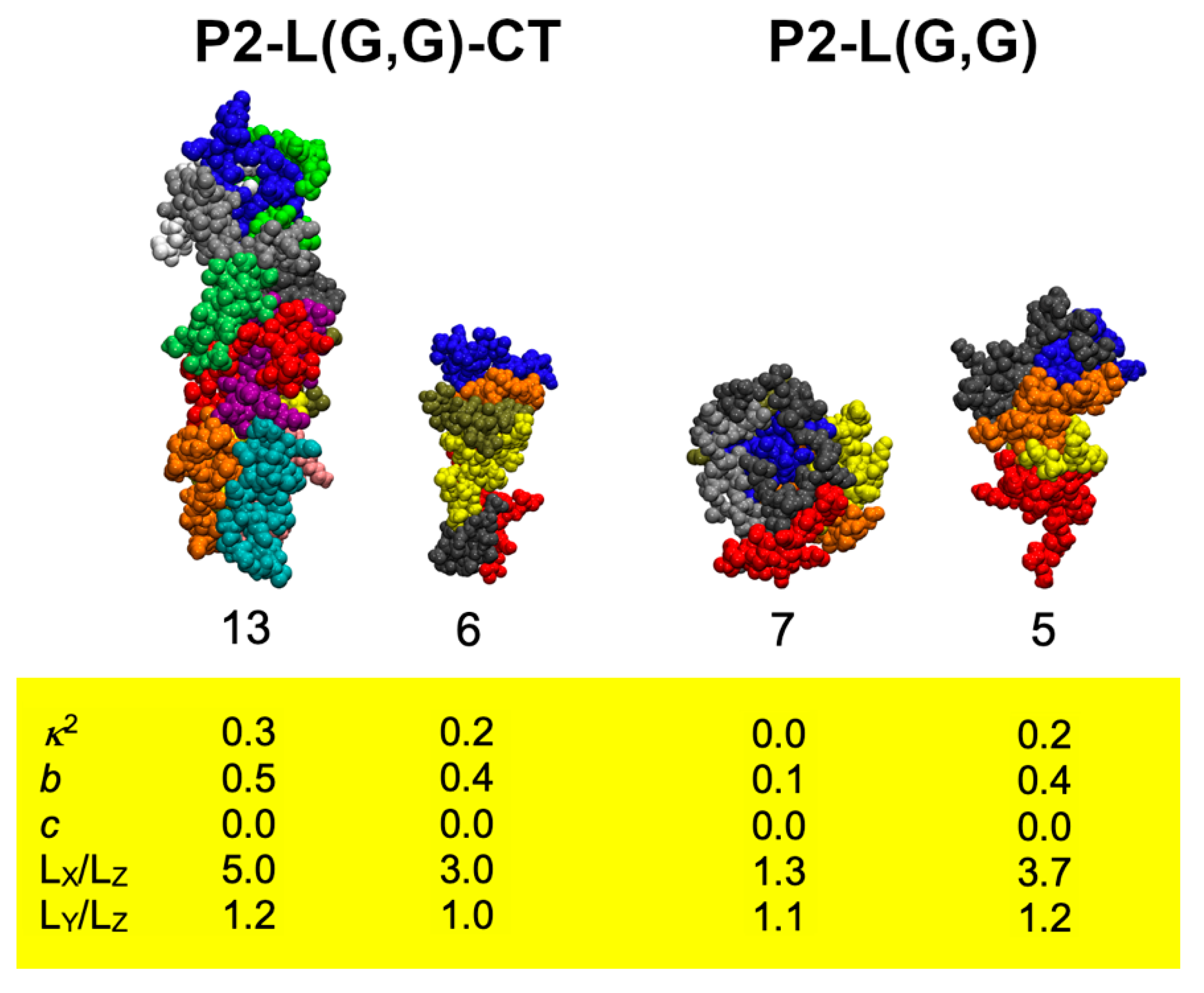
2.1. Molecular Dynamics Simulations Suggest the Formation of Stable P2-L(G,G)-CT Peptide Aggregates
2.2. Synthesised P2-L(G,G)-CT Fibrils Demonstrated Structural Heterogeneity
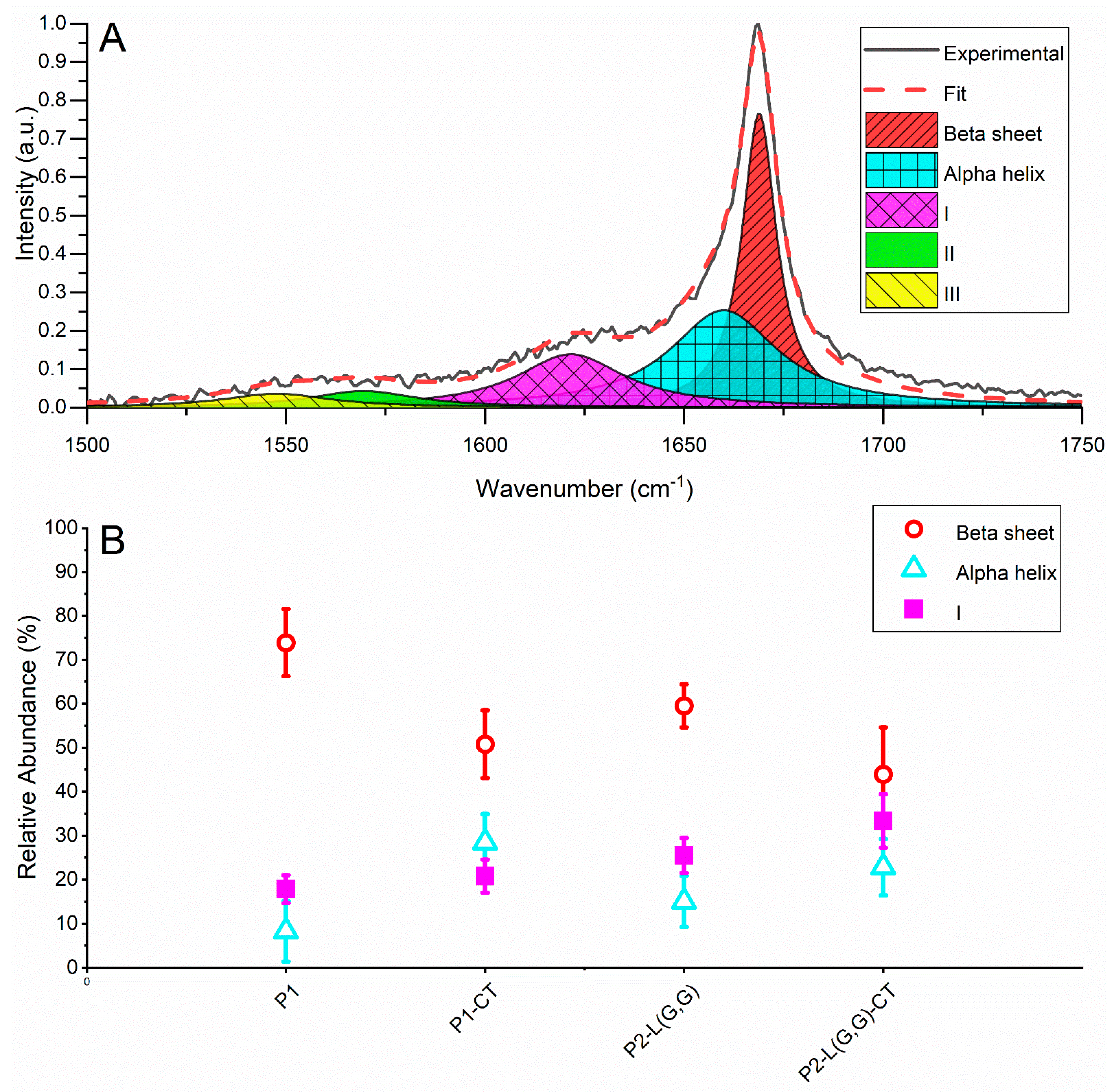
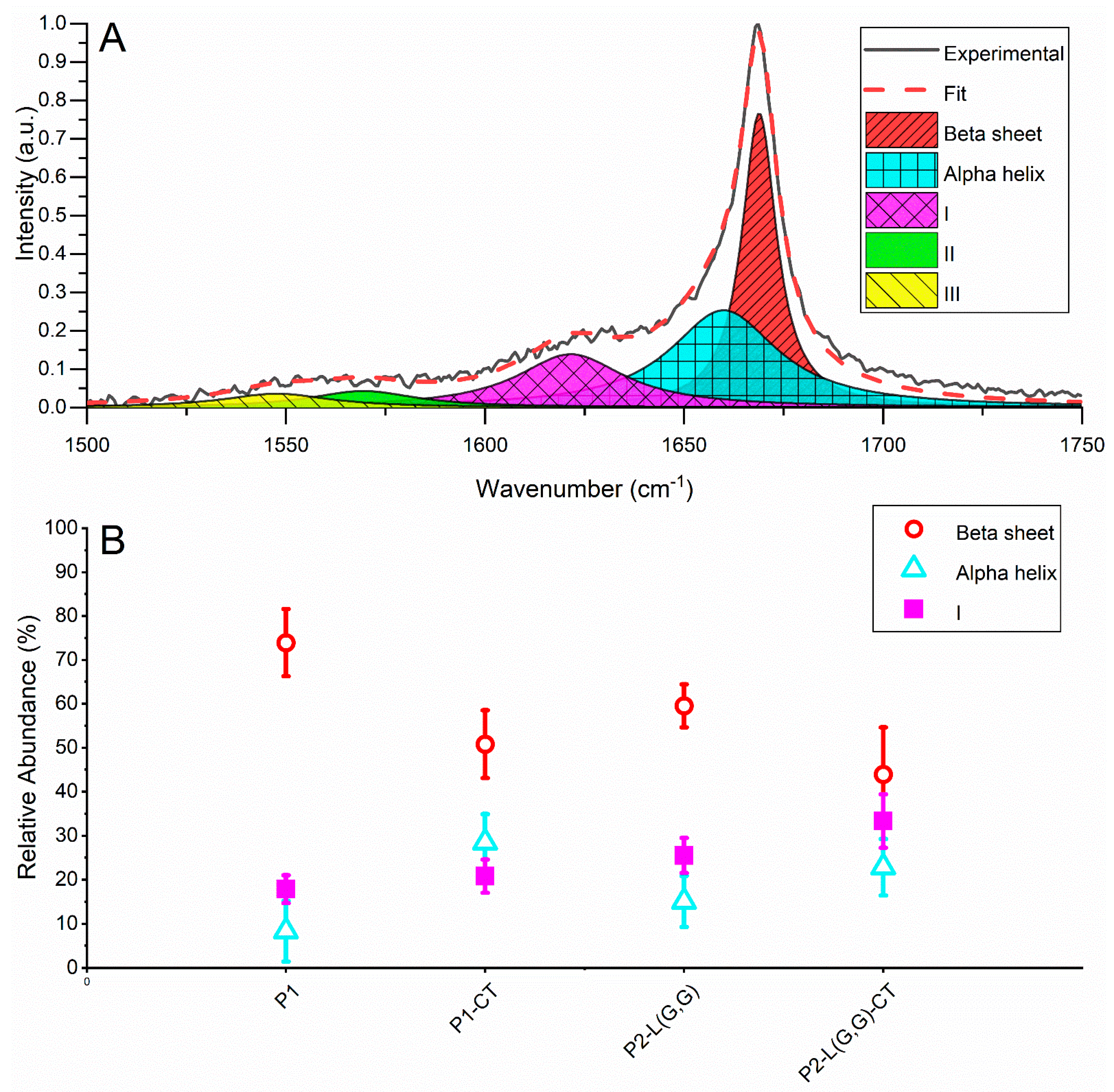
2.3. Raman Spectroscopy Revealed β-Sheet Structure in Double-Fragment NAC 71-82 Peptide Fibrils
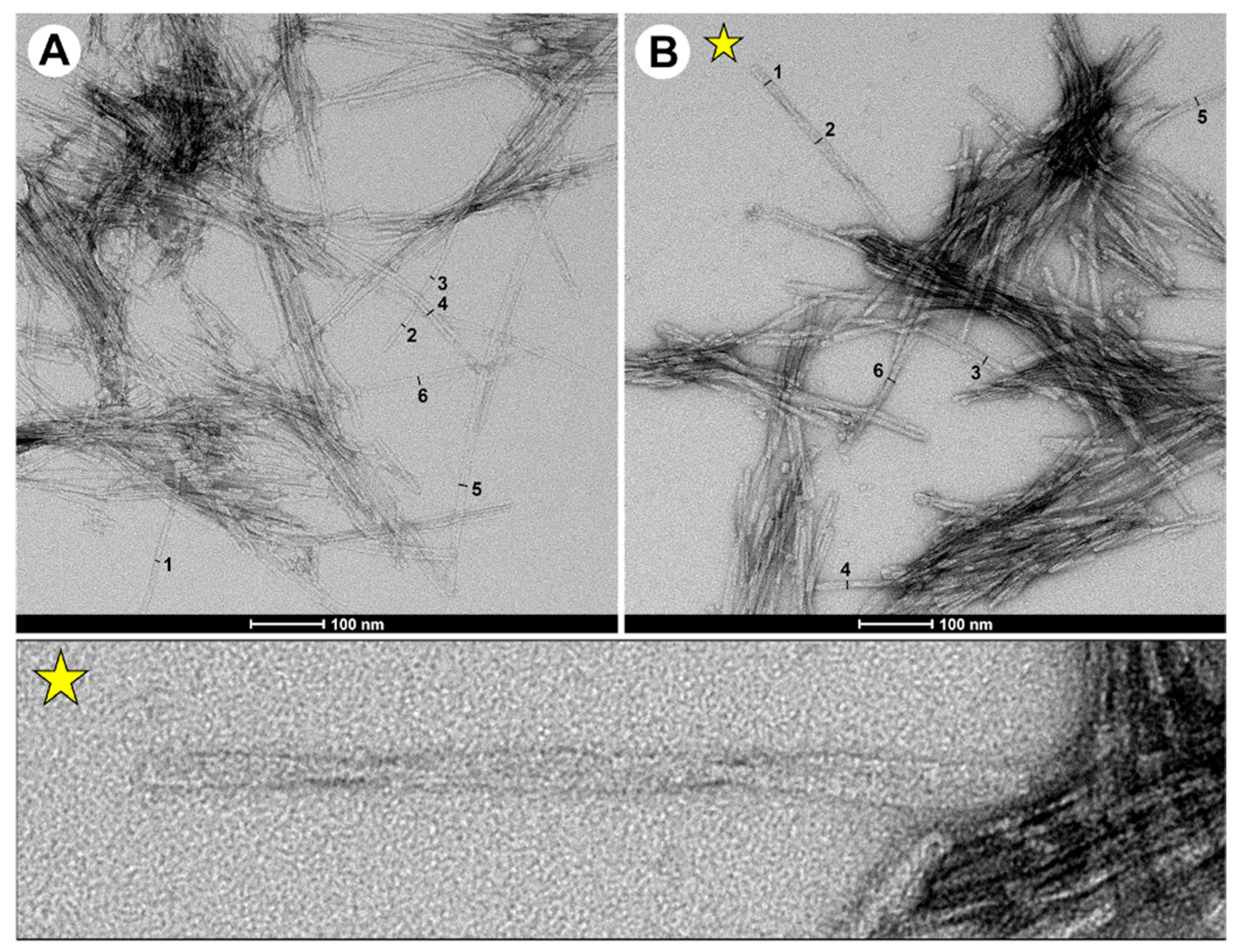
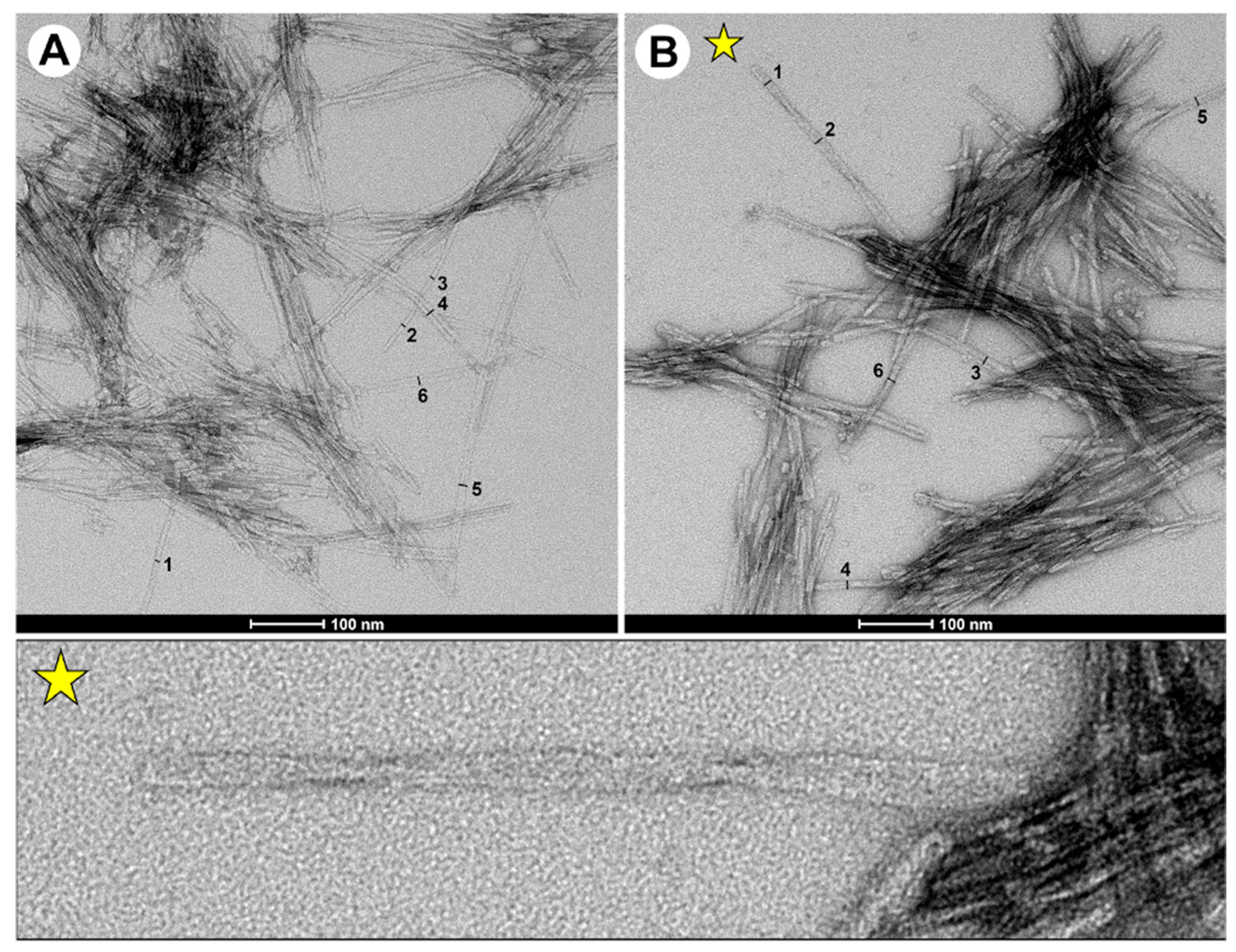
2.4. TEM Data Showed the Presence of Elongated Fibrils in Double-Fragment NAC 71-82 Peptide Fibrils
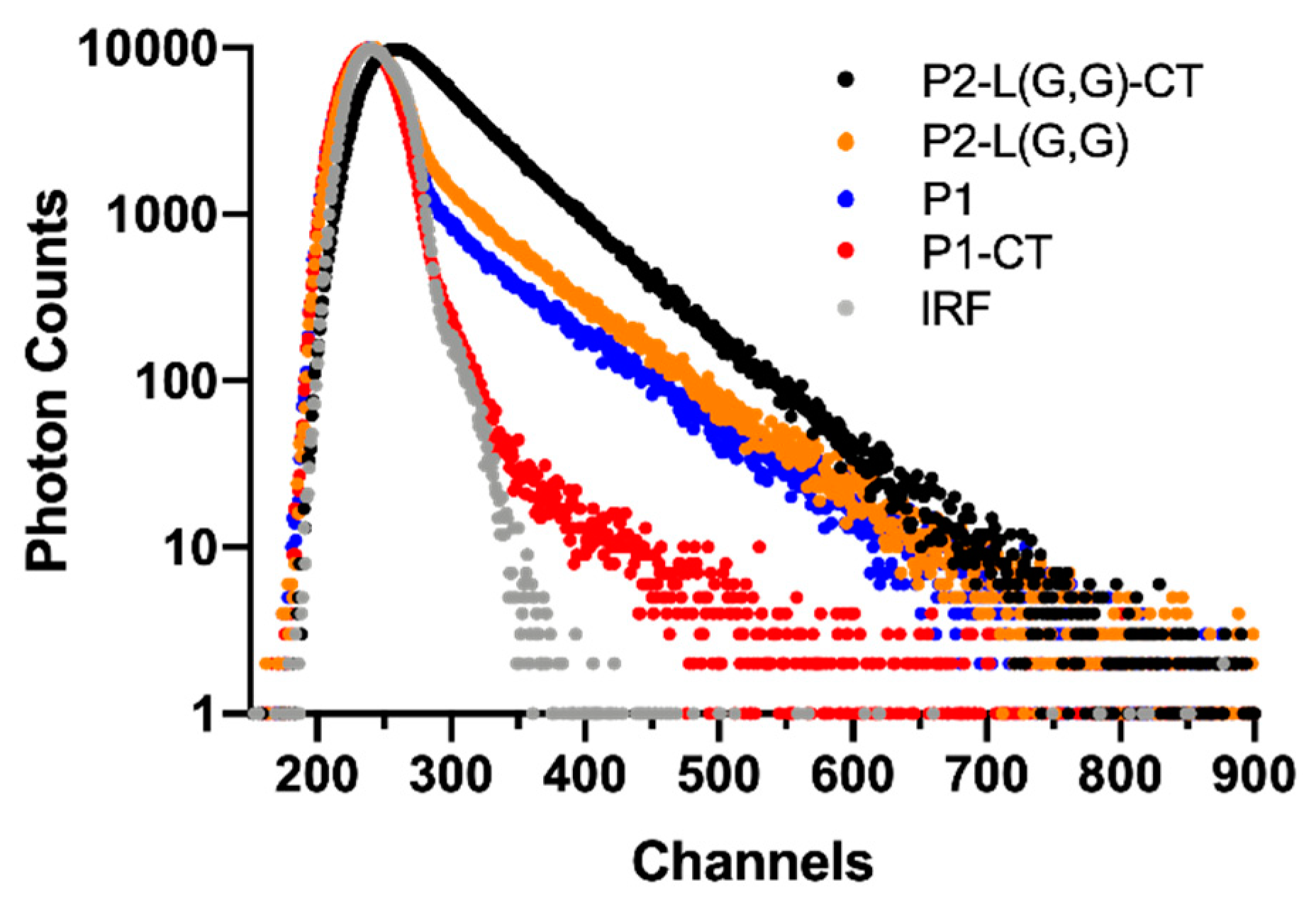
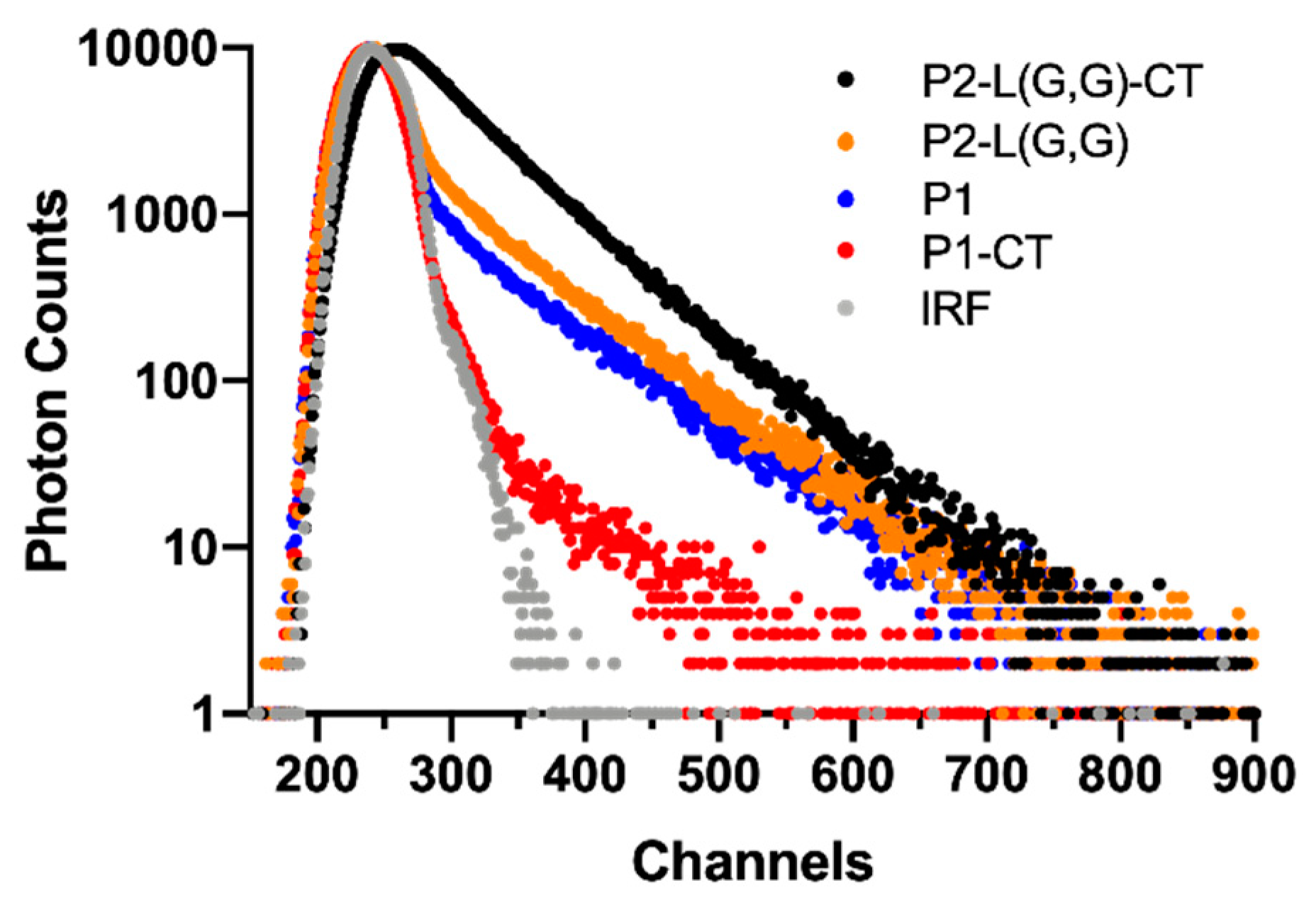
2.5. TCSPC Spectroscopy Can Detect Structural Differences in Double-Fragment NAC 71-82 Peptide Fibrils
3. Materials and Methods
3.1. Chemicals
3.2. Instruments
3.3. Molecular Dynamics Simulations
3.4. Preparation of Fibrils
3.5. Bicinchoninic Acid (BCA) Assay
3.6. Circular Dichroism (CD) Spectroscopy
3.7. Raman Spectroscopy
3.8. Transmission Electron Microscopy (TEM)
3.9. Time-Correlated Single Photon Counting (TCSPC) Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The Structural Differences between Patient-Derived α-Synuclein Strains Dictate Characteristics of Parkinson’s Disease, Multiple System Atrophy and Dementia with Lewy Bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, J.; Escalona-Noguero, C.; Sot, B. Role of α-Synuclein Regions in Nucleation and Elongation of Amyloid Fiber Assembly. ACS Chem. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Stöckl, M.; Fischer, P.; Wanker, E.; Herrmann, A. α-Synuclein Selectively Binds to Anionic Phospholipids Embedded in Liquid-Disordered Domains. J. Mol. Biol. 2008, 375, 1394–1404. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-Bound Human-Synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational Behavior and Aggregation of α-Synuclein in Organic Solvents: Modeling the Effects of Membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef]
- Iwai, A.; Yoshimoto, M.; Masliah, E.; Saitoh, T. Non-a Beta Component of Alzheimer’s Disease Amyloid (NAC) Is Amyloidogenic. Biochemistry 1995, 34, 10139–10145. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.J.; Trojanowski, J.Q.; Lee, V.M.-Y. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of Full-Length α-Synuclein Reveals Fibril Polymorphs with a Common Structural Kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM Structure of Alpha-Synuclein Fibrils. eLife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid Fibril Structure of α-Synuclein Determined by Cryo-Electron Microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurouski, D.; Duyne, R.P.V.; Lednev, I.K. Exploring the Structure and Formation Mechanism of Amyloid Fibrils by Raman Spectroscopy: A Review. Analyst 2015, 140, 4967–4980. [Google Scholar] [CrossRef] [PubMed]
- Näsström, T.; Andersson, P.O.; Lejon, C.; Karlsson, B.C.G. Amyloid Fibrils Prepared Using an Acetylated and Methyl Amidated Peptide Model of the α-Synuclein NAC 71-82 Amino Acid Stretch Contain an Additional Cross-β Structure Also Found in Prion Proteins. Sci. Rep. 2019, 9, 15949. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, A.; Vaneyck, J.; Blum, C.; Segers-Nolten, I.; Subramaniam, V. Polymorph-Specific Distribution of Binding Sites Determines Thioflavin-T Fluorescence Intensity in α-Synuclein Fibrils. Amyloid 2018, 25, 189–196. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Rodina, N.P.; Sulatsky, M.I.; Povarova, O.I.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K. Investigation of α-Synuclein Amyloid Fibrils Using the Fluorescent Probe Thioflavin T. Int. J. Mol. Sci. 2018, 19, 2486. [Google Scholar] [CrossRef] [Green Version]
- Frederix, P.W.J.M.; Scott, G.G.; Abul-Haija, Y.M.; Kalafatovic, D.; Pappas, C.G.; Javid, N.; Hunt, N.T.; Ulijn, R.V.; Tuttle, T. Exploring the Sequence Space for (Tri-)Peptide Self-Assembly to Design and Discover New Hydrogels. Nat. Chem. 2015, 7, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campioni, S.; Carret, G.; Jordens, S.; Nicoud, L.; Mezzenga, R.; Riek, R. The Presence of an Air–Water Interface Affects Formation and Elongation of α-Synuclein Fibrils. J. Am. Chem. Soc. 2014, 136, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Näsström, T.; Ådén, J.; Shibata, F.; Andersson, P.O.; Karlsson, B.C.G. A Capped Peptide of the Aggregation Prone NAC 71–82 Amino Acid Stretch of α-Synuclein Folds into Soluble β-Sheet Oligomers at Low and Elevated Peptide Concentrations. Int. J. Mol. Sci. 2020, 21, 1629. [Google Scholar] [CrossRef] [Green Version]
- Dahlberg, T.; Andersson, M. Optical Design for Laser Tweezers Raman Spectroscopy Setups for Increased Sensitivity and Flexible Spatial Detection. Appl. Opt. 2021, 60, 4519–4523. [Google Scholar] [CrossRef]
- Malyshev, D.; Dahlberg, T.; Wiklund, K.; Andersson, P.O.; Henriksson, S.; Andersson, M. Mode of Action of Disinfection Chemicals on the Bacterial Spore Structure and Their Raman Spectra. Anal. Chem. 2021, 93, 3146–3153. [Google Scholar] [CrossRef]
- Devitt, G.; Howard, K.; Mudher, A.; Mahajan, S. Raman Spectroscopy: An Emerging Tool in Neurodegenerative Disease Research and Diagnosis. ACS Chem. Neurosci. 2018, 9, 404–420. [Google Scholar] [CrossRef]
- Green, E.; Ellis, R.; Winlove, P. The Molecular Structure and Physical Properties of Elastin Fibers as Revealed by Raman Microspectroscopy. Biopolymers 2008, 89, 931–940. [Google Scholar] [CrossRef]
- Huang, K.; Maiti, N.C.; Phillips, N.B.; Carey, P.R.; Weiss, M.A. Structure-Specific Effects of Protein Topology on Cross-β Assembly: Studies of Insulin Fibrillation. Biochemistry 2006, 45, 10278–10293. [Google Scholar] [CrossRef] [PubMed]
- Gullekson, C.; Lucas, L.; Hewitt, K.; Kreplak, L. Surface-Sensitive Raman Spectroscopy of Collagen I Fibrils. Biophys. J. 2011, 100, 1837–1845. [Google Scholar] [CrossRef] [Green Version]
- Apetri, M.M.; Maiti, N.C.; Zagorski, M.G.; Carey, P.R.; Anderson, V.E. Secondary Structure of α-Synuclein Oligomers: Characterization by Raman and Atomic Force Microscopy. J. Mol. Biol. 2006, 355, 63–71. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun. 2013, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.D.; McGlinchey, R.P.; Walker, R.L.; Lee, J.C. Structural Features of α-Synuclein Amyloid Fibrils Revealed by Raman Spectroscopy. J. Biol. Chem. 2018, 293, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeVine, H. Quantification of Beta-Sheet Amyloid Fibril Structures with Thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar] [PubMed]
- Malmos, K.G.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A Primer on the Use of Thioflavin T to Investigate Amyloid Formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef]
- Eilers, P.H.; Boelens, H.F. Baseline Correction with Asymmetric Least Squares Smoothing. Leiden Univ. Med. Cent. Rep. 2005, 1, 5. [Google Scholar]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- MATLAB Version 7.10.0 (R2010a); The MathWorks Inc.: Natick, MA, USA, 2010.
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- GIMP; The GIMP Development Team: Berkeley, CA, USA, 2018.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lifetime/ns | Amplitudes/% | ||||
|---|---|---|---|---|---|
| Fibril Sample | T1 | T2 | A1 | A2 | χ2 |
| P1-CT | 0.189 ± 0.002 | 100 | 2.11 | ||
| P1 | 0.196 ± 0.003 | 2.190 ± 0.008 | 79 | 21 | 0.94 |
| P2-L(G,G) | 0.223 ± 0.003 | 2.031 ± 0.006 | 67 | 33 | 0.91 |
| P2-L(G,G)-CT | 0.991 ± 0.025 | 1.816 ± 0.006 | 32 | 68 | 1.15 |
| ID a | Amino Acid Sequence b | Purity/% |
|---|---|---|
| P1 | Ac-VTGVTAVAQKTV-NMe | ≥95.3 |
| P1-CT | VTGVTAVAQKTV | ≥96.3 |
| P2-L(G,G) | Ac-VTGVTAVAQKTVGGVTGVTAVAQKTV-NH2 | ≥97.3 |
| P2-L(G,G)-CT | VTGVTAVAQKTVGGVTGVTAVAQKTV | ≥95.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Näsström, T.; Dahlberg, T.; Malyshev, D.; Ådén, J.; Andersson, P.O.; Andersson, M.; Karlsson, B.C.G. Synthetic NAC 71-82 Peptides Designed to Produce Fibrils with Different Protofilament Interface Contacts. Int. J. Mol. Sci. 2021, 22, 9334. https://doi.org/10.3390/ijms22179334
Näsström T, Dahlberg T, Malyshev D, Ådén J, Andersson PO, Andersson M, Karlsson BCG. Synthetic NAC 71-82 Peptides Designed to Produce Fibrils with Different Protofilament Interface Contacts. International Journal of Molecular Sciences. 2021; 22(17):9334. https://doi.org/10.3390/ijms22179334
Chicago/Turabian StyleNäsström, Thomas, Tobias Dahlberg, Dmitry Malyshev, Jörgen Ådén, Per Ola Andersson, Magnus Andersson, and Björn C. G. Karlsson. 2021. "Synthetic NAC 71-82 Peptides Designed to Produce Fibrils with Different Protofilament Interface Contacts" International Journal of Molecular Sciences 22, no. 17: 9334. https://doi.org/10.3390/ijms22179334
APA StyleNäsström, T., Dahlberg, T., Malyshev, D., Ådén, J., Andersson, P. O., Andersson, M., & Karlsson, B. C. G. (2021). Synthetic NAC 71-82 Peptides Designed to Produce Fibrils with Different Protofilament Interface Contacts. International Journal of Molecular Sciences, 22(17), 9334. https://doi.org/10.3390/ijms22179334