
Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis



Abstract
1. Introduction
2. Acetylcholinesterase in Neurodegenerative Diseases
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
2.3. Huntington’s Disease
2.4. Multiple Sclerosis
2.5. Amyotrophic Lateral Sclerosis
2.6. Olivopontocerebellar Atrophy, Spinocerebellar Ataxia and Progressive Supranuclear Palsy
2.7. Depressive Disorders
3. AChE Inhibitors in the Treatment of Neurodegenerative Diseases
3.1. Rivastigmine
3.2. Donepezil
3.3. Galantamine
3.4. Huperzine A
3.5. Phenserine and Its Enantiomer
4. The Use of AChE Inhibitors for MDD and BPAD
5. Multi-Target Directed Ligands
- Modifications of the A ring in which it is replaced by a pyranopyrazole grouping (pyranopyrazole Tacrines);
- Introduction of a hydroxypyranone group into the molecule (pyranopyranone Tacrines);
- Introduction of a naphthalene, quinoline or naphthoquinone moiety into the molecule (pentacylcic pyranotacrines);
- Modifications of A ring, where it is replaced by nitrogen heterocycles (pyridine-, indole- and quinoxalinotacrines);
- Modifications of A ring, where it was replaced by nitrogen heterocycles and oxygen (pyrrolo-, pyrazolo-, furanotacrines and pyrazolophthalazine tacrines);
- Modification of the A ring, where it was replaced by other heterocycles (urea and thiourea tacrines);
- Addition of an amide group at the C2 position of cyclohexyl tacrine (amido-, amino- and iminotacrine);
- Modification of the aromatic ring A where it is replaced by pyranonapthalene or pyranonaphthoquinone and simultaneously modification of the aromatic center in ring B from aminopyridine to a pyrimidinone or pyrimidinimine.
6. Plants Extracts and Essential Oils
6.1. Hydrangea spp.
6.2. Salvia spp.
6.3. Prunus spp.
6.4. Citrus spp.
6.5. Xanthones
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Enzyme Nomenclature. Available online: https://www.qmul.ac.uk/sbcs/iubmb/enzyme/ (accessed on 15 March 2021).
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic Structure of Acetylcholinesterase from Torpedo Californica: A Prototypic Acetylcholine-Binding Protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Taylor, P. Anticholinesterase Agents|Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13th ed.; Shanahan, J.F., Lebowitz, H., Eds.; McGraw-Hill Education: New York, NY, USA, 2017; ISBN 978-1-259-58473-2. [Google Scholar]
- Barril, X.; Orozco, M.; Luque, F.J. Towards Improved Acetylcholinesterase Inhibitors: A Structural and Computational Approach. Mini. Rev. Med. Chem. 2001, 1, 255–266. [Google Scholar] [CrossRef]
- Ordentlich, A.; Barak, D.; Kronman, C.; Ariel, N.; Segall, Y.; Velan, B.; Shafferman, A. Functional Characteristics of the Oxyanion Hole in Human Acetylcholinesterase. J. Biol. Chem. 1998, 273, 19509–19517. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary Ligand Binding to Aromatic Residues in the Active-Site Gorge of Acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Ordentlich, A.; Barak, D.; Kronman, C.; Flashner, Y.; Leitner, M.; Segall, Y.; Ariel, N.; Cohen, S.; Velan, B.; Shafferman, A. Dissection of the Human Acetylcholinesterase Active Center Determinants of Substrate Specificity. Identification of Residues Constituting the Anionic Site, the Hydrophobic Site, and the Acyl Pocket. J. Biol. Chem. 1993, 268, 17083–17095. [Google Scholar] [CrossRef]
- Sanson, B.; Colletier, J.P.; Xu, Y.; Lang, P.T.; Jiang, H.; Silman, I.; Sussman, J.L.; Weik, M. Backdoor Opening Mechanism in Acetylcholinesterase Based on X-Ray Crystallography and Molecular Dynamics Simulations. Protein Sci. 2011, 20, 1114–1118. [Google Scholar] [CrossRef]
- Ruz, C.; Alcantud, J.L.; Montero, F.V.; Duran, R.; Bandres-Ciga, S. Proteotoxicity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5646. [Google Scholar] [CrossRef]
- Van Enkhuizen, J.; Janowsky, D.S.; Olivier, B.; Minassian, A.; Perry, W.; Young, J.W.; Geyer, M.A. The Catecholaminergic-Cholinergic Balance Hypothesis of Bipolar Disorder Revisited. Eur. J. Pharmacol. 2015, 753, 114–126. [Google Scholar] [CrossRef]
- ACHE (Acetylcholinesterase). Available online: http://atlasgeneticsoncology.org/Genes/GC_ACHE.html (accessed on 18 March 2021).
- Rao, N.; Whitsett, C.F.; Oxendine, S.M.; Telen, M.J. Human Erythrocyte Acetylcholinesterase Bears the Yta Blood Group Antigen and Is Reduced or Absent in the Yt(a-b-) Phenotype. Blood 1993, 81, 815–819. [Google Scholar] [CrossRef]
- Spring, F.; Gardner, B.; Anstee, D. Evidence That the Antigens of the Yt Blood Group System Are Located on Human Erythrocyte Acetylcholinesterase. Blood 1992, 80, 2136–2141. [Google Scholar] [CrossRef]
- Sternfeld, M.; Shoham, S.; Klein, O.; Flores-Flores, C.; Evron, T.; Idelson, G.H.; Kitsberg, D.; Patrick, J.W.; Soreq, H. Excess “Read-through” Acetylcholinesterase Attenuates but the “Synaptic” Variant Intensifies Neurodeterioration Correlates. Proc. Natl. Acad. Sci. USA 2000, 97, 8647–8652. [Google Scholar] [CrossRef]
- Meshorer, E.; Soreq, H. Virtues and Woes of AChE Alternative Splicing in Stress-Related Neuropathologies. Trends Neurosci. 2006, 29, 216–224. [Google Scholar] [CrossRef]
- Mor, I.; Sklan, E.H.; Podoly, E.; Pick, M.; Kirschner, M.; Yogev, L.; Bar-Sheshet Itach, S.; Schreiber, L.; Geyer, B.; Mor, T.; et al. Acetylcholinesterase-R Increases Germ Cell Apoptosis but Enhances Sperm Motility. J. Cell. Mol. Med. 2008, 12, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Toiber, D.; Berson, A.; Greenberg, D.; Melamed-Book, N.; Diamant, S.; Soreq, H. N-Acetylcholinesterase-Induced Apoptosis in Alzheimer’s Disease. PLoS ONE 2008, 3, e3108. [Google Scholar] [CrossRef] [PubMed]
- Grisaru, D.; Sternfeld, M.; Eldor, A.; Glick, D.; Soreq, H. Structural Roles of Acetylcholinesterase Variants in Biology and Pathology. Eur. J. Biochem. 1999, 264, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.T. A Morphogenic Role for Transiently Expressed Acetylcholinesterase in Developing Thalamocortical Systems? Neurosci. Lett. 1987, 75, 259–264. [Google Scholar] [CrossRef]
- Bigbee, J.W.; Sharma, K.V.; Gupta, J.J.; Dupree, J.L. Morphogenic Role for Acetylcholinesterase in Axonal Outgrowth during Neural Development. Environ. Health Perspect. 1999, 107, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Layer, P.G.; Weikert, T.; Alber, R. Cholinesterases Regulate Neurite Growth of Chick Nerve Cells in Vitro by Means of a Non-Enzymatic Mechanism. Cell Tissue Res. 1993, 273, 219–226. [Google Scholar] [CrossRef]
- Grifman, M.; Galyam, N.; Seidman, S.; Soreq, H. Functional Redundancy of Acetylcholinesterase and Neuroligin in Mammalian Neuritogenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13935–13940. [Google Scholar] [CrossRef]
- García, R.R.; Montiel, J.F.; Villalón, A.U.; Gatica, M.A.; Aboitiz, F. AChE-Rich Magnopyramidal Neurons Have a Left-Right Size Asymmetry in Broca’s Area. Brain Res. 2004, 1026, 313–316. [Google Scholar] [CrossRef]
- Zhang, X.J.; Yang, L.; Zhao, Q.; Caen, J.P.; He, H.Y.; Jin, Q.H.; Guo, L.H.; Alemany, M.; Zhang, L.Y.; Shi, Y.F. Induction of Acetylcholinesterase Expression during Apoptosis in Various Cell Types. Cell Death Differ. 2002, 9, 790–800. [Google Scholar] [CrossRef]
- Pegan, K.; Matkovic, U.; Mars, T.; Mis, K.; Pirkmajer, S.; Brecelj, J.; Grubic, Z. Acetylcholinesterase Is Involved in Apoptosis in the Precursors of Human Muscle Regeneration. Chem. Biol. Interact. 2010, 187, 96–100. [Google Scholar] [CrossRef]
- Hu, T.; Fu, Q.; Liu, X.; Zhang, H.; Dong, M. Increased Acetylcholinesterase and Capase-3 Expression in the Brain and Peripheral Immune System of Focal Cerebral Ischemic Rats. J. Neuroimmunol. 2009, 211, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jiang, H.; Wan, Y.H.; Du, A.Y.; Guo, K.J.; Liu, T.; Ye, W.Y.; Niu, X.; Wu, J.; Dong, X.Q.; et al. Induction of a 55 KDa Acetylcholinesterase Protein during Apoptosis and Its Negative Regulation by the Akt Pathway. J. Mol. Cell Biol. 2011, 3, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Greenberg, D.S. Acetylcholinesterase Involvement in Apoptosis. Front. Mol. Neurosci. 2012, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.H.; He, H.Y.; Shi, Y.F.; Lu, H.; Zhang, X.J. Overexpression of Acetylcholinesterase Inhibited Cell Proliferation and Promoted Apoptosis in NRK Cells. Acta Pharmacol. Sin. 2004, 25, 1013–1021. [Google Scholar]
- Park, S.E.; Jeong, S.H.; Yee, S.B.; Kim, T.H.; Soung, Y.H.; Ha, N.C.; Kim, N.D.; Park, J.Y.; Bae, H.R.; Park, B.S.; et al. Interactions of Acetylcholinesterase with Caveolin-1 and Subsequently with Cytochrome c Are Required for Apoptosome Formation. Carcinogenesis 2008, 29, 729–737. [Google Scholar] [CrossRef]
- Perry, C.; Sklan, E.H.; Birikh, K.; Shapira, M.; Trejo, L.; Eldor, A.; Soreq, H. Complex Regulation of Acetylcholinesterase Gene Expression in Human Brain Tumors. Oncogene 2002, 21, 8428–8441. [Google Scholar] [CrossRef]
- Park, S.E.; Kim, N.D.; Yoo, Y.H. Acetylcholinesterase Plays a Pivotal Role in Apoptosome Formation. Cancer Res. 2004, 64, 2652–2655. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, X.J. Acetylcholinesterase and Apoptosis: A Novel Perspective for an Old Enzyme. FEBS J. 2008, 275, 612–617. [Google Scholar] [CrossRef]
- Paraoanu, L.E.; Layer, P.G. Acetylcholinesterase in Cell Adhesion, Neurite Growth and Network Formation. FEBS J. 2008, 275, 618–624. [Google Scholar] [CrossRef]
- Sharma, K.V.; Koenigsberger, C.; Brimijoin, S.; Bigbee, J.W. Direct Evidence for an Adhesive Function in the Noncholinergic Role of Acetylcholinesterase in Neurite Outgrowth. J. Neurosci. Res. 2001, 63, 165–175. [Google Scholar] [CrossRef]
- Anderson, A.A.; Ushakov, D.S.; Ferenczi, M.A.; Mori, R.; Martin, P.; Saffell, J.L. Morphoregulation by Acetylcholinesterase in Fibroblasts and Astrocytes. J. Cell. Physiol. 2008, 215, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association, 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer Disease in the United States (2010–2050) Estimated Using the 2010 Census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef]
- Kuźnicki, J.; Rossa, A.; Sadowska, A. Sytuacja Osób Chorych Na Chorobę Alzheimera w Polsce Raport RPO; Biuro Rzecznika Praw Obywatelskich: Warsaw, Poland, 2016. [Google Scholar]
- Bilkiewicz, A. Psychiatria Podręcznik Dla Studentów Medycyny; PZWL Wydawnictwo Lekarskie: Warsaw, Poland, 2011. [Google Scholar]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and Progress of Hypotheses and Clinical Trials for Alzheimer’s Disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimer’s Dis. 2019, 6, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic System during the Progression of Alzheimer’s Disease: Therapeutic Implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef]
- Rinne, J.O.; Kaasinen, V.; Järvenpää, T.; Någren, K.; Roivainen, A.; Yu, M.; Oikonen, V.; Kurki, T. Brain Acetylcholinesterase Activity in Mild Cognitive Impairment and Early Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Shinotoh, H.; Namba, H.; Fukushi, K.; Nagatsuka, S.-I.; Tanaka, N.; Aotsuka, A.; Ota, T.; Tanada, S.; Irie, T. Progressive Loss of Cortical Acetylcholinesterase Activity in Association with Cognitive Decline in Alzheimer’s Disease: A Positron Emission Tomography Study. Ann. Neurol. 2000, 48, 194–200. [Google Scholar] [CrossRef]
- Blotnick-Rubin, E.; Anglister, L. Fine Localization of Acetylcholinesterase in the Synaptic Cleft of the Vertebrate Neuromuscular Junction. Front. Mol. Neurosci. 2018, 11, 123. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Musiek, E.S.; Schindler, S.E. Alzheimer Disease: Current Concepts & Future Directions. Mo. Med. 2013, 110, 395–400. [Google Scholar]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Apostolova, L.G. Alzheimer Disease. Contin. Lifelong Learn. Neurol. 2016, 22, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and Neurotoxic Effects of Amyloid β Protein: Reversal by Tachykinin Neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Periz, G.; Fortini, M.E. Proteolysis in Alzheimer’s Disease: Can Plasmin Tip the Balance? EMBO Rep. 2000, 1, 477–478. [Google Scholar] [CrossRef]
- Vassar, R.; Citron, M. Aβ-Generating Enzymes: Recent Advances in β- and γ-Secretase Research. Neuron 2000, 27, 419–422. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and Proteolytic Processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Campanari, M.L.; García-Ayllón, M.S.; Belbin, O.; Galcerán, J.; Lleó, A.; Sáez-Valero, J. Acetylcholinesterase Modulates Presenilin-1 Levels and γ-Secretase Activity. J. Alzheimer’s Dis. JAD 2014, 41, 911–924. [Google Scholar] [CrossRef]
- Silveyra, M.X.; García-Ayllón, M.S.; Serra-Basante, C.; Mazzoni, V.; García-Gutierrez, M.S.; Manzanares, J.; Culvenor, J.G.; Sáez-Valero, J. Changes in Acetylcholinesterase Expression Are Associated with Altered Presenilin-1 Levels. Neurobiol. Aging 2012, 33, 627.e27–627.e37. [Google Scholar] [CrossRef]
- Hicks, D.A.; Makova, N.Z.; Gough, M.; Parkin, E.T.; Nalivaeva, N.N.; Turner, A.J. The Amyloid Precursor Protein Represses Expression of Acetylcholinesterase in Neuronal Cell Lines. J. Biol. Chem. 2013, 288, 26039–26051. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Mesulam, M. Special Properties of Cholinesterases in the Cerebral Cortex of Alzheimer’s Disease. Brain Res. 1989, 498, 185–189. [Google Scholar] [CrossRef]
- Ciro, A.; Park, J.; Burkhard, G.; Yan, N.; Geula, C. Biochemical Differentiation of Cholinesterases from Normal and Alzheimers Disease Cortex. Curr. Alzheimer Res. 2012, 9, 138–143. [Google Scholar] [CrossRef]
- Alvarez, A.; Alarcón, R.; Opazo, C.; Campos, E.O.; Muñoz, F.J.; Calderón, F.H.; Dajas, F.; Gentry, M.K.; Doctor, B.P.; De Mello, F.G.; et al. Stable Complexes Involving Acetylcholinesterase and Amyloid-β Peptide Change the Biochemical Properties of the Enzyme and Increase the Neurotoxicity of Alzheimer’s Fibrils. J. Neurosci. 1998, 18, 3213–3223. [Google Scholar] [CrossRef]
- Reyes, A.E.; Chacón, M.A.; Dinamarca, M.C.; Cerpa, W.; Morgan, C.; Inestrosa, N.C. Acetylcholinesterase-Aβ Complexes Are More Toxic than Aβ Fibrils in Rat Hippocampus: Effect on Rat β-Amyloid Aggregation, Laminin Expression, Reactive Astrocytosis, and Neuronal Cell Loss. Am. J. Pathol. 2004, 164, 2163–2174. [Google Scholar] [CrossRef]
- Jean, L.; Brimijoin, S.; Vaux, D.J. In Vivo Localization of Human Acetylcholinesterase-Derived Species in a β-Sheet Conformation at the Core of Senile Plaques in Alzheimer’s Disease. J. Biol. Chem. 2019, 294, 6253–6272. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase Accelerates Assembly of Amyloid-β-Peptides into Alzheimer’s Fibrils: Possible Role of the Peripheral Site of the Enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Rees, T.; Hammond, P.I.; Soreq, H.; Younkin, S.; Brimijoin, S. Acetylcholinesterase Promotes Beta-Amyloid Plaques in Cerebral Cortex. Neurobiol. Aging 2003, 24, 777–787. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A Structural Motif of Acetylcholinesterase That Promotes Amyloid β-Peptide Fibril Formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef]
- Hou, L.N.; Xu, J.R.; Zhao, Q.N.; Gao, X.L.; Cui, Y.Y.; Xu, J.; Wang, H.; Chen, H.Z. A New Motif in the N-Terminal of Acetylcholinesterase Triggers Amyloid-β Aggregation and Deposition. CNS Neurosci. Ther. 2014, 20, 59–66. [Google Scholar] [CrossRef]
- Jean, L.; Thomas, B.; Tahiri-Alaoui, A.; Shaw, M.; Vaux, D.J. Heterologous Amyloid Seeding: Revisiting the Role of Acetylcholinesterase in Alzheimer’s Disease. PLoS ONE 2007, 2, e652. [Google Scholar] [CrossRef]
- Alkalay, A.; Rabinovici, G.D.; Zimmerman, G.; Agarwal, N.; Kaufer, D.; Miller, B.L.; Jagust, W.J.; Soreq, H. Plasma Acetylcholinesterase Activity Correlates with Intracerebral β-Amyloid Load. Curr. Alzheimer Res. 2013, 10, 48–56. [Google Scholar] [CrossRef]
- Shaked, I.; Meerson, A.; Wolf, Y.; Avni, R.; Greenberg, D.; Gilboa-Geffen, A.; Soreq, H. MicroRNA-132 Potentiates Cholinergic Anti-Inflammatory Signaling by Targeting Acetylcholinesterase. Immunity 2009, 31, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Parrish, W.R.; Rosas-Ballina, M.; Ochani, M.; Puerta, M.; Ochani, K.; Chavan, S.; Al-Abed, Y.; Tracey, K.J. Brain Acetylcholinesterase Activity Controls Systemic Cytokine Levels through the Cholinergic Anti-Inflammatory Pathway. Brain Behav. Immun. 2009, 23, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Schegg, K.M.; Harrington, L.S.; Neilsen, S.; Zweig, R.M.; Peacock, J.H. Soluble and Membrane-Bound Forms of Brain Acetylcholinesterase in Alzheimer’s Disease. Neurobiol. Aging 1992, 13, 697–704. [Google Scholar] [CrossRef]
- Catarina Silva, M.; Haggarty, S.J. Tauopathies: Deciphering Disease Mechanisms to Develop Effective Therapies. Int. J. Mol. Sci. 2020, 21, 8948. [Google Scholar]
- Silveyra, M.X.; García-Ayllón, M.S.; de Barreda, E.G.; Small, D.H.; Martínez, S.; Avila, J.; Sáez-Valero, J. Altered Expression of Brain Acetylcholinesterase in FTDP-17 Human Tau Transgenic Mice. Neurobiol. Aging 2012, 33, 624.e23–624.e34. [Google Scholar] [CrossRef] [PubMed]
- García-Ayllón, M.S.; Small, D.H.; Avila, J.; Sáez-Valero, J. Revisiting the Role of Acetylcholinesterase in Alzheimer¬s Disease: Cross-Talk with β-Tau and p-Amyloid. Front. Mol. Neurosci. 2011, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Gómez, M.; Llorens-Álvarez, E.; Alom, J.; del Ser, T.; Avila, J.; Sáez-Valero, J.; García-Ayllón, M. Tau Phosphorylation by Glycogen Synthase Kinase 3β Modulates Enzyme Acetylcholinesterase Expression. J. Neurochem. 2021, 157, 2091–2105. [Google Scholar] [CrossRef]
- Guo, P.; Wang, R.D.; Lian, T.H.; Ding, D.Y.; Zhang, Y.N.; Zhang, W.J.; Li, D.N.; Li, L.X.; Li, J.H.; Guan, H.Y.; et al. Olfactory Dysfunction and Its Association with Neuropathologic Proteins in Cerebrospinal Fluid from Patients with Parkinson Disease. Front. Aging Neurosci. 2020, 12, 594324. [Google Scholar] [CrossRef]
- Bergamino, M.; Keeling, E.G.; Mishra, V.R.; Stokes, A.M.; Walsh, R.R. Assessing White Matter Pathology in Early-Stage Parkinson Disease Using Diffusion Mri: A Systematic Review. Front. Neurol. 2020, 11, 314. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Ivanco, L.S.; Lopresti, B.; Koeppe, R.A.; Davis, J.G.; Mathis, C.A.; Moore, R.Y.; DeKosky, S.T. Cortical Cholinergic Function Is More Severely Affected in Parkinsonian Dementia Than in Alzheimer Disease: An In Vivo Positron Emission Tomographic Study. Arch. Neurol. 2003, 60, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Sławek, J.; Kozubski, W.N.; Liberski, P. (Eds.) Neurologia; PZWL Wydawnictwo Lekarskie: Warsaw, Poland, 2013; pp. 285–323. [Google Scholar]
- Christopher, L.; Strafella, A.P. Neuroimaging of Brain Changes Associated with Cognitive Impairment in Parkinson’s Disease. Proc. J. Neuropsychol. 2013, 7, 225–240. [Google Scholar] [CrossRef]
- Ziegler, D.A.; Corkin, S. New Magnetic Resonance Imaging Biomarkers Advance the Characterisation of Parkinson’s Disease. Eur. Neurol. Rev. 2013, 8, 85–89. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bohnen, N.I.; Müller, M.L.T.M.; Kotagal, V.; Koeppe, R.A.; Kilbourn, M.R.; Gilman, S.; Albin, R.L.; Frey, K.A. Heterogeneity of Cholinergic Denervation in Parkinson’s Disease without Dementia. J. Cereb. Blood Flow Metab. 2012, 32, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Albin, R.L. Cholinergic Denervation Occurs Early in Parkinson Disease. Neurology 2009, 73, 256–257. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Albin, R.L.; Müller, M.L.T.M.; Petrou, M.; Kotagal, V.; Koeppe, R.A.; Scott, P.J.H.; Frey, K.A. Frequency of Cholinergic and Caudate Nucleus Dopaminergic Deficits across the Predemented Cognitive Spectrum of Parkinson Disease and Evidence of Interaction Effects. JAMA Neurol. 2015, 72, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Kotagal, V.; Albin, R.L.; Müller, M.L.T.M.; Koeppe, R.A.; Frey, K.A.; Bohnen, N.I. Gender Differences in Cholinergic and Dopaminergic Deficits in Parkinson Disease. J. Neural Transm. 2013, 120, 1421–1424. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Albin, R.L. The Cholinergic System and Parkinson Disease. Behav. Brain Res. 2011, 221, 564–573. [Google Scholar] [CrossRef]
- Hilker, R.; Thomas, A.V.; Klein, J.C.; Weisenbach, S.; Kalbe, E.; Burghaus, L.; Jacobs, A.H.; Herholz, K.; Heiss, W.D. Dementia in Parkinson Disease: Functional Imaging of Cholinergic and Dopaminergic Pathways. Neurology 2005, 65, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Hirano, S.; Shinotoh, H.; Aotsuka, A.; Sato, K.; Tanaka, N.; Ota, T.; Asahina, M.; Fukushi, K.; Kuwabara, S.; et al. Mapping of Brain Acetylcholinesterase Alterations in Lewy Body Disease by PET. Neurology 2009, 73, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Kaufer, D.I.; Hendrickson, R.; Constantine, G.M.; Mathis, C.A.; Moore, R.Y. Cortical Cholinergic Denervation Is Associated with Depressive Symptoms in Parkinson’s Disease and Parkinsonian Dementia. J. Neurol. Neurosurg. Psychiatry 2007, 78, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.A.; Petrou, M. Imaging Amyloidopathy in Parkinson Disease and Parkinsonian Dementia Syndromes. Clin. Transl. Imaging 2015, 3, 57–64. [Google Scholar] [CrossRef]
- Akhtar, R.S.; Xie, S.X.; Brennan, L.; Pontecorvo, M.J.; Hurtig, H.I.; Trojanowski, J.Q.; Weintraub, D.; Siderowf, A.D. Amyloid-Beta Positron Emission Tomography Imaging of Alzheimer’s Pathology in Parkinson’s Disease Dementia. Mov. Disord. Clin. Pract. 2016, 3, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.L.T.M.; Frey, K.A.; Petrou, M.; Kotagal, V.; Koeppe, R.A.; Albin, R.L.; Bohnen, N.I. β-Amyloid and Postural Instability and Gait Difficulty in Parkinson’s Disease at Risk for Dementia. Mov. Disord. 2013, 28, 296–301. [Google Scholar] [CrossRef]
- Zhou, Z.; Müller, M.L.T.M.; Kanel, P.; Chua, J.; Kotagal, V.; Kaufer, D.I.; Albin, R.L.; Frey, K.A.; Bohnen, N.I. Apathy Rating Scores and β-Amyloidopathy in Patients with Parkinson Disease at Risk for Cognitive Decline. Neurology 2020, 94, e376–e383. [Google Scholar] [CrossRef]
- Shah, N.; Frey, K.A.; Müller, M.L.T.M.; Petrou, M.; Kotagal, V.; Koeppe, R.A.; Scott, P.J.H.; Albin, R.L.; Bohnen, N.I. Striatal and Cortical β-Amyloidopathy and Cognition in Parkinson’s Disease. Mov. Disord. 2016, 31, 111–117. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Frey, K.A.; Studenski, S.; Kotagal, V.; Koeppe, R.A.; Scott, P.J.H.; Albin, R.L.; Müller, M.L.T.M. Gait Speed in Parkinson Disease Correlates with Cholinergic Degeneration. Neurology 2013, 81, 1611–1616. [Google Scholar] [CrossRef]
- Rochester, L.; Yarnall, A.J.; Baker, M.R.; David, R.V.; Lord, S.; Galna, B.; Burn, D.J. Cholinergic Dysfunction Contributes to Gait Disturbance in Early Parkinson’s Disease. Brain 2012, 135, 2779–2788. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Müller, M.L.T.M.; Koeppe, R.A.; Studenski, S.A.; Kilbourn, M.A.; Frey, K.A.; Albin, R.L. History of Falls in Parkinson Disease Is Associated with Reduced Cholinergic Activity. Neurology 2009, 73, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.L.T.M.; Albin, R.L.; Kotagal, V.; Koeppe, R.A.; Scott, P.J.H.; Frey, K.A.; Bohnen, N.I. Thalamic Cholinergic Innervation and Postural Sensory Integration Function in Parkinson’s Disease. Brain 2013, 136, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Müller, M.L.T.M.; Kotagal, V.; Koeppe, R.A.; Kilbourn, M.A.; Albin, R.L.; Frey, K.A. Olfactory Dysfunction, Central Cholinergic Integrity and Cognitive Impairment in Parkinson’s Disease. Brain 2010, 133, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Kotagal, V.; Albin, R.L.; Müller, M.L.T.M.; Koeppe, R.A.; Chervin, R.D.; Frey, K.A.; Bohnen, N.I. Symptoms of Rapid Eye Movement Sleep Behavior Disorder Are Associated with Cholinergic Denervation in Parkinson Disease. Ann. Neurol. 2012, 71, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Chan, P.; Stoessl, A.J. The Underlying Mechanism of Prodromal PD: Insights from the Parasympathetic Nervous System and the Olfactory System. Transl. Neurodegener. 2017, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Barrantes, F.J. Deficits in Cholinergic Neurotransmission and Their Clinical Correlates in Parkinson’s Disease. NPJ Parkinson’s Dis. 2016, 2, 16001. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Wszolek, Z.K.; Graff-Radford, N.R.; Cookson, N.; Dickson, D.W. Tau Pathology in the Olfactory Bulb Correlates with Braak Stage, Lewy Body Pathology and Apolipoprotein Ε4. Neuropathol. Appl. Neurobiol. 2003, 29, 503–510. [Google Scholar] [CrossRef]
- Mundiñano, I.C.; Caballero, M.C.; Ordóñez, C.; Hernandez, M.; DiCaudo, C.; Marcilla, I.; Erro, M.E.; Tuñon, M.T.; Luquin, M.R. Increased Dopaminergic Cells and Protein Aggregates in the Olfactory Bulb of Patients with Neurodegenerative Disorders. Acta Neuropathol. 2011, 122, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Goris, A.; Williams-Gray, C.H.; Clark, G.R.; Foltynie, T.; Lewis, S.J.G.; Brown, J.; Ban, M.; Spillantini, M.G.; Compston, A.; Burn, D.J.; et al. Tau and α-Synuclein in Susceptibility to, and Dementia in, Parkinson’s Disease. Ann. Neurol. 2007, 62, 145–153. [Google Scholar] [CrossRef]
- Yi, Y.M.; Cai, L.; Shao, Y.; Xu, M.; Yi, J.L. The Protective Role of Tacrine and Donepezil in the Retina of Acetylcholinesterase Knockout Mice. Int. J. Ophthalmol. 2015, 8, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; MacIntyre, J.N.; Pouliot, M.; Casanova, C.; Vaucher, E.; Kelly, M.E.M.; Di Polo, A. Acetylcholinesterase Inhibition Promotes Retinal Vasoprotection and Increases Ocular Blood Flow in Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3171–3183. [Google Scholar] [CrossRef]
- Borm, C.D.J.M.; Visser, F.; Werkmann, M.; De Graaf, D.; Putz, D.; Seppi, K.; Poewe, W.; Vlaar, A.M.M.; Hoyng, C.; Bloem, B.R.; et al. Seeing Ophthalmologic Problems in Parkinson Disease: Results of a Visual Impairment Questionnaire. Neurology 2020, 94, E1539–E1547. [Google Scholar] [CrossRef]
- Fedorova, T.; Knudsen, C.S.; Mouridsen, K.; Nexo, E.; Borghammer, P. Salivary Acetylcholinesterase Activity Is Increased in Parkinson’s Disease: A Potential Marker of Parasympathetic Dysfunction. Parkinson’s Dis. 2015, 2015, 156479. [Google Scholar] [CrossRef]
- Liu, S.Y.; Wile, D.J.; Fu, J.F.; Valerio, J.; Shahinfard, E.; McCormick, S.; Mabrouk, R.; Vafai, N.; McKenzie, J.; Neilson, N.; et al. The Effect of LRRK2 Mutations on the Cholinergic System in Manifest and Premanifest Stages of Parkinson’s Disease: A Cross-Sectional PET Study. Lancet Neurol. 2018, 17, 309–316. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Albin, R.L. Hypercholinergic Activity in LRRK2 Parkinson’s Disease. Lancet Neurol. 2018, 17, 290–291. [Google Scholar] [CrossRef]
- Clément, C.; Lalonde, R.; Strazielle, C. Acetylcholinesterase Activity in the Brain of Dystonia Musculorum (Dst Dt-J) Mutant Mice. Neurosci. Res. 2012, 72, 79–86. [Google Scholar] [CrossRef]
- Percário, S.; da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; de Nazaré Araújo Moreira, T.; Dolabela, M.F. Oxidative Stress in Parkinson’s Disease: Potential Benefits of Antioxidant Supplementation. Oxidative Med. Cell. Longev. 2020, 2020, 2360872. [Google Scholar] [CrossRef]
- Bond, C.E.; Patel, P.; Crouch, L.; Tetlow, N.; Day, T.; Abu-Hayyeh, S.; Williamson, C.; Greenfield, S.A. Astroglia Up-Regulate Transcription and Secretion of “readthrough” Acetylcholinesterase Following Oxidative Stress. Eur. J. Neurosci. 2006, 24, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shaul, Y.; BenMoyal-Segal, L.; Ben-Ari, S.; Bergman, H.; Soreq, H. Adaptive Acetylcholinesterase Splicing Patterns Attenuate 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinsonism in Mice. Eur. J. Neurosci. 2006, 23, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Benmoyal-Segal, L.; Vander, T.; Shifman, S.; Bryk, B.; Ebstein, R.; Marcus, E.-L.; Stessman, J.; Darvasi, A.; Herishanu, Y.; Friedman, A.; et al. Acetylcholinesterase/Paraoxonase Interactions Increase the Risk of Insecticide-induced Parkinson’s Disease. FASEB J. 2005, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, L.; Liu, S.; Ye, W.; Wu, J.; Zhang, X. Acetylcholinesterase Deficiency Decreases Apoptosis in Dopaminergic Neurons in the Neurotoxin Model of Parkinson’s Disease. Int. J. Biochem. Cell Biol. 2013, 45, 265–272. [Google Scholar] [CrossRef]
- Ahmad Aziz, N.; Van Der Burg, J.M.M.; Tabrizi, S.J.; Bernhard Landwehrmeyer, G. Overlap between Age-at-Onset and Disease-Progression Determinants in Huntington Disease. Neurol. 2018, 90, 1127. [Google Scholar] [CrossRef]
- Raymond, L.A.; André, V.M.; Cepeda, C.; Gladding, C.M.; Milnerwood, A.J.; Levine, M.S. Pathophysiology of Huntington’s Disease: Time-Dependent Alterations in Synaptic and Receptor Function. Neurosci. 2011, 198, 252–273. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Choi, D.W. Cultured Striatal Neurons Containing NADPH-Diaphorase or Acetylcholinesterase Are Selectively Resistant to Injury by NMDA Receptor Agonists. Brain Res. 1988, 446, 374–378. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kowall, N.W.; Cipolloni, P.B.; Storey, E.; Beal, M.F. Excitotoxin Lesions in Primates as a Model for Huntington’s Disease: Histopathologic and Neurochemical Characterization. Exp. Neurol. 1993, 119, 46–71. [Google Scholar] [CrossRef] [PubMed]
- Schippling, S.; Schneider, S.A.; Bhatia, K.P.; Münchau, A.; Rothwell, J.C.; Tabrizi, S.J.; Orth, M. Abnormal Motor Cortex Excitability in Preclinical and Very Early Huntington’s Disease. Biol. Psychiatry 2009, 65, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Farrar, A.M.; Callahan, J.W.; Abercrombie, E.D. Reduced Striatal Acetylcholine Efflux in the R6/2 Mouse Model of Huntington’s Disease: An Examination of the Role of Altered Inhibitory and Excitatory Mechanisms. Exp. Neurol. 2011, 232, 119–125. [Google Scholar] [CrossRef] [PubMed]
- de Aragão, B.C.; Rodrigues, H.A.; Valadão, P.A.C.; Camargo, W.; Naves, L.A.; Ribeiro, F.M.; Guatimosim, C. Changes in Structure and Function of Diaphragm Neuromuscular Junctions from BACHD Mouse Model for Huntington’s Disease. Neurochem. Int. 2016, 93, 64–72. [Google Scholar] [CrossRef]
- Smith, R.; Chung, H.; Rundquist, S.; Maat-Schieman, M.L.C.; Colgan, L.; Englund, E.; Liu, Y.J.; Roos, R.A.C.; Faull, R.L.M.; Brundin, P.; et al. Cholinergic Neuronal Defect without Cell Loss in Huntington’s Disease. Hum. Mol. Genet. 2006, 15, 3119–3131. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Desmond, T.J.; Albin, R.L.; Frey, K.A. Vesicular Neurotransmitter Transporters in Huntington’s Disease: Initial Observations and Comparison with Traditional Synaptic Markers. Synapse 2001, 41, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.M.; Wild, E.J. Cerebrospinal Fluid Biomarkers for Huntington’s Disease. J. Huntingt. Dis. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Menze, E.T.; Esmat, A.; Tadros, M.G.; Abdel-Naim, A.B.; Khalifa, A.E. Genistein Improves 3-NPA-Induced Memory Impairment in Ovariectomized Rats: Impact of Its Antioxidant, Anti-Inflammatory and Acetylcholinesterase Modulatory Properties. PLoS ONE 2015, 10, e0117223. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, C.; Garcia-Martinez, V.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Gutierrez-Merino, C. Early Reactive A1 Astrocytes Induction by the Neurotoxin 3-Nitropropionic Acid in Rat Brain. Int. J. Mol. Sci. 2020, 21, 3609. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial Activation in Presymptomatic Huntington’s Disease Gene Carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Imaging Microglial Activation in Huntington’s Disease. Brain Res. Bull. 2007, 72, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Hsiao, H.Y.; Chen, Y.C.; Chen, H.M.; Tu, P.H.; Chern, Y. A Critical Role of Astrocyte-Mediated Nuclear Factor-ΚB-Dependent Inflammation in Huntington’s Disease. Hum. Mol. Genet. 2013, 22, 1826–1842. [Google Scholar] [CrossRef]
- Crevier-Sorbo, G.; Rymar, V.V.; Crevier-Sorbo, R.; Sadikot, A.F. Thalamostriatal Degeneration Contributes to Dystonia and Cholinergic Interneuron Dysfunction in a Mouse Model of Huntington’s Disease. Acta Neuropathol. Commun. 2020, 8, 1–19. [Google Scholar] [CrossRef]
- Aquilonius, S.M.; Sjöström, R. Cholinergic and Dopaminergic Mechanisms in Huntington’s Chorea. Life Sci. 1971, 10, 405–414. [Google Scholar] [CrossRef]
- Vetter, J.M.; Jehle, T.; Heinemeyer, J.; Franz, P.; Behrens, P.F.; Jackisch, R.; Landwehrmeyer, G.B.; Feuerstein, T.J. Mice Transgenic for Exon 1 of Huntington’s Disease: Properties of Cholinergic and Dopaminergic Pre-Synaptic Function in the Striatum. J. Neurochem. 2003, 85, 1054–1063. [Google Scholar] [CrossRef]
- Adam, O.R.; Jankovic, J. Symptomatic Treatment of Huntington Disease. Neurotherapeutics 2008, 5, 181–197. [Google Scholar] [CrossRef]
- Giralt, A.; Saavedra, A.; Alberch, J.; Pérez-Navarro, E. Cognitive Dysfunction in Huntington’s Disease: Humans, Mouse Models and Molecular Mechanisms. J. Huntingt. Dis. 2012, 1, 155–173. [Google Scholar] [CrossRef]
- Epidemiologia|PTSR. Available online: https://www.ptsr.org.pl/stwardnienie_rozsiane,sm_w_liczbach,107.asp (accessed on 9 April 2021).
- Leray, E.; Moreau, T.; Fromont, A.; Edan, G. Epidemiology of Multiple Sclerosis. Rev. Neurol. 2016, 172, 3–13. [Google Scholar] [CrossRef]
- Karussis, D. The Diagnosis of Multiple Sclerosis and the Various Related Demyelinating Syndromes: A Critical Review. J. Autoimmun. 2014, 48–49, 134–142. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple Sclerosis—A Review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Physiological Functions of the Cholinergic System in Immune Cells. J. Pharmacol. Sci. 2017, 134, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Costantini, E.; Di Nicola, M.; D’Angelo, C.; Franchi, S.; D’Aurora, M.; Di Bari, M.; Orlando, V.; Galizia, S.; Ruggieri, S.; et al. Butyrylcholinesterase and Acetylcholinesterase Polymorphisms in Multiple Sclerosis Patients: Implication in Peripheral Inflammation. Sci. Rep. 2018, 8, 1319. [Google Scholar] [CrossRef]
- Reale, M.; de Angelis, F.; di Nicola, M.; Capello, E.; di Ioia, M.; Luca, G.; Lugaresi, A.; Tata, A. Relation between Pro-Inflammatory Cytokines and Acetylcholine Levels in Relapsing-Remitting Multiple Sclerosis Patients. Int. J. Mol. Sci. 2012, 13, 12656. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Reale, M.; Di Nicola, M.; Orlando, V.; Galizia, S.; Porfilio, I.; Costantini, E.; D’Angelo, C.; Ruggieri, S.; Biagioni, S.; et al. Dysregulated Homeostasis of Acetylcholine Levels in Immune Cells of RR-Multiple Sclerosis Patients. Int. J. Mol. Sci. 2016, 17, 2009. [Google Scholar] [CrossRef]
- Polachini, C.R.N.; Spanevello, R.M.; Casali, E.A.; Zanini, D.; Pereira, L.B.; Martins, C.C.; Baldissareli, J.; Cardoso, A.M.; Duarte, M.F.; da Costa, P.; et al. Alterations in the Cholinesterase and Adenosine Deaminase Activities and Inflammation Biomarker Levels in Patients with Multiple Sclerosis. Neuroscience 2014, 266, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Gatta, V.; Mengod, G.; Reale, M.; Tata, A.M. Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis. Biomedicines 2020, 8, 153. [Google Scholar] [CrossRef]
- Kooi, E.J.; Prins, M.; Bajic, N.; Beliën, J.A.M.; Gerritsen, W.H.; Van Horssen, J.; Aronica, E.; Van Dam, A.M.; Hoozemans, J.J.M.; Francis, P.T.; et al. Cholinergic Imbalance in the Multiple Sclerosis Hippocampus. Acta Neuropathol. 2011, 122, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Virta, J.R.; Laatu, S.; Parkkola, R.; Oikonen, V.; Rinne, J.O.; Ruutiainen, J. Cerebral Acetylcholinesterase Activity Is Not Decreased in MS Patients with Cognitive Impairment. Mult. Scler. 2011, 17, 931–938. [Google Scholar] [CrossRef]
- Di Pinto, G.; Di Bari, M.; Martin-Alvarez, R.; Sperduti, S.; Serrano-Acedo, S.; Gatta, V.; Tata, A.M.; Mengod, G. Comparative Study of the Expression of Cholinergic System Components in the CNS of Experimental Autoimmune Encephalomyelitis Mice: Acute vs Remitting Phase. Eur. J. Neurosci. 2018, 48, 2165–2181. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, C.M.; Spanevello, R.; Ahmed, M.; Pereira, L.B.; Gonçalves, J.F.; Corrêa, M.; Schmatz, R.; Stefanello, N.; Leal, D.B.R.; Mazzanti, A.; et al. Pre-Treatment with Ebselen and Vitamin E Modulate Acetylcholinesterase Activity: Interaction with Demyelinating Agents. Int. J. Dev. Neurosci. 2009, 27, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hanieh, H.; Alzahrani, A. MicroRNA-132 Suppresses Autoimmune Encephalomyelitis by Inducing Cholinergic Anti-Inflammation: A New Ahr-Based Exploration. Eur. J. Immunol. 2013, 43, 2771–2782. [Google Scholar] [CrossRef]
- Melø, T.M.; Larsen, C.; White, L.R.; Aasly, J.; Sjøbakk, T.E.; Flaten, T.P.; Sonnewald, U.; Syversen, T. Manganese, Copper, and Zinc in Cerebrospinal Fluid from Patients with Multiple Sclerosis. Biol. Trace Elem. Res. 2003, 93, 1–8. [Google Scholar] [CrossRef]
- Dales, J.P.; Desplat-Jégo, S. Metal Imbalance in Neurodegenerative Diseases with a Specific Concern to the Brain of Multiple Sclerosis Patients. Int. J. Mol. Sci. 2020, 21, 9105. [Google Scholar] [CrossRef]
- Santos, D.; Milatovic, D.; Andrade, V.; Batoreu, M.C.; Aschner, M.; Marreilha dos Santos, A.P. The Inhibitory Effect of Manganese on Acetylcholinesterase Activity Enhances Oxidative Stress and Neuroinflammation in the Rat Brain. Toxicology 2012, 292, 90–98. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Hardiman, O.; Van Den Berg, L.H.; Kiernan, M.C. Clinical Diagnosis and Management of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The Glutamate Hypothesis in ALS: Pathophysiology and Drug Development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ithurralde, D.; Maruri, A.; Rodríguez, X. Motor Neurone Acetylcholinesterase Release Precedes Neurotoxicity Caused by Systemic Administration of Excitatory Amino Acids and Strychnine. Proc. J. Neurol. Sci. 1998, 160, S80–S86. [Google Scholar] [CrossRef]
- Rodríguez-Ithurralde, D.; Olivera, S.; Vincent, O.; Maruri, A. In Vivo and in Vitro Studies of Glycine- and Glutamate-Evoked Acetylcholinesterase Release from Spinal Motor Neurones: Implications for Amyotrophic Lateral Sclerosis/Motor Neurone Disease Pathogenesis. J. Neurol. Sci. 1997, 152, s54–s61. [Google Scholar] [CrossRef]
- Marc, G.; Leah, R.; Ofira, E.; Oded, A.; Zohar, A.; Hanna, R. Presymptomatic Treatment with Acetylcholinesterase Antisense Oligonucleotides Prolongs Survival in ALS (G93A-SOD1) Mice. BioMed Res. Int. 2013, 2013, 845345. [Google Scholar] [CrossRef]
- Maniatis, S.; Äijö, T.; Vickovic, S.; Braine, C.; Kang, K.; Mollbrink, A.; Fagegaltier, D.; Andrusivová, Ž.; Saarenpää, S.; Saiz-Castro, G.; et al. Spatiotemporal Dynamics of Molecular Pathology in Amyotrophic Lateral Sclerosis. Science 2019, 364, 89–93. [Google Scholar] [CrossRef]
- Campanari, M.L.; Marian, A.; Ciura, S.; Kabashi, E. TDP-43 Regulation of AChE Expression Can Mediate ALS-Like Phenotype in Zebrafish. Cells 2021, 10, 221. [Google Scholar] [CrossRef]
- Dzieciolowska, S.; Drapeau, P.; Armstrong, G.A.B. Augmented Quantal Release of Acetylcholine at the Vertebrate Neuromuscular Junction Following Tdp-43 Depletion. PLoS ONE 2017, 12, e0177005. [Google Scholar] [CrossRef]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral Factors in ALS Patients during Disease Progression. J. Neuroinflamm. 2015, 12, s12974–s13015. [Google Scholar] [CrossRef]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef]
- Gerber, Y.N.; Sabourin, J.C.; Rabano, M.; Vivanco, M.D.M.; Perrin, F.E. Early Functional Deficit and Microglial Disturbances in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e36000. [Google Scholar] [CrossRef]
- Prusiński, A. Neurologia Praktyczna, 3rd ed.; PZWL Wydawnictwo Lekarskie: Warsaw, Poland, 2011; pp. 287–289. [Google Scholar]
- Kish, S.J.; Schut, L.; Simmons, J.; Gilbert, J.; Chang, L.J.; Rebbetoy, M. Brain Acetylcholinesterase Activity Is Markedly Reduced in Dominantly-Inherited Olivopontocerebellar Atrophy. J. Neurol. Neurosurg. Psychiatry 1988, 51, 544–548. [Google Scholar] [CrossRef]
- Hirano, S.; Shinotoh, H.; Arai, K.; Aotsuka, A.; Yasuno, F.; Tanaka, N.; Ota, T.; Sato, K.; Fukushi, K.; Tanada, S.; et al. PET Study of Brain Acetylcholinesterase in Cerebellar Degenerative Disorders. Mov. Disord. 2008, 23, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, D.G.; Litvan, I. Progressive Supranuclear Palsy: Advances in Diagnosis and Management. Parkinsonism Relat. Disord. 2020, 73, 105–116. [Google Scholar] [CrossRef]
- Warren, N.M.; Piggott, M.A.; Perry, E.K.; Burn, D.J. Cholinergic Systems in Progressive Supranuclear Palsy. Brain 2005, 128, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, K.; Dickson, D.W. Microglial Activation Parallels System Degeneration in Progressive Supranuclear Palsy and Corticobasal Degeneration. J. Neuropathol. Exp. Neurol. 2001, 60, 647–657. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Depression and Other Common Mental Disorders Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Centrala Narodowego Funduszu Zdrowia, Departament Analiz i Innowacji NFZ o Zdrowiu. Depresja; Narodowy Fundusz Zdrowia: Warsaw, Poland, 2020. [Google Scholar]
- Jarema, M. Psychiatria w Praktyce; Oficyna Wydawnicza Medical Education sp. z o.o.: Warsaw, Poland, 2011; pp. 35–59. [Google Scholar]
- Jarema, M. Psychiatria, 2nd ed.; PZWL Wydawnictwo Lekarskie: Warsaw, Poland, 2016; pp. 153–163. [Google Scholar]
- Dagyte, G.; Den Boer, J.A.; Trentani, A. The Cholinergic System and Depression. Behav. Brain Res. 2011, 221, 574–582. [Google Scholar] [CrossRef]
- Hannestad, J.O.; Cosgrove, K.P.; Dellagioia, N.F.; Perkins, E.; Bois, F.; Bhagwagar, Z.; Seibyl, J.P.; McClure-Begley, T.D.; Picciotto, M.R.; Esterlis, I. Changes in the Cholinergic System between Bipolar Depression and Euthymia as Measured with [123I]5IA Single Photon Emission Computed Tomography. Biol. Psychiatry 2013, 74, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Warner-Schmidt, J.L.; Schmidt, E.F.; Marshall, J.J.; Rubin, A.J.; Arango-Lievano, M.; Kaplitt, M.G.; Ibañez-Tallon, I.; Heintz, N.; Greengard, P. Cholinergic Interneurons in the Nucleus Accumbens Regulate Depression-like Behavior. Proc. Natl. Acad. Sci. USA 2012, 109, 11360–11365. [Google Scholar] [CrossRef]
- Cheng, J.; Umschweif, G.; Leung, J.; Sagi, Y.; Greengard, P. HCN2 Channels in Cholinergic Interneurons of Nucleus Accumbens Shell Regulate Depressive Behaviors. Neuron 2019, 101, 662–672.e5. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Colasante, C.; Skirzewski, M.; Hernandez, L.; Hoebel, B. Behavioral Depression in the Swim Test Causes a Biphasic, Long-Lasting Change in Accumbens Acetylcholine Release, with Partial Compensation by Acetylcholinesterase and Muscarinic-1 Receptors. Neuroscience 2006, 141, 67–76. [Google Scholar] [CrossRef]
- Lafmejani, R.N.; Zare, K.; Fazollahi, Z.; Roghani, M. Changes of Serum Level of Acetylcholinesterase Enzyme in Lipopolysaccharide-Induced Model of Depression in Mice. J. Basic Clin. Pathophysiol. 2018, 6, 23–26. [Google Scholar] [CrossRef]
- Dulawa, S.C.; Janowsky, D.S. Cholinergic Regulation of Mood: From Basic and Clinical Studies to Emerging Therapeutics. Mol. Psychiatry 2019, 24, 694–709. [Google Scholar] [CrossRef]
- Chau, D.T.; Rada, P.V.; Kim, K.; Kosloff, R.A.; Hoebel, B.G. Fluoxetine Alleviates Behavioral Depression While Decreasing Acetylcholine Release in the Nucleus Accumbens Shell. Neuropsychopharmacology 2011, 36, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.U.; Chaturvedi, A.; Yerrapragada, D.B.; Kundapura, N.; Amin, N.; Devaramane, V. Increased Levels of Acetylcholinesterase, Paraoxonase 1, and Copper in Patients with Moderate Depression- A Preliminary Study. Rep. Biochem. Mol. Biol. 2019, 7, 174–180. [Google Scholar]
- MacHado, D.G.; Cunha, M.P.; Neis, V.B.; Balen, G.O.; Colla, A.; Grando, J.; Brocardo, P.S.; Bettio, L.E.B.; Capra, J.C.; Rodrigues, A.L.S. Fluoxetine Reverses Depressive-like Behaviors and Increases Hippocampal Acetylcholinesterase Activity Induced by Olfactory Bulbectomy. Pharmacol. Biochem. Behav. 2012, 103, 220–229. [Google Scholar] [CrossRef]
- Wang, X.; Li, P.; Ding, Q.; Wu, C.; Zhang, W.; Tang, B. Observation of Acetylcholinesterase in Stress-Induced Depression Phenotypes by Two-Photon Fluorescence Imaging in the Mouse Brain. J. Am. Chem. Soc. 2019, 141, 2061–2068. [Google Scholar] [CrossRef]
- Suarez-Lopez, J.; Suarez-Torres, J.; Sheila, G.; Lopez-Paredes, D.; Noble, M. Acetylcholinesterase Inhibition and Symptoms of Depression and Anxiety among Adolescents in Agricultural Communities in Ecuador. Environ. Epidemiol. 2019, 3, 386. [Google Scholar] [CrossRef]
- Suarez-Lopez, J.R.; Hood, N.; Suárez-Torres, J.; Gahagan, S.; Gunnar, M.R.; López-Paredes, D. Associations of Acetylcholinesterase Activity with Depression and Anxiety Symptoms among Adolescents Growing up near Pesticide Spray Sites. Int. J. Hyg. Environ. Health 2019, 222, 981–990. [Google Scholar] [CrossRef]
- Altinyazar, V.; Sirin, F.B.; Sutcu, R.; Eren, I.; Omurlu, I.K. The Red Blood Cell Acetylcholinesterase Levels of Depressive Patients with Suicidal Behavior in an Agricultural Area. Indian J. Clin. Biochem. 2016, 31, 473–479. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Obayemi, A.; Wigestrand, M.B.; Fote, G.M.; Calarco, C.A.; Li, A.M.; Picciotto, M.R. Cholinergic Signaling in the Hippocampus Regulates Social Stress Resilience and Anxiety- and Depression-like Behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 3573–3578. [Google Scholar] [CrossRef] [PubMed]
- Salas, R.; Main, A.; Gangitano, D.A.; Zimmerman, G.; Ben-Ari, S.; Soreq, H.; De Biasi, M. Nicotine Relieves Anxiogenic-like Behavior in Mice That Overexpress the Read-through Variant of Acetylcholinesterase but Not in Wild-Type Mice. Mol. Pharmacol. 2008, 74, 1641–1648. [Google Scholar] [CrossRef]
- Rao, B.S.; Raju, T.R. Restraint Stress-Induced Alterations in the Levels of Biogenic Amines, Amino Acids, and AChE Activity in the Hippocampus. Neurochem. Res. 2000, 25, 1547–1552. [Google Scholar] [CrossRef]
- Das, A.; Kapoor, K.; Sayeepriyadarshini, A.T.; Dikshit, M.; Palit, G.; Nath, C. Immobilization Stress-Induced Changes in Brain Acetylcholinesterase Activity and Cognitive Function in Mice. Pharmacol. Res. 2000, 42, 213–217. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thase, M.E. Depression and Sleep: Pathophysiology and Treatment. Dialogues Clin. Neurosci. 2006, 8, 217–226. [Google Scholar] [PubMed]
- Benedito, M.A.C.; Camarini, R. Rapid Eye Movement Sleep Deprivation Induces an Increase in Acetylcholinesterase Activity in Discrete Rat Brain Regions. Braz. J. Med Biol. Res. 2001, 34, 103–109. [Google Scholar] [CrossRef]
- Thakkar, M.; Nath Mallick, B. Effect of REM Sleep Deprivation on Rat Brain Acetylcholinesterase. Pharmacol. Biochem. Behav. 1991, 39, 211–214. [Google Scholar] [CrossRef]
- Vazquez, J.; Baghdoyan, H.A. Basal Forebrain Acetylcholine Release during REM Sleep Is Significantly Greater than during Waking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R598–R601. [Google Scholar] [CrossRef]
- Fernandes, S.S.; Koth, A.P.; Parfitt, G.M.; Cordeiro, M.F.; Peixoto, C.S.; Soubhia, A.; Moreira, F.P.; Wiener, C.D.; Oses, J.P.; Kaszubowski, E.; et al. Enhanced Cholinergic-Tone during the Stress Induce a Depressive-like State in Mice. Behav. Brain Res. 2018, 347, 17–25. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Chang, D.; Zhang, X.; Lian, H.; Du, X.; Gao, L. Low-Dose Ketamine Improves LPS-Induced Depression-like Behavior in Rats by Activating Cholinergic Anti-Inflammatory Pathways. ACS Chem. Neurosci. 2020, 11, 752–762. [Google Scholar] [CrossRef]
- Pomara, N.; Bruno, D.; Plaska, C.R.; Pillai, A.; Ramos-Cejudo, J.; Osorio, R.; Imbimbo, B.P.; Heslegrave, A.; Zetterberg, H.; Blennow, K. Evidence of Upregulation of the Cholinergic Anti-Inflammatory Pathway in Late-Life Depression. J. Affect. Disord. 2021, 286, 275–281. [Google Scholar] [CrossRef]
- McCloskey, M.C.; Young, T.J.; Anderson, S.M. The Influence of Acetylcholinesterase on Anxiety- and Depression-like Behaviors in Fluoxetine-Treated Male Mice. BIOS 2017, 88, 29–38. [Google Scholar] [CrossRef]
- Clarke, Z. Rivastigmine. In xPharm: The Comprehensive Pharmacology Reference; Elsevier Inc.: London, UK, 2007; pp. 1–4. [Google Scholar]
- Nguyen, K.; Hoffman, H.; Chakkamparambil, B.; Grossberg, G.T. Evaluation of Rivastigmine in Alzheimer’s Disease. Neurodegener. Dis. Manag. 2021, 11, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Summary of Product Characteristic- RYWASTYGMINE MYLAN. Available online: https://www.mylan.com.pl/-/media/mylanpl/documents/product-pils/rivastygmine-mylan/rivastigmine-mylan_3mg_smpc.pdf (accessed on 25 April 2021).
- Birks, J.S.; Grimley Evans, J. Rivastigmine for Alzheimer’s Disease. Cochrane Database Syst. Rev. 2015, 2015, CD001191. [Google Scholar]
- Small, G.W.; Kaufer, D.; Mendiondo, M.S.; Quarg, P.; Spiegel, R. Cognitive Performance in Alzheimer’s Disease Patients Receiving Rivastigmine for up to 5 Years. Int. J. Clin. Pract. 2005, 59, 473–477. [Google Scholar] [CrossRef]
- Ferris, S.; Karantzoulis, S.; Somogyi, M.; Meng, X. Rivastigmine in Moderately Severe-to-Severe Alzheimer’s Disease: Severe Impairment Battery Factor Analysis. Alzheimer’s Res. Ther. 2013, 5, 63. [Google Scholar] [CrossRef]
- Farlow, M.R.; Grossberg, G.T.; Sadowsky, C.H.; Meng, X.; Somogyi, M. A 24-Week, Randomized, Controlled Trial of Rivastigmine Patch 13.3 Mg/24 h versus 4.6 Mg/24 h in Severe Alzheimer’s Dementia. CNS Neurosci. Ther. 2013, 19, 745–752. [Google Scholar] [CrossRef]
- Emre, M.; Bernabei, R.; Blesa, R.; Bullock, R.; Cunha, L.; Daniëls, H.; Dziadulewicz, E.; Förstl, H.; Frölich, L.; Gabryelewicz, T.; et al. Drug Profile: Transdermal Rivastigmine Patch in the Treatment of Alzheimer Disease. CNS Neurosci. Ther. 2010, 16, 246–253. [Google Scholar] [CrossRef]
- Sadeghi, M.; Ganji, F.; Taghizadeh, S.M.; Daraei, B. Preparation and Characterization of Rivastigmine Transdermal Patch Based on Chitosan Microparticles. Iran. J. Pharm. Res. 2016, 15, 283–294. [Google Scholar] [CrossRef]
- Tsuno, N.; Mori, T.; Ishikawa, I.; Bando, N.; Park, H.; Matsumoto, Y.; Mori, I.; Tanaka, M.; Hirano, T.; Nakamura, Y. Efficacy of Rivastigmine Transdermal Therapy on Low Food Intake in Patients with Alzheimer’s Disease: The Attitude Towards Food Consumption in Alzheimer’s Disease Patients Revive with Rivastigmine Effects Study. Geriatr. Gerontol. Int. 2019, 19, 571–576. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Keller, J.N.; Kaddoumi, A. Role of P-Glycoprotein in Mediating Rivastigmine Effect on Amyloid-β Brain Load and Related Pathology in Alzheimer’s Disease Mouse Model. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 778–787. [Google Scholar] [CrossRef]
- Ray, B.; Maloney, B.; Sambamurti, K.; Karnati, H.K.; Nelson, P.T.; Greig, N.H.; Lahiri, D.K. Rivastigmine Modifies the α-Secretase Pathway and Potentially Early Alzheimer’s Disease. Transl. Psychiatry 2020, 10, 47. [Google Scholar] [CrossRef]
- Bailey, J.A.; Ray, B.; Greig, N.H.; Lahiri, D.K. Rivastigmine Lowers Aβ and Increases SAPPα Levels, Which Parallel Elevated Synaptic Markers and Metabolic Activity in Degenerating Primary Rat Neurons. PLoS ONE 2011, 6, 21954. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Qosa, H.; Kaddoumi, A. Age-Related Decline in Brain and Hepatic Clearance of Amyloid-Beta Is Rectified by the Cholinesterase Inhibitors Donepezil and Rivastigmine in Rats. ACS Chem. Neurosci. 2015, 6, 725–736. [Google Scholar] [CrossRef]
- Farlow, M.R.; Doraiswamy, P.M.; Meng, X.; Cooke, K.; Somogyi, M. The Effect of Vascular Risk Factors on the Efficacy of Rivastigmine Patch and Capsule Treatment in Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. Extra 2011, 1, 150–162. [Google Scholar] [CrossRef]
- Nour, J.M.; Chouliaras, L.; Hickey, L. High Dose Rivastigmine in the Symptom Management of Lewy Body Dementia. BMJ Case Rep. 2016, 2016, bcr2016217240. [Google Scholar] [CrossRef]
- Wesnes, K.A.; McKeith, I.; Edgar, C.; Emre, M.; Lane, R. Benefits of Rivastigmine on Attention in Dementia Associated with Parkinson Disease. Neurology 2005, 65, 1654–1656. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.A.; Farlow, M.R.; Meng, X.; Tekin, S.; Olin, J.T. Efficacy of Rivastigmine on Executive Function in Patients with Parkinson’s Disease Dementia. CNS Neurosci. Ther. 2010, 16, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Possin, K.L.; Kang, G.A.; Guo, C.; Fine, E.M.; Trujillo, A.J.; Racine, C.A.; Wilheim, R.; Johnson, E.T.; Witt, J.L.; Seeley, W.W.; et al. Rivastigmine Is Associated with Restoration of Left Frontal Brain Activity in Parkinson’s Disease. Mov. Disord. 2013, 28, 1384–1390. [Google Scholar] [CrossRef]
- Reading, P.J.; Luce, A.K.; McKeith, I.G. Rivastigmine in the Treatment of Parkinsonian Psychosis and Cognitive Impairment: Preliminary Findings from an Open Trial. Mov. Disord. 2001, 16, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.-S.; Kim, J.-S.; Lee, P.H. Effect of Rivastigmine on Behavioral and Psychiatric Symptoms of Parkinson’s Disease Dementia. J. Mov. Disord. 2015, 8, 98–102. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P.; Dal Ben, M. Rivastigmine as a Symptomatic Treatment for Apathy in Parkinson’s Dementia Complex: New Aspects for This Riddle. Parkinson’s Dis. 2017, 2017, 6219851. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Maltête, D.; Lefaucheur, R.; Kreisler, A.; Eusebio, A.; Defer, G.; Ouk, T.; Azulay, J.P.; Krystkowiak, P.; et al. Rivastigmine in Apathetic but Dementia and Depression-Free Patients with Parkinson’s Disease: A Double-Blind, Placebo-Controlled, Randomised Clinical Trial. J. Neurol. Neurosurg. Psychiatry 2014, 85, 668–674. [Google Scholar] [CrossRef]
- Espay, A.J.; Marsili, L.; Mahajan, A.; Sturchio, A.; Pathan, R.; Pilotto, A.; Elango, D.S.; Pezous, N.; Masellis, M.; Gomez-Mancilla, B. Rivastigmine in Parkinson’s Disease Dementia with Orthostatic Hypotension. Ann. Neurol. 2021, 89, 91–98. [Google Scholar] [CrossRef]
- Li, Z.; Yu, Z.; Zhang, J.; Wang, J.; Sun, C.; Wang, P.; Zhang, J. Impact of Rivastigmine on Cognitive Dysfunction and Falling in Parkinson’s Disease Patients. Eur. Neurol. 2015, 74, 86–91. [Google Scholar] [CrossRef]
- Šešok, S.; Bolle, N.; Kobal, J.; Bucik, V.; Vodušek, D.B. Cognitive Function in Early Clinical Phase Huntington Disease after Rivastigmine Treatment. Psychiatr Danub. 2014, 26, 239–248. [Google Scholar]
- De Tommaso, M.; Difruscolo, O.; Sciruicchio, V.; Specchio, N.; Livrea, P. Two Years’ Follow-up of Rivastigmine Treatment in Huntington Disease. Clin. Neuropharmacol. 2007, 30, 43–46. [Google Scholar] [CrossRef]
- De Tommaso, M.; Specchio, N.; Sciruicchio, V.; Difruscolo, O.; Specchio, L.M. Effects of Rivastigmine on Motor and Cognitive Impairment in Huntington’s Disease. Mov. Disord. 2004, 19, 1516–1518. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Protective Effect of Rivastigmine against 3-Nitropropionic Acid-Induced Huntington’s Disease like Symptoms: Possible Behavioural, Biochemical and Cellular Alterations. Eur. J. Pharmacol. 2009, 615, 91–101. [Google Scholar] [CrossRef]
- Huolman, S.; Hämäläinen, P.; Vorobyev, V.; Ruutiainen, J.; Parkkola, R.; Laine, T.; Hämäläinen, H. The Effects of Rivastigmine on Processing Speed and Brain Activation in Patients with Multiple Sclerosis and Subjective Cognitive Fatigue. Mult. Scler. J. 2011, 17, 1351–1361. [Google Scholar] [CrossRef]
- Mäurer, M.; Ortler, S.; Baier, M.; Meergans, M.; Scherer, P.; Hofmann, W.E.; Tracik, F. Randomised Multicentre Trial on Safety and Efficacy of Rivastigmine in Cognitively Impaired Multiple Sclerosis Patients. Mult. Scler. J. 2013, 19, 631–638. [Google Scholar] [CrossRef]
- Shaygannejad, V.; Janghorbani, M.; Ashtari, F.; Zanjani, H.A.; Zakizade, N. Effects of Rivastigmine on Memory and Cognition in Multiple Sclerosis. Can. J. Neurol. Sci. 2008, 35, 476–481. [Google Scholar] [CrossRef]
- Cucurachi, L.; Immovilli, P.; Granella, F.; Pavesi, G.; Cattaneo, L. Short-Latency Afferent Inhibition Predicts Verbal Memory Performance in Patients with Multiple Sclerosis. J. Neurol. 2008, 255, 1949–1956. [Google Scholar] [CrossRef]
- Nizri, E.; Irony-Tur-Sinai, M.; Faranesh, N.; Lavon, I.; Lavi, E.; Weinstock, M.; Brenner, T. Suppression of Neuroinflammation and Immunomodulation by the Acetylcholinesterase Inhibitor Rivastigmine. J. Neuroimmunol. 2008, 203, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Grobe-Einsler, M.; Vogt, I.R.; Schaprian, T.; Hurlemann, R.; Klockgether, T.; Kaut, O. Effects of Rivastigmine on Patients with Spinocerebellar Ataxia Type 3: A Case Series of Five Patients. Neurodegener. Dis. 2020, 20, 104–109. [Google Scholar] [CrossRef]
- Liepelt, I.; Gaenslen, A.; Godau, J.; Di Santo, A.; Schweitzer, K.J.; Gasser, T.; Berg, D. Rivastigmine for the Treatment of Dementia in Patients with Progressive Supranuclear Palsy: Clinical Observations as a Basis for Power Calculations and Safety Analysis. Alzheimer’s Dement. 2010, 6, 70–74. [Google Scholar] [CrossRef]
- Summary of Product Characteristic- Donepezil Polfarmex. Available online: http://leki.urpl.gov.pl/files/Donepezil_Polfarmex_tabl_powl_5.pdf (accessed on 25 April 2021).
- Summary of Product Characteristic-Donepezil Bluefish. Available online: http://leki.urpl.gov.pl/files/26_DonepezilBluefish_ALL.pdf (accessed on 25 April 2021).
- Adlimoghaddam, A.; Neuendorff, M.; Roy, B.; Albensi, B.C. A Review of Clinical Treatment Considerations of Donepezil in Severe Alzheimer’s Disease. CNS Neurosci. Ther. 2018, 24, 876–888. [Google Scholar] [CrossRef]
- Cummings, J.L.; Geldmacher, D.; Farlow, M.; Sabbagh, M.; Christensen, D.; Betz, P. High-Dose Donepezil (23 Mg/Day) for the Treatment of Moderate and Severe Alzheimer’s Disease: Drug Profile and Clinical Guidelines. CNS Neurosci. Ther. 2013, 19, 294–301. [Google Scholar] [CrossRef]
- Sabbagh, M.; Cummings, J.; Christensen, D.; Doody, R.; Farlow, M.; Liu, L.; Mackell, J.; Fain, R. Evaluating the Cognitive Effects of Donepezil 23 Mg/d in Moderate and Severe Alzheimer’s Disease: Analysis of Effects of Baseline Features on Treatment Response. BMC Geriatr. 2013, 13, 56. [Google Scholar] [CrossRef]
- Tariot, P.; Salloway, S.; Yardley, J.; MacKell, J.; Moline, M. Long-Term Safety and Tolerability of Donepezil 23mg in Patients with Moderate to Severe Alzheimers Disease. BMC Res. Notes 2012, 5, 283. [Google Scholar] [CrossRef]
- Farlow, M.R.; Salloway, S.; Tariot, P.N.; Yardley, J.; Moline, M.L.; Wang, Q.; Brand-Schieber, E.; Zou, H.; Hsu, T.; Satlin, A. Effectiveness and Tolerability of High-Dose (23 Mg/d) versus Standard-Dose (10 Mg/d) Donepezil in Moderate to Severe Alzheimer’s Disease: A 24-Week, Randomized, Double-Blind Study. Clin. Ther. 2010, 32, 1234–1251. [Google Scholar] [CrossRef]
- Sabbagh, M.; Cummings, J. Progressive Cholinergic Decline in Alzheimer’s Disease: Consideration for Treatment with Donepezil 23 Mg in Patients with Moderate to Severe Symptomatology. BMC Neurol. 2011, 11, 21. [Google Scholar] [CrossRef]
- Cheng, J.; Yang, H.; Zhang, J. Donepezil’s Effects on Brain Functions of Patients with Alzheimer Disease: A Regional Homogeneity Study Based on Resting-State Functional Magnetic Resonance Imaging. Clin. Neuropharmacol. 2019, 42, 42–48. [Google Scholar] [CrossRef]
- Winblad, B.; Kilander, L.; Eriksson, S.; Minthon, L.; Båtsman, S.; Wetterholm, A.L.; Jansson-Blixt, C.; Haglund, A. Donepezil in Patients with Severe Alzheimer’s Disease: Double-Blind, Parallel-Group, Placebo-Controlled Study. Lancet 2006, 367, 1057–1065. [Google Scholar] [CrossRef]
- Dubois, B.; Chupin, M.; Hampel, H.; Lista, S.; Cavedo, E.; Croisile, B.; Louis Tisserand, G.; Touchon, J.; Bonafe, A.; Ousset, P.J.; et al. Donepezil Decreases Annual Rate of Hippocampal Atrophy in Suspected Prodromal Alzheimer’s Disease. Alzheimer’s Dement. 2015, 11, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lai, T.J.; Hemrungrojn, S.; Mohandas, E.; Yun Kim, S.; Nair, G.; Dash, A. Role of Donepezil in the Management of Neuropsychiatric Symptoms in Alzheimer’s Disease and Dementia with Lewy Bodies. CNS Neurosci. Ther. 2016, 22, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Franceschi, M.; D’Onofrio, G.; Bizzarro, A.; Mangialasche, F.; Cascavilla, L.; Paris, F.; Matera, M.G.; Pilotto, A.; Daniele, A.; et al. Effect of a Cyp2d6 Polymorphism on the Efficacy of Donepezil in Patients with Alzheimer Disease. Neurology 2009, 73, 761–767. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, S.Y.; Na, H.R.; Kim, B.K.; Yang, D.W.; Kwon, J.C.; Park, M.Y. Effect of ApoE Genotype on Response to Donepezil in Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2008, 25, 445–450. [Google Scholar] [CrossRef]
- Xiao, T.; Jiao, B.; Zhang, W.; Tang, B.; Shen, L. Effect of the CYP2D6 and APOE Polymorphisms on the Efficacy of Donepezil in Patients with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. CNS Drugs 2016, 30, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Najar-Ahmadi, S.; Haghaei, H.; Farajnia, S.; Yekta, R.; Ezzati Nazhad Dolatabadi, J.; Rashidi, M.R. Interaction of Donepezil with Tau Protein: Insights from Surface Plasmon Resonance and Molecular Modeling Methods. J. Mol. Liq. 2021, 333, 115924. [Google Scholar] [CrossRef]
- Ma, Y.; Ji, J.; Li, G.; Yang, S.; Pan, S. Effects of Donepezil on Cognitive Functions and the Expression Level of β-Amyloid in Peripheral Blood of Patients with Alzheimer’s Disease. Exp. Ther. Med. 2018, 15, 1875–1878. [Google Scholar] [CrossRef]
- Baik, K.; Kim, S.M.; Jung, J.H.; Lee, Y.H.; Chung, S.J.; Yoo, H.S.; Ye, B.S.; Lee, P.H.; Sohn, Y.H.; Kang, S.W.; et al. Donepezil for Mild Cognitive Impairment in Parkinson’s Disease. Sci. Rep. 2021, 11, 4734. [Google Scholar] [CrossRef]
- Hiraoka, K.; Okamura, N.; Funaki, Y.; Hayashi, A.; Tashiro, M.; Hisanaga, K.; Fujii, T.; Takeda, A.; Yanai, K.; Iwata, R.; et al. Cholinergic Deficit and Response to Donepezil Therapy in Parkinson’s Disease with Dementia. Eur. Neurol. 2012, 68, 137–143. [Google Scholar] [CrossRef]
- Sawada, H.; Oeda, T.; Kohsaka, M.; Umemura, A.; Tomita, S.; Park, K.; Mizoguchi, K.; Matsuo, H.; Hasegawa, K.; Fujimura, H.; et al. Early Use of Donepezil against Psychosis and Cognitive Decline in Parkinson’s Disease: A Randomised Controlled Trial for 2 Years. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1332–1340. [Google Scholar] [CrossRef]
- Dubois, B.; Tolosa, E.; Katzenschlager, R.; Emre, M.; Lees, A.J.; Schumann, G.; Pourcher, E.; Gray, J.; Thomas, G.; Swartz, J.; et al. Donepezil in Parkinson’s Disease Dementia: A Randomized, Double-Blind Efficacy and Safety Study. Mov. Disord. 2012, 27, 1230–1238. [Google Scholar] [CrossRef]
- Ishikawa, K.-I.; Motoi, Y.; Mizuno, Y.; Kubo, S.-I.; Hattori, N. Effects of Donepezil Dose Escalation in Parkinson’s Patients with Dementia Receiving Long-Term Donepezil Treatment: An Exploratory Study. Psychogeriatrics 2014, 14, 93–100. [Google Scholar] [CrossRef]
- Ivanco, L.S.; Bohnen, N.I. Effects of Donepezil on Compulsive Hypersexual Behavior in Parkinson Disease. Am. J. Ther. 2005, 12, 467–468. [Google Scholar] [CrossRef]
- Shahpouri, M.M.; Barekatain, M.; Tavakoli, M.; Mirmosayyeb, O.; Safaei, A.; Shaygannejad, V. Comparison of Cognitive Rehabilitation versus Donepezil Therapy on Memory Performance, Attention, Quality of Life, and Depression among Multiple Sclerosis Patients. Neurol. Res. Int. 2020, 2020, 8874424. [Google Scholar] [CrossRef]
- Shahpouri, M.; Barekatain, M.; Tavakoli, M.; Badihian, S.; Shaygannejad, V. Effect of Donepezil on Cognitive Impairment, Quality of Life, and Depression in Multiple Sclerosis Patients: A Randomized Clinical Trial. Int. J. Prev. Med. 2020, 11, 69. [Google Scholar] [CrossRef]
- Christodoulou, C.; Melville, P.; Scherl, W.F.; MacAllister, W.S.; Elkins, L.E.; Krupp, L.B. Effects of Donepezil on Memory and Cognition in Multiple Sclerosis. J. Neurol. Sci. 2006, 245, 127–136. [Google Scholar] [CrossRef]
- Krupp, L.B.; Christodoulou, C.; Melville, P.; Scherl, W.F.; Pai, L.Y.; Muenz, L.R.; He, D.; Benedict, R.H.B.; Goodman, A.; Rizvi, S.; et al. Multicenter Randomized Clinical Trial of Donepezil for Memory Impairment in Multiple Sclerosis. Neurology 2011, 76, 1500–1507. [Google Scholar] [CrossRef]
- O’Carroll, C.B.; Woodruff, B.K.; Locke, D.E.; Hoffman-Snyder, C.R.; Wellik, K.E.; Thaera, G.M.; Demaerschalk, B.M.; Wingerchuk, D.M. Is Donepezil Effective for Multiple Sclerosis-Related Cognitive Dysfunction? Neurologist 2012, 18, 51–54. [Google Scholar] [CrossRef]
- Brunner, D.; Murphy, C.A.; Paterson, N.E.; Chen, A.; Arias, W.; He, D.; Alosio, W.; Oakeshott, S.; Farrar, A.; Menalled, L.; et al. Cognitive Deficits in the R6/2 Mouse Model of Huntington’s Disease and Their Amelioration with Donepezil. Int. J. Comp.Psychol. 2014, 27, 397–407. [Google Scholar]
- Fernandez, H.H.; Friedman, J.H.; Grace, J.; Beason-Hazen, S. Donepezil for Huntington’s Disease. Mov. Disord. 2000, 15, 173–176. [Google Scholar] [CrossRef]
- Cubo, E.; Shannon, K.M.; Tracy, D.; Jaglin, J.A.; Bernard, B.A.; Wuu, J.; Leurgans, S.E. Effect of Donepezil on Motor and Cognitive Function in Huntington Disease. Neurology 2006, 67, 1268–1271. [Google Scholar] [CrossRef]
- Litvan, I.; Phipps, M.; Pharr, V.L.; Hallett, M.; Grafman, J.; Salazar, A. Randomized Placebo-Controlled Trial of Donepezil in Patients with Progressive Supranuclear Palsy. Neurology 2001, 57, 467–473. [Google Scholar] [CrossRef]
- Scott, L.J.; Goa, K.L. Galantamine: A Review of Its Use in Alzheimer’s Disease. Drugs 2000, 60, 1095–1122. [Google Scholar] [CrossRef]
- Farlow, M.R. Clinical Pharmacokinetics of Galantamine. Clin. Pharmacokinet. 2003, 42, 1383–1392. [Google Scholar] [CrossRef]
- Prvulovic, D.; Hampel, H.; Pantel, J. Galantamine for Alzheimer’s Disease. Expert Opin. Drug Metab. Toxicol. 2010, 6, 345–354. [Google Scholar] [CrossRef]
- Kowal, N.M.; Ahring, P.K.; Liao, V.W.Y.; Indurti, D.C.; Harvey, B.S.; O’Connor, S.M.; Chebib, M.; Olafsdottir, E.S.; Balle, T. Galantamine Is Not a Positive Allosteric Modulator of Human A4β2 or A7 Nicotinic Acetylcholine Receptors. Br. J. Pharmacol. 2018, 175, 2911–2925. [Google Scholar] [CrossRef]
- Seltzer, B. Galantamine-ER for the Treatment of Mild-to-Moderate Alzheimer’s Disease. Clin. Interv. Aging 2010, 5, 1–6. [Google Scholar]
- Burns, A.; Bernabei, R.; Bullock, R.; Jentoft, A.J.C.; Frölich, L.; Hock, C.; Raivio, M.; Triau, E.; Vandewoude, M.; Wimo, A.; et al. Safety and Efficacy of Galantamine (Reminyl) in Severe Alzheimer’s Disease (the SERAD Study): A Randomised, Placebo-Controlled, Double-Blind Trial. Lancet Neurol. 2009, 8, 39–47. [Google Scholar] [CrossRef]
- Raskind, M.A.; Peskind, E.R.; Truyen, L.; Kershaw, P.; Damaraju, C.R.V. The Cognitive Benefits of Galantamine Are Sustained for at Least 36 Months: A Long-Term Extension Trial. Arch. Neurol. 2004, 61, 252–256. [Google Scholar] [CrossRef]
- Lin, Y.T.; Chou, M.C.; Wu, S.J.; Yang, Y.H. Galantamine Plasma Concentration and Cognitive Response in Alzheimer’s Disease. PeerJ 2019, 2019, e6887. [Google Scholar] [CrossRef]
- Hager, K.; Baseman, A.S.; Nye, J.S.; Brashear, H.R.; Han, J.; Sano, M.; Davis, B.; Richards, H.M. Effects of Galantamine in a 2-Year, Randomized, Placebo-Controlled Study in Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2014, 10, 391–401. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Zheng, X.; Fang, T.; Yang, X.; Luo, X.; Guo, A.; Newell, K.A.; Huang, X.F.; Yu, Y. Galantamine Improves Cognition, Hippocampal Inflammation, and Synaptic Plasticity Impairments Induced by Lipopolysaccharide in Mice. J. Neuroinflamm. 2018, 15, s12974–s13018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhao, Y.; Zhang, T.; Lan, J.; Yang, J.; Yuan, L.; Zhang, Q.; Pan, K.; Zhang, K. Galantamine Inhibits β-Amyloid-Induced Cytostatic Autophagy in PC12 Cells through Decreasing ROS Production. Cell Prolif. 2018, 51, e12427. [Google Scholar] [CrossRef]
- Freund-Levi, Y.; Jedenius, E.; Tysen-Bäckström, A.C.; Lärksäter, M.; Wahlund, L.O.; Eriksdotter, M. Galantamine versus Risperidone Treatment of Neuropsychiatric Symptoms in Patients with Probable Dementia: An Open Randomized Trial. Am. J. Geriatr. Psychiatry 2014, 22, 341–348. [Google Scholar] [CrossRef]
- Kavanagh, S.; Howe, I.; R Brashear, H.; Wang, D.; Van Baelen, B.; Todd, M.; Schwalen, S. Long-Term Response to Galantamine in Relation to Short-Term Efficacy Data: Pooled Analysis in Patients with Mild to Moderate Alzheimers Disease. Curr. Alzheimer Res. 2012, 8, 175–186. [Google Scholar] [CrossRef]
- Ohnishi, T.; Sakiyama, Y.; Okuri, Y.; Kimura, Y.; Sugiyama, N.; Saito, T.; Takahashi, M.; Kobayashi, T. The Prediction of Response to Galantamine Treatment in Patients with Mild to Moderate Alzheimer’s Disease. Curr. Alzheimer Res. 2014, 11, 110–118. [Google Scholar] [CrossRef]
- Wattmo, C.; Jedenius, E.; Blennow, K.; Wallin, Å.K. Dose and Plasma Concentration of Galantamine in Alzheimer’s Diseas—Clinical Application. Alzheimer’s Res. Ther. 2013, 5, 2. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Haertel, C.; Maelicke, A.; Montag, D. Galantamine Slows down Plaque Formation and Behavioral Decline in the 5XFAD Mouse Model of Alzheimer’s Disease. PLoS ONE 2014, 9, e89454. [Google Scholar] [CrossRef]
- Rao, P.P.N.; Mohamed, T.; Osman, W. Investigating the Binding Interactions of Galantamine with β-Amyloid Peptide. Bioorganic Med. Chem. Lett. 2013, 23, 239–243. [Google Scholar] [CrossRef]
- Grace, J.; Amick, M.M.; Friedman, J.H. A Double-Blind Comparison of Galantamine Hydrobromide ER and Placebo in Parkinson Disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 18–23. [Google Scholar] [CrossRef]
- Litvinenko, I.V.; Odinak, M.M.; Mogil’naya, V.I.; Emelin, A.Y.U. Efficacy and Safety of Galantamine (Reminyl) for Dementia in Patients with Parkinson’s Disease (an Open Controlled Trial). Neurosci. Behav. Physiol. 2008, 38, 937–945. [Google Scholar] [CrossRef]
- Aarsland, D.; Hutchinson, M.; Larsen, J.P. Cognitive, Psychiatric and Motor Response to Galantamine in Parkinson’s Disease with Dementia. Int. J. Geriatr. Psychiatry 2003, 18, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lee, S.T.; Im, W.S.; Chu, K.; Kim, M. Galantamine Reduces Striatal Degeneration in 3-Nitropropionic Acid Model of Huntington’s Disease. Neurosci. Lett. 2008, 448, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Petrikis, P.; Andreou, C.; Piachas, A.; Bozikas, V.P.; Karavatos, A. Treatment of Huntington’s Disease with Galantamine. Int. Clin. Psychopharmacol. 2004, 19, 49–50. [Google Scholar] [CrossRef]
- Herzon, S.; Tun, M.K.M. The Pharmacology and Therapeutic Potential of (-)-Huperzine A. J. Exp. Pharmacol. 2012, 4, 113. [Google Scholar] [CrossRef][Green Version]
- Kim Thu, D.; Vui, D.T.; Ngoc Huyen, N.T.; Duyen, D.K.; Thanh Tung, B. The Use of Huperzia Species for the Treatment of Alzheimer’s Disease. J. Basic Clin. Physiol. Pharmacol. 2020, 31, 0159. [Google Scholar] [CrossRef]
- Zhang, H.Y. New Insights into Huperzine A for the Treatment of Alzheimer’s Disease. Acta Pharmacol. Sin. 2012, 33, 1170–1175. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.P. Huperzine A for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. PLoS ONE 2013, 8, e74916. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.H.; Zhu, C.X.; Zhang, R.; An, L. Huperzine A in the Treatment of Alzheimer’s Disease and Vascular Dementia: A Meta-Analysis. Evid. Based Complementary Altern. Med. 2014, 2014, 363985. [Google Scholar] [CrossRef] [PubMed]
- Gul, A.; Bakht, J.; Mehmood, F. Huperzine-A Response to Cognitive Impairment and Task Switching Deficits in Patients with Alzheimer’s Disease. J. Chin. Med Assoc. 2019, 82, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Walsh, S.; Little, J.T.; Behan, K.; Reynolds, B.; Ward, C.; Jin, S.; Thomas, R.; Aisen, P.S. A Phase II Trial of Huperzine A in Mild to Moderate Alzheimer Disease. Neurology 2011, 76, 1389–1394. [Google Scholar] [CrossRef]
- Damar, U.; Gersner, R.; Johnstone, J.T.; Schachter, S.; Rotenberg, A. Huperzine A: A Promising Anticonvulsant, Disease Modifying, and Memory Enhancing Treatment Option in Alzheimer’s Disease. Med. Hypotheses 2017, 99, 57–62. [Google Scholar] [CrossRef]
- Qian, Z.M.; Ke, Y. Huperzine A: Is It an Effective Disease-Modifying Drug for Alzheimer’s Disease? Front. Aging Neurosci. 2014, 6, 216. [Google Scholar] [CrossRef]
- Darreh-Shori, T.; Hosseini, S.M.; Nordberg, A. Pharmacodynamics of Cholinesterase Inhibitors Suggests Add-on Therapy with a Low-Dose Carbamylating Inhibitor in Patients on Long-Term Treatment with Rapidly Reversible Inhibitors. J. Alzheimer’s Dis. 2014, 39, 423–440. [Google Scholar] [CrossRef]
- Winblad, B.; Giacobini, E.; Frölich, L.; Friedhoff, L.T.; Bruinsma, G.; Becker, R.E.; Greig, N.H. Phenserine Efficacy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 22, 1201–1208. [Google Scholar] [CrossRef]
- Marutle, A.; Ohmitsu, M.; Nilbratt, M.; Greig, N.H.; Nordberg, A.; Sugaya, K. Modulation of Human Neural Stem Cell Differentiation in Alzheimer (APP23) Transgenic Mice by Phenserine. Proc. Natl. Acad. Sci. USA 2007, 104, 12506–12511. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.R.; Greig, H.N. Was Phenserine a Failure or Were Investigators Mislead by Methods? Curr. Alzheimer Res. 2012, 9, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, S.C.; Lecca, D.; Greig, N.H.; Wang, J.Y.; Selman, W.; Hoffer, B.J.; Miller, J.P.; Chiang, Y.H. (-)-Phenserine Ameliorates Contusion Volume, Neuroinflammation, and Behavioral Impairments Induced by Traumatic Brain Injury in Mice. Cell Transplant. 2019, 28, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, D.; Fukui, K.; Li, Y.; Yu, Q.S.; Barak, S.; Tamargo, I.A.; Rubovitch, V.; Holloway, H.W.; Lehrmann, E.; Wood, W.H.; et al. Cognitive Impairments Induced by Concussive Mild Traumatic Brain Injury in Mouse Are Ameliorated by Treatment with Phenserine via Multiple Non-Cholinergic and Cholinergic Mechanisms. PLoS ONE 2016, 11, e0156493. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, B.J.; Pick, C.G.; Hoffer, M.E.; Becker, R.E.; Chiang, Y.H.; Greig, N.H. Repositioning Drugs for Traumatic Brain Injury—N-Acetyl Cysteine and Phenserine. J. Biomed. Sci. 2017, 24, 71. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Bader, M.; Tweedie, D.; Hoffman, A.F.; Jung, Y.J.; Hsueh, S.C.; Hoffer, B.J.; Becker, R.E.; Pick, C.G.; Lupica, C.R.; et al. (-)-Phenserine and the Prevention of Pre-Programmed Cell Death and Neuroinflammation in Mild Traumatic Brain Injury and Alzheimer’s Disease Challenged Mice. Neurobiol. Dis. 2019, 130, 104528. [Google Scholar] [CrossRef]
- Reale, M.; Di Nicola, M.; Velluto, L.; D’angelo, C.; Costantini, E.; Lahiri, D.K.; Kamal, M.A.; Yu, Q.-S.; Greig, N.H. Selective Acetyl-and Butyrylcholinesterase Inhibitors Reduce Amyloid-β Ex Vivo Activation of Peripheral Chemo-Cytokines from Alzheimer’s Disease Subjects: Exploring the Cholinergic Anti-Inflammatory Pathway; Bentham Science Publishers: Sharjah, United Arab Emirates, 2014. [Google Scholar]
- Kadir, A.; Andreasen, N.; Almkvist, O.; Wall, A.; Forsberg, A.; Engler, H.; Hagman, G.; Lärksäter, M.; Winblad, B.; Zetterberg, H.; et al. Effect of Phenserine Treatment on Brain Functional Activity and Amyloid in Alzheimer’s Disease. Ann. Neurol. 2008, 63, 621–631. [Google Scholar] [CrossRef]
- Chen, J.; Pan, H.; Chen, C.; Wu, W.; Iskandar, K.; He, J.; Piermartiri, T.; Jacobowitz, D.M.; Yu, Q.S.; McDonough, J.H.; et al. (-)-Phenserine Attenuates Soman-Induced Neuropathology. PLoS ONE 2014, 9, e99818. [Google Scholar] [CrossRef]
- Lilja, A.M.; Luo, Y.; Yu, Q.S.; Röjdner, J.; Li, Y.; Marini, A.M.; Marutle, A.; Nordberg, A.; Greig, N.H. Neurotrophic and Neuroprotective Actions of (-)- and (+)-Phenserine, Candidate Drugs for Alzheimer’s Disease. PLoS ONE 2013, 8, e0054887. [Google Scholar] [CrossRef]
- Chang, C.F.; Lai, J.H.; Wu, J.C.C.; Greig, N.H.; Becker, R.E.; Luo, Y.; Chen, Y.H.; Kang, S.J.; Chiang, Y.H.; Chen, K.Y. (−)-Phenserine Inhibits Neuronal Apoptosis Following Ischemia/Reperfusion Injury. Brain Res. 2017, 1677, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Mikkilineni, S.; Cantuti-Castelvetri, I.; Cahill, C.M.; Balliedier, A.; Greig, N.H.; Rogers, J.T. The Anticholinesterase Phenserine and Its Enantiomer Posiphen as 5′untranslated-Region-Directed Translation Blockers of the Parkinson’s Alpha Synuclein Expression. Parkinson’s Dis. 2012, 2012, 142372. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Chen, D.M.; Maloney, B.; Holloway, H.W.; Yu, Q.S.; Utsuki, T.; Giordano, T.; Sambamurti, K.; Greig, N.H. The Experimental Alzheimer’s Disease Drug Posiphen [(+)-Phenserine] Lowers Amyloid-β Peptide Levels in Cell Culture and Mice. J. Pharmacol. Exp. Ther. 2007, 320, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Teich, A.F.; Sharma, E.; Barnwell, E.; Zhang, H.; Staniszewski, A.; Utsuki, T.; Padmaraju, V.; Mazell, C.; Tzekou, A.; Sambamurti, K.; et al. Translational Inhibition of APP by Posiphen: Efficacy, Pharmacodynamics, and Pharmacokinetics in the APP/PS1 Mouse. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 37–45. [Google Scholar] [CrossRef]
- Maccecchini, M.L.; Chang, M.Y.; Pan, C.; John, V.; Zetterberg, H.; Greig, N.H. Posiphen as a Candidate Drug to Lower CSF Amyloid Precursor Protein, Amyloid-β Peptide and τ Levels: Target Engagement, Tolerabilityand Pharmacokinetics in Humans. J. Neurolog. Neurosurg. Psychiatry 2012, 83, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Reale, M.; Kamal, M.A.; Holloway, H.W.; Luo, W.; Sambamurti, K.; Ray, B.; Lahiri, D.K.; Rogers, J.T.; Greig, N.H. Synthesis of the Alzheimer Drug Posiphen into Its Primary Metabolic Products (+)-N1-NorPosiphen, (+)-N8-NorPosiphen and (+)-N1, N8-BisnorPosiphen, Their Inhibition of Amyloid Precursor Protein, α -Synuclein Synthesis, Interleukin-1β Release, and Cholinergic Action. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2013, 12, 117–128. [Google Scholar] [CrossRef]
- Rogers, J.T.; Mikkilineni, S.; Cantuti-Castelvetri, I.; Smith, D.H.; Huang, X.; Bandyopadhyay, S.; Cahill, C.M.; Maccecchini, M.L.; Lahiri, D.K.; Greig, N.H. The Alpha-Synuclein 5′untranslated Region Targeted Translation Blockers: Anti-Alpha Synuclein Efficacy of Cardiac Glycosides and Posiphen. J. Neural Transm. 2011, 118, 493–507. [Google Scholar] [CrossRef]
- Kuo, Y.-M.; Nwankwo, E.I.; Nussbaum, R.L.; Rogers, J.; Maccecchini, M.L. Translational Inhibition of α-Synuclein by Posiphen Normalizes Distal Colon Motility in Transgenic Parkinson Mice. Am. J. Neurodegener. Dis. 2019, 8, 1–15. [Google Scholar]
- Annovis Bio Inc. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Dose-Finding Study to Evaluate the Safety, Tolerability, Pharmacokinetic and Pharmacodynamic Effects of Posiphen® in Subjects with Early Alzheimer’s Disease (AD) or Early Parkinson’s Disease (PD). 2021. Available online: Clinicaltrials.gov (accessed on 24 August 2021).
- Annovis Bio Inc. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Ascending Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics (PK) and Pharmacodynamic (PD) Effects of Posiphen® in Subjects with Early Alzheimer’s Disease (AD). 2021. Available online: Clinicaltrials.gov (accessed on 24 August 2021).
- Fitzgerald, P.J.; Hale, P.J.; Ghimire, A.; Watson, B.O. Repurposing Cholinesterase Inhibitors as Antidepressants? Dose and Stress-Sensitivity May Be Critical to Opening Possibilities. Front. Behav. Neurosci. 2021, 14, 620119. [Google Scholar] [CrossRef]
- Reynolds, C.F.; Butters, M.A.; Lopez, O.; Pollock, B.G.; Dew, M.A.; Mulsant, B.H.; Lenze, E.J.; Holm, M.; Rogers, J.C.; Mazumdar, S.; et al. Maintenance Treatment of Depression in Old Age: A Randomized, Double-Blind, Placebo-Controlled Evaluation of the Efficacy and Safety of Donepezil Combined with Antidepressant Pharmacotherapy. Arch. Gen. Psychiatry 2011, 68, 51–60. [Google Scholar] [CrossRef]
- Akechi, T.; Suzuki, M.; Hashimoto, N.; Yamada, T.; Yamada, A.; Nakaaki, S. Different Pharmacological Responses in Late-Life Depression with Subsequent Dementia: A Case Supporting the Reserve Threshold Theory. Psychogeriatrics 2017, 17, 500–501. [Google Scholar] [CrossRef]
- Amara, G.; Saada, W.; Ben Nasr, S.; Ben Hadj Ali, B. Cholinesterase Inhibitors and Depression in the Elderly. Encephale 2010, 36, 77–81. [Google Scholar] [CrossRef]
- Jawaid, A.; Pawlowicz, E.; Schulz, P.E. Do Acetylcholinesterase Inhibitors Increase Anxiety and Depression in Elderly Adults with Dementia? J. Am. Geriatr. Soc. 2015, 63, 1702–1704. [Google Scholar] [CrossRef]
- Devanand, D.P.; Pelton, G.H.; D’Antonio, K.; Ciarleglio, A.; Scodes, J.; Andrews, H.; Lunsford, J.; Beyer, J.L.; Petrella, J.R.; Sneed, J.; et al. Donepezil Treatment in Patients with Depression and Cognitive Impairment on Stable Antidepressant Treatment: A Randomized Controlled Trial. Am. J. Geriatr. Psychiatry 2018, 26, 1050–1060. [Google Scholar] [CrossRef]
- Fishback, J.A.; Robson, M.J.; Xu, Y.T.; Matsumoto, R.R. Sigma Receptors: Potential Targets for a New Class of Antidepressant Drug. Pharmacol. Ther. 2010, 127, 271–282. [Google Scholar] [CrossRef]
- Ramakrishnan, N.K.; Visser, A.K.D.; Schepers, M.; Luurtsema, G.; Nyakas, C.J.; Elsinga, P.H.; Ishiwata, K.; Dierckx, R.A.J.O.; Van Waarde, A. Dose-Dependent Sigma-1 Receptor Occupancy by Donepezil in Rat Brain Can Be Assessed with 11C-SA4503 and MicroPET. Psychopharmacology 2014, 231, 3997–4006. [Google Scholar] [CrossRef]
- Maurice, T.; Meunier, J.; Feng, B.; Ieni, J.; Monaghan, D.T. Interaction with Σ1 Protein, but Not N-Methyl-D-Aspartate Receptor, Is Involved in the Pharmacological Activity of Donepezil. J. Pharmacol. Exp. Ther. 2006, 317, 606–614. [Google Scholar] [CrossRef]
- Perlis, M.L.; Smith, M.T.; Orff, H.J.; Andrews, P.J.; Gillin, J.C.; Giles, D.E. The Effects of an Orally Administered Cholinergic Agonist on REM Sleep in Major Depression. Biol. Psychiatry 2002, 51, 457–462. [Google Scholar] [CrossRef]
- Fitzgerald, P.J.; Hale, P.J.; Ghimire, A.; Watson, B.O. The Cholinesterase Inhibitor Donepezil Has Antidepressant-like Properties in the Mouse Forced Swim Test. Transl. Psychiatry 2020, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, F.; Nadi, M.; Kiani, M.; Shahzeidi, S. Effect of Donepezil on Cognitive Disorders Due to the Selective Serotonin Reuptake Inhibitors in the Patients with Major Depressive Disorder. J. Shahid Sadoughi Univ. Med Sci. 2020, 22, 114–119. [Google Scholar] [CrossRef]
- Fitzgerald, P.J.; Hale, P.J.; Ghimire, A.; Watson, B.O. Multiple Cholinesterase Inhibitors Have Antidepressant-like Properties in the Mouse Forced Swim Test. Behav. Brain Res. 2021, 409, 113323. [Google Scholar] [CrossRef] [PubMed]
- Rozzini, L.; Chilovi, B.V.; Bertoletti, E.; Trabucchi, M.; Padovani, A. Acetylcholinesterase Inhibitors and Depressive Symptoms in Patients with Mild to Moderate Alzheimer’s Disease. Aging Clin. Exp. Res. 2007, 19, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.; Gruca, P.; Lason-Tyburkiewicz, M.; Willner, P. Antidepressant, Anxiolytic and Procognitive Effects of Rivastigmine and Donepezil in the Chronic Mild Stress Model in Rats. Psychopharmacology 2016, 233, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Spalletta, G.; Gianni, W.; Giubilei, F.; Casini, A.R.; Sancesario, G.; Caltagirone, C.; Cravello, L. Rivastigmine Patch Ameliorates Depression in Mild AD: Preliminary Evidence from a 6-Month Open-Label Observational Study. Alzheimer Dis. Assoc. Disord. 2013, 27, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Moriguchi, S.; Tagashira, H.; Fukunaga, K. Rivastigmine Improves Hippocampal Neurogenesis and Depression-like Behaviors via 5-HT1A Receptor Stimulation in Olfactory Bulbectomized Mice. Neuroscience 2014, 272, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Fayyazi Bordbar, M.R.; Talaei, A. Rivastigmine as an Effective Add-on to Standard Treatment of Veterans with Chronic Posttraumatic Stress Disorder: A Case Series. J. Clin. Psychopharmacol. 2013, 33, 706–709. [Google Scholar] [CrossRef]
- Rezaei Ardani, A.; Hosseini, G.; Fayyazi Bordbar, M.R.; Talaei, A.; Mostafavi Toroghi, H. Effect of Rivastigmine Augmentation in Treatment of Male Patients with Combat-Related Chronic Posttraumatic Stress Disorder: A Randomized Controlled Trial. J. Clin. Psychopharmacol. 2017, 37, 54–60. [Google Scholar] [CrossRef]
- McAllister, T.W.; Zafonte, R.; Jain, S.; Flashman, L.A.; George, M.S.; Grant, G.A.; He, F.; Lohr, J.B.; Andaluz, N.; Summerall, L.; et al. Randomized Placebo-Controlled Trial of Methylphenidate or Galantamine for Persistent Emotional and Cognitive Symptoms Associated with PTSD and/or Traumatic Brain Injury. Neuropsychopharmacology 2016, 41, 1191–1198. [Google Scholar] [CrossRef]
- Holtzheimer, P.E.; Meeks, T.W.; Kelley, M.E.; Mufti, M.; Young, R.; McWhorter, K.; Vito, N.; Chismar, R.; Quinn, S.; Dey, S.; et al. A Double Blind, Placebo-Controlled Pilot Study of Galantamine Augmentation of Antidepressant Treatment in Older Adults with Major Depression. Int. J. Geriatr. Psychiatry 2008, 23, 625–631. [Google Scholar] [CrossRef]
- Elgamal, S.; MacQueen, G. Galantamine as an Adjunctive Treatment in Major Depression. J. Clin. Psychopharmacol. 2008, 28, 357–359. [Google Scholar] [CrossRef]
- Alagiakrishnan, K. Galantamine in the Treatment of Minor Depression with Mild to Moderate Alzheimer’s Dementia in an Elderly Woman. Prim. Care Companion J. Clin. Psychiatry 2010, 12, PCC.09l00905. [Google Scholar] [CrossRef]
- Elgamal, S.A.; Marriott, M.; MacQueen, G.M. Electroencephalographic Effects of Galantamine in Major Depressive Disorder. J. Clin. Neurophysiol. 2009, 26, 192–197. [Google Scholar] [CrossRef]
- Du, Y.; Liang, H.; Zhang, L.; Fu, F. Administration of Huperzine a Exerts Antidepressant-like Activity in a Rat Model of Post-Stroke Depression. Pharmacol. Biochem. Behav. 2017, 158, 32–38. [Google Scholar] [CrossRef]
- Zheng, W.; Xiang, Y.Q.; Ungvari, G.S.; Chiu, H.F.K.; Ng, C.H.; Wang, Y.; Xiang, Y.T. Huperzine A for Treatment of Cognitive Impairment in Major Depressive Disorder: A Systematic Review of Randomized Controlled Trials. Shanghai Arch. Psychiatry 2016, 28, 64–71. [Google Scholar] [CrossRef]
- Smart, C.; McAllister-Williams, H.; Cousins, D.A. Acetylcholinesterase Inhibitors in Treatment-Resistant Psychotic Depression. Ther. Adv. Psychopharmacol. 2018, 8, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Burt, T.; Sachs, G.S.; Demopulos, C. Donepezil in Treatment-Resistant Bipolar Disorder. Biol. Psychiatry 1999, 45, 959–964. [Google Scholar] [CrossRef]
- Eden Evins, A.; Demopulos, C.; Nierenberg, A.; Culhane, M.A.; Eisner, L.; Sachs, G. A Double-Blind, Placebo-Controlled Trial of Adjunctive Donepezil in Treatment-Resistant Mania. Bipolar Disord. 2006, 8, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lu, Z.; Zhang, M.; Zhang, J.; Ni, X.; Jiang, X.; Jiang, X.; Xu, H.; Heeramun-Aubeeluck, A.; Hu, Q.; et al. A Randomized, 4-Week Double-Blind Placebo Control Study on the Efficacy of Donepezil Augmentation of Lithium for Treatment of Acute Mania. Neuropsychiatr. Dis. Treat. 2013, 9, 839–845. [Google Scholar] [CrossRef]
- Wicklund, S.; Wright, M. Donepezil-Induced Mania. J. Neuropsychiatry Clin. Neurosci. 2012, 24, 191–195. [Google Scholar] [CrossRef]
- Benazzi, F. Mania Associated with Donepezil. J. Psychiatry Neurosci. 1999, 24, 468–469. [Google Scholar] [CrossRef]
- Hategan, A.; Bourgeois, J.A. Donepezil-Associated Manic Episode with Psychotic Features: A Case Report and Review of the Literature. Gen. Hosp. Psychiatry 2016, 38, 115.e1–115.e4. [Google Scholar] [CrossRef] [PubMed]
- Bateman, D.R.; Bonacci, D.D.; Bartels, S. Mania, Bipolar Disorder and Cholinesterase Inhibitors. Am. J. Geriatr. Psychiatry 2013, 21, S74–S75. [Google Scholar] [CrossRef]
- Collins, C.; Copeland, B.; Croucher, M. Bipolar Affective Disorder, Type II, Apparently Precipitated by Donepezil. Int. Psychogeriatr. 2011, 23, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T. Is Donepezil Useful for Improving Cognitive Dysfunction in Bipolar Disorder? J. Affect. Disord. 2008, 107, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Gildengers, A.G.; Butters, M.A.; Chisholm, D.; Reynolds, C.F.; Mulsant, B.H. A 12-Week Open-Label Pilot Study of Donepezil for Cognitive Functioning and Instrumental Activities of Daily Living in Late-Life Bipolar Disorder. Int. J. Geriatr. Psychiatry 2008, 23, 693–698. [Google Scholar] [CrossRef]
- Tseng, W.S.; Tzeng, N.-S. A Rivastigmine-Precipitated Manic Episode in a Patient with Alzheimer-Type Dementia. Int. Psychogeriatr. 2012, 24, 1697–1699. [Google Scholar] [CrossRef]
- Keshavrzi, A.; Rezaei, H.; Haghighi, M.; Jahangard, L. Effect of Rivastigmine (Acetyl Cholinesterase Inhibitor) versus Placebo on Manic Episodes in Patients with Bipolar Disorders: Results from a Double Blind, Randomized, Placebo-Controlled Clinical Trial. Neuropsychobiology 2019, 78, 200–208. [Google Scholar] [CrossRef]
- Morana, P.; Mucci, F.; Baroni, S.; Della Vecchia, A.; Piccinni, A.; Morana, B.; Marazziti, D. Effectiveness of Clozapine, Oxcarbazepine and Rivastigmine Combination in a Bipolar Disorder Patient with Initial Cerebral Atrophy. Clin. Case Rep. 2020, 8, 254–257. [Google Scholar] [CrossRef]
- Ehrt, U.; Fritze, F.; Aarsland, D. Mania after Administration of Cholinesterase Inhibitors in Patients with Dementia and Comorbid Bipolar Disorder: Two Case Reports. J. Clin. Psychopharmacol. 2011, 31, 254–256. [Google Scholar] [CrossRef]
- Ghaemi, S.N.; Gilmer, W.S.; Dunn, R.T.; Hanlon, R.E.; Kemp, D.E.; Bauer, A.D.; Chriki, L.; Filkowski, M.M.; Harvey, P.D. A Double-Blind, Placebo-Controlled Pilot Study of Galantamine to Improve Cognitive Dysfunction in Minimally Symptomatic Bipolar Disorder. J. Clin. Psychopharmacol. 2009, 29, 291–295. [Google Scholar] [CrossRef]
- Iosifescu, D.V.; Moore, C.M.; Deckersbach, T.; Tilley, C.A.; Ostacher, M.J.; Sachs, G.S.; Nierenberg, A.A. Galantamine-ER for Cognitive Dysfunction in Bipolar Disorder and Correlation with Hippocampal Neuronal Viability: A Proof-of-Concept Study. CNS Neurosci. Ther. 2009, 15, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, E.; Ghaemi, S.N. Galantamine Treatment of Cognitive Impairment in Bipolar Disorder: Four Cases. Bipolar Disord. 2006, 8, 196–199. [Google Scholar] [CrossRef]
- Dias, V.V.; Brissos, S.; Gorman, J.M. Adjuvant Galantamine for Cognitive Dysfunction in a Patient with Bipolar Disorder. J. Psychiatr. Pract. 2006, 12, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.C.; Rocha, J.B.T.; Morsch, V.M.; Neis, R.T.; Schetinger, M.R.C. Antidepressants Inhibit Human Acetylcholinesterase and Butyrylcholinesterase Activity. Biochim. Biophys. Acta Mol. Basis Dis. 2002, 1587, 92–98. [Google Scholar] [CrossRef]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From Combinations to Multitarget-Directed Ligands: A Continuum in Alzheimer’s Disease Polypharmacology. Med. Res. Rev. 2020, 41, 2606–2633. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Mìnarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-Target Drugs for Alzheimer’s Disease. Trends Pharm. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Zagórska, A.; Jaromin, A. Perspectives for New and More Efficient Multifunctional Ligands for Alzheimer’s Disease Therapy. Molecules 2020, 25, 3337. [Google Scholar] [CrossRef]
- Donepezil|C24H29NO3—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/3152#section=2D-Structure (accessed on 25 April 2021).
- Zarei, S.; Shafiei, M.; Firouzi, M.; Firoozpour, L.; Divsalar, K.; Asadipour, A.; Akbarzadeh, T.; Foroumadi, A. Design, Synthesis and Biological Assessment of New 1-Benzyl-4-((4-Oxoquinazolin-3(4H)-Yl)Methyl) Pyridin-1-Ium Derivatives (BOPs) as Potential Dual Inhibitors of Acetylcholinesterase and Butyrylcholinesterase. Heliyon 2021, 7, e06683. [Google Scholar] [CrossRef]
- Angona, I.P.; Martin, H.; Daniel, S.; Moraleda, I.; Bonet, A.; Wnorowski, A.; Maj, M.; Jozwiak, K.; Iriepa, I.; Refouvelet, B.; et al. Synthesis of Hantzsch Adducts as Cholinesterases and Calcium Flux Inhibitors, Antioxidants and Neuroprotectives. Int. J. Mol. Sci. 2020, 21, 7652. [Google Scholar] [CrossRef]
- Lecoutey, C.; Legay, R.; Davis, A.; Santos, J.S.D.O.; Dallemagne, P.; Rochais, C. Development of Novel Potential Pleiotropic Compounds of Interest in Alzheimer’s Disease Treatment through Rigidification Strategy. Molecules 2021, 26, 2536. [Google Scholar] [CrossRef]
- Tacrine|C13H14N2—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tacrine#section=Computed-Properties (accessed on 27 June 2021).
- Eckroat, T.J.; Manross, D.L.; Cowan, S.C. Merged Tacrine-Based, Multitarget-Directed Acetylcholinesterase Inhibitors 2015–Present: Synthesis and Biological Activity. Int. J. Mol. Sci. 2020, 21, 5965. [Google Scholar] [CrossRef]
- Wan, L.X.; Zhen, Y.Q.; He, Z.X.; Zhang, Y.; Zhang, L.; Li, X.; Gao, F.; Zhou, X.L. Late-Stage Modification of Medicine: Pd-Catalyzed Direct Synthesis and Biological Evaluation of N-Aryltacrine Derivatives. ACS Omega 2021, 6, 9960–9972. [Google Scholar] [CrossRef]
- Rajeshwari, R.; Chand, K.; Candeias, E.; Cardoso, S.M.; Chaves, S.; Amélia Santos, M. New Multitarget Hybrids Bearing Tacrine and Phenylbenzothiazole Motifs as Potential Drug Candidates for Alzheimer’s Disease. Molecules 2019, 24, 587. [Google Scholar] [CrossRef]
- Fancellu, G.; Chand, K.; Tomás, D.; Orlandini, E.; Piemontese, L.; Silva, D.F.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel Tacrine–Benzofuran Hybrids as Potential Multi-Target Drug Candidates for the Treatment of Alzheimer’s Disease. J. Enzym. Inhib. Med. Chem. 2020, 35, 211–226. [Google Scholar] [CrossRef]
- Rampa, A.; Bartolini, M.; Pruccoli, L.; Naldi, M.; Iriepa, I.; Moraleda, I.; Belluti, F.; Gobbi, S.; Tarozzi, A.; Bisi, A. Exploiting the Chalcone Scaffold to Develop Multifunctional Agents for Alzheimer’s Disease. Molecules 2018, 23, 1902. [Google Scholar] [CrossRef]
- Hwang, J.; Youn, K.; Lim, G.; Lee, J.; Kim, D.H.; Jun, M. Discovery of Natural Inhibitors of Cholinesterases from Hydrangea: In Vitro and in Silico Approaches. Nutrients 2021, 13, 254. [Google Scholar] [CrossRef]
- Wang, Q.; Matsuda, H.; Matsuhira, K.; Nakamura, S.; Yuan, D.; Yoshikawa, M. Inhibitory Effects of Thunberginols A, B, and F on Degranulations and Releases of TNF-α and IL-4 in RBL-2H3 Cells. Biol. Pharm. Bull. 2007, 30, 388–392. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, C.H.; Jayasooriya, R.G.P.T.; Dilshara, M.G.; Lee, S.; Choi, Y.H.; Seo, Y.T.; Kim, G.Y. Hydrangenol Inhibits Lipopolysaccharide-Induced Nitric Oxide Production in BV2 Microglial Cells by Suppressing the NF-ΚB Pathway and Activating the Nrf2-Mediated HO-1 Pathway. Int. Immunopharmacol. 2016, 35, 61–69. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, J.; Meng, A. Protective Effects of Hydrangenol on PC12 Cells Injury Induced by Oxygen and Glucose Deprivation-Reoxygenation. Am. J. Pharm 2017, 36, 479–485. [Google Scholar]
- Akanda, M.R.; Tae, H.J.; Kim, I.S.; Ahn, D.; Tian, W.; Islam, A.; Nam, H.H.; Choo, B.K.; Park, B.Y. Hepatoprotective Role of Hydrangea Macrophylla against Sodium Arsenite-Induced Mitochondrial-Dependent Oxidative Stress via the Inhibition of MAPK/Caspase-3 Pathways. Int. J. Mol. Sci. 2017, 18, 1482. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, J.; Sheng, L.; Zhang, D.; Chen, X.; Yang, J.; Wang, D. Total Coumarins from Hydrangea Paniculata Show Renal Protective Effects in Lipopolysaccharide-Induced Acute Kidney Injury via Anti-Inflammatory and Antioxidant Activities. Front. Pharmacol. 2017, 8, 872. [Google Scholar] [CrossRef]
- Frandsen, J.R.; Narayanasamy, P. Neuroprotection through Flavonoid: Enhancement of the Glyoxalase Pathway. Redox Biol. 2018, 14, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Esmaeilizadeh, M. Pharmacological Properties of Salvia officinalis and Its Components. J. Tradit. Complementary Med. 2017, 7, 433–440. [Google Scholar] [CrossRef]
- Bahadori, M.B.; Dinparast, L.; Zengin, G.; Sarikurkcu, C.; Bahadori, S.; Asghari, B.; Movahhedin, N. Functional Components, Antidiabetic, Anti-Alzheimer’s Disease, and Antioxidant Activities of Salvia syriaca L. Int. J. Food Prop. 2017, 20, 1761–1772. [Google Scholar] [CrossRef]
- Smach, M.A.; Hafsa, J.; Charfeddine, B.; Dridi, H.; Limem, K. Effects of Sage Extract on Memory Performance in Mice and Acetylcholinesterase Activity. Ann. Pharm. Fr. 2015, 73, 281–288. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Noroozian, M.; Mohammadi, M.; Ohadinia, S.; Jamshidi, A.H.; Khani, M. Salvia officinalis Extract in the Treatment of Patients with Mild to Moderate Alzheimer’s Disease: A Double Blind, Randomized and Placebo-Controlled Trial. J. Clin. Pharm. Ther. 2003, 28, 53–59. [Google Scholar] [CrossRef]
- Kennedy, D.O.; Pace, S.; Haskell, C.; Okello, E.J.; Milne, A.; Scholey, A.B. Effects of Cholinesterase Inhibiting Sage (Salvia officinalis) on Mood, Anxiety and Performance on a Psychological Stressor Battery. Neuropsychopharmacology 2006, 31, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Tober, C.; Schoop, R. Modulation of Neurological Pathways by Salvia officinalis and Its Dependence on Manufacturing Process and Plant Parts Used. BMC Complementary Altern. Med. 2019, 19, s12906–s13019. [Google Scholar] [CrossRef]
- Leporini, M.; Bonesi, M.; Loizzo, M.R.; Passalacqua, N.G.; Tundis, R. The Essential Oil of Salvia Rosmarinus Spenn. From Italy as a Source of Health-Promoting Compounds: Chemical Profile and Antioxidant and Cholinesterase Inhibitory Activity. Plants 2020, 9, 798. [Google Scholar] [CrossRef]
- Adsersen, A.; Gauguin, B.; Gudiksen, L.; Jäger, A.K. Screening of Plants Used in Danish Folk Medicine to Treat Memory Dysfunction for Acetylcholinesterase Inhibitory Activity. J. Ethnopharmacol. 2006, 104, 418–422. [Google Scholar] [CrossRef]
- Cutillas, A.B.; Carrasco, A.; Martinez-Gutierrez, R.; Tomas, V.; Tudela, J. Rosmarinus officinalis L. Essential Oils from Spain: Composition, Antioxidant Capacity, Lipoxygenase and Acetylcholinesterase Inhibitory Capacities, and Antimicrobial Activities. Plant Biosyst. 2018, 152, 1282–1292. [Google Scholar] [CrossRef]
- Meng, P.; Yoshida, H.; Matsumiya, T.; Imaizumi, T.; Tanji, K.; Xing, F.; Hayakari, R.; Dempoya, J.; Tatsuta, T.; Aizawa-Yashiro, T.; et al. Carnosic Acid Suppresses the Production of Amyloid-β 1-42 by Inducing the Metalloprotease Gene TACE/ADAM17 in SH-SY5Y Human Neuroblastoma Cells. Neurosci. Res. 2013, 75, 94–102. [Google Scholar] [CrossRef]
- Cornejo, A.; Aguilar Sandoval, F.; Caballero, L.; Machuca, L.; Muñoz, P.; Caballero, J.; Perry, G.; Ardiles, A.; Areche, C.; Melo, F. Rosmarinic Acid Prevents Fibrillization and Diminishes Vibrational Modes Associated to β Sheet in Tau Protein Linked to Alzheimer’s Disease. J. Enzym. Inhib. Med. Chem. 2017, 32, 945–953. [Google Scholar] [CrossRef]
- Perry, N.S.L.; Bollen, C.; Perry, E.K.; Ballard, C. Salvia for Dementia Therapy: Review of Pharmacological Activity and Pilot Tolerability Clinical Trial. Pharmacol. Biochem. Behav. 2003, 75, 651–659. [Google Scholar] [CrossRef]
- Kennedy, D.O.; Dodd, F.L.; Robertson, B.C.; Okello, E.J.; Reay, J.L.; Scholey, A.B.; Haskell, C.F. Monoterpenoid Extract of Sage (Salvia lavandulaefolia) with Cholinesterase Inhibiting Properties Improves Cognitive Performance and Mood in Healthy Adults. J. Psychopharmacol. 2011, 25, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Gürbüz, P.; Dokumacı, A.H.; Gündüz, M.G.; Perez, C.; Göger, F.; Paksoy, M.Y.; Yerer, M.B.; Ömür Demirezer, L. In Vitro Biological Activity of Salvia Fruticosa Mill. Infusion against Amyloid β-Peptide-Induced Toxicity and Inhibition of GSK-3β, CK-1δ, and BACE-1 Enzymes Relevant to Alzheimer’s Disease. Saudi Pharm. J. 2021, 29, 236–243. [Google Scholar] [CrossRef]
- Ververis, A.; Savvidou, G.; Ioannou, K.; Nicolaou, P.; Christodoulou, K.; Plioukas, M. Greek Sage Exhibits Neuroprotective Activity against Amyloid Beta-Induced Toxicity. Evid. Based Complementary Altern. Med. 2020, 2020, 2975284. [Google Scholar] [CrossRef] [PubMed]
- Şenol, F.S.; Orhan, I.E.; Erdem, S.A.; Kartal, M.; Şener, B.; Kan, Y.; Celep, F.; Kahraman, A.; Dogan, M. Evaluation of Cholinesterase Inhibitory and Antioxidant Activities of Wild and Cultivated Samples of Sage (Salvia fruticosa) by Activity-Guided Fractionation. J. Med. Food 2011, 14, 1476–1483. [Google Scholar] [CrossRef]
- Tel, G.; Öztürk, M.; Duru, M.E.; Harmandar, M.; Topçu, G. Chemical Composition of the Essential Oil and Hexane Extract of Salvia Chionantha and Their Antioxidant and Anticholinesterase Activities. Food Chem. Toxicol. 2010, 48, 3189–3193. [Google Scholar] [CrossRef]
- Bahadori, M.B.; Salehi, P.; Sonboli, A. Comparative Study of the Essential Oil Composition of Salvia Urmiensis and Its Enzyme Inhibitory Activities Linked to Diabetes Mellitus and Alzheimer’s Disease. Int. J. Food Prop. 2017, 20, 2974–2981. [Google Scholar] [CrossRef]
- Çulhaoglu, B.; Yapara, G.; Dirmencic, T.; Topçu, G. Bioactive Constituents of Salvia Chrysophylla Stapf. Nat. Prod. Res. 2013, 27, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Kumar Durairajan, S.S.; Chirasani, V.R.; Shetty, S.G.; Iyaswamy, A.; Malampati, S.; Song, J.; Liu, L.; Huang, J.; Senapati, S.; Li, M. Decrease in the Generation of Amyloid-β Due to Salvianolic Acid B by Modulating BACE1 Activity. Curr. Alzheimer Res. 2017, 14, 1229–1237. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, X.; Tang, M.; Zhang, J.; Zhang, B.; Zhao, Q.; Han, Y.; Yan, W.; Peng, C.; You, Z. Salvianolic Acid B Promotes Microglial M2-Polarization and Rescues Neurogenesis in Stress-Exposed Mice. Brain Behav. Immun. 2017, 66, 111–124. [Google Scholar] [CrossRef]
- Liao, D.; Chen, Y.; Guo, Y.; Wang, C.; Liu, N.; Gong, Q.; Fu, Y.; Fu, Y.; Cao, L.; Yao, D.; et al. Salvianolic Acid b Improves Chronic Mild Stress-Induced Depressive Behaviors in Rats: Involvement of AMPK/SIRT1 Signaling Pathway. J. Inflamm. Res. 2020, 13, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, F.X.; Li, N.; Cao, Y.; Lei, Y.; Wang, J.Z.; Tian, Q.; Zhou, X.W. AMPK Ameliorates Tau Acetylation and Memory Impairment Through Sirt1. Mol. Neurobiol. 2020, 57, 5011–5025. [Google Scholar] [CrossRef] [PubMed]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson’s Disease. J. Parkinson’s Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, C.; Kong, P.; Bian, G.; Sun, Z.; Sun, Y.; Guo, L.; Li, B. Methylene Blue Alleviates Experimental Autoimmune Encephalomyelitis by Modulating AMPK/SIRT1 Signaling Pathway and Th17/Treg Immune Response. J. Neuroimmunol. 2016, 299, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Tulino, R.; Benjamin, A.C.; Jolinon, N.; Smith, D.L.; Chini, E.N.; Carnemolla, A.; Bates, G.P. SIRT1 Activity Is Linked to Its Brain Region-Specific Phosphorylation and Is Impaired in Huntington’s Disease Mice. PLoS ONE 2016, 11, e0145425. [Google Scholar] [CrossRef]
- Yun, Y.C.; Jeong, S.G.; Kim, S.H.; Cho, G.W. Reduced Sirtuin 1/Adenosine Monophosphate-Activated Protein Kinase in Amyotrophic Lateral Sclerosis Patient-Derived Mesenchymal Stem Cells Can Be Restored by Resveratrol. J. Tissue Eng. Regen. Med. 2019, 13, 110–115. [Google Scholar] [CrossRef]
- Lee, Y.W.; Kim, D.H.; Jeon, S.J.; Park, S.J.; Kim, J.M.; Jung, J.M.; Lee, H.E.; Bae, S.G.; Oh, H.K.; Son, K.H.H.; et al. Neuroprotective Effects of Salvianolic Acid B on an Aβ25-35 Peptide-Induced Mouse Model of Alzheimer’s Disease. Eur. J. Pharmacol. 2013, 704, 70–77. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, S.J.; Kim, J.M.; Jeon, S.J.; Kim, D.H.; Cho, Y.W.; Son, K.H.; Lee, H.J.; Moon, J.H.; Cheong, J.H.; et al. Cognitive Dysfunctions Induced by a Cholinergic Blockade and Aβ 25-35 Peptide Are Attenuated by Salvianolic Acid B. Neuropharmacology 2011, 61, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Ghanem, S.S.; Abdulla, S.A.; Lv, G.; Emara, M.M.; Paleologou, K.E.; Vaikath, N.N.; Lu, J.H.; Li, M.; Vekrellis, K.; et al. Inhibition of Alpha-Synuclein Seeded Fibril Formation and Toxicity by Herbal Medicinal Extracts. BMC Complementary Med. Ther. 2020, 20, 73. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Zhao, Y.; Yu, T.; Wang, Z.; Gong, H.; Yang, X.; Liu, Y.; Huang, K. Inhibition Effects of Tanshinone on the Aggregation of α-Synuclein. Food Funct. 2016, 7, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Li, C.; Xiang, Z.; Jiao, B. Tanshinone IIA Reduces the Risk of Alzheimer’s Disease by Inhibiting INOS, MMP-2 and NF-ΚBp65 Transcription and Translation in the Temporal Lobes of Rat Models of Alzheimer’s Disease. Mol. Med. Rep. 2014, 10, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Liu, W.; Chen, Y. Tanshinone IIA Attenuates Aβ-Induced Neurotoxicity by down-Regulating COX-2 Expression and PGE2 Synthesis via Inactivation of NF-ΚB Pathway in SH-SY5Y Cells. J. Biol. Res. 2019, 26, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, Y.; Zha, S.; Liu, M.; Wang, Y.; Yang, G.; Ma, K.; Fei, Y.; Zhang, Y.; Hu, X.; et al. Treatment Effects of Tanshinone IIA against Intracerebroventricular Streptozotocin Induced Memory Deficits in Mice. Brain Res. 2016, 1631, 137–146. [Google Scholar] [CrossRef]
- Santos, J.A.; Piccinelli, A.C.; Formagio, M.D.; Oliveira, C.S.; Santos, E.P.; Dos Stefanello, M.É.A.; Junior, U.L.; Oliveira, R.J.; Sugizaki, M.M.; Kassuya, C.A.L. Antidepressive and Antinociceptive Effects of Ethanolic Extract and Fruticuline a from Salvia Lachnostachys Benth Leaves on Rodents. PLoS ONE 2017, 12, e0172151. [Google Scholar] [CrossRef]
- Sul, D.; Kim, H.S.; Lee, D.; Joo, S.S.; Hwang, K.W.; Park, S.Y. Protective Effect of Caffeic Acid against Beta-Amyloid-Induced Neurotoxicity by the Inhibition of Calcium Influx and Tau Phosphorylation. Life Sci. 2009, 84, 257–262. [Google Scholar] [CrossRef]
- Wightman, E.L.; Jackson, P.A.; Spittlehouse, B.; Heffernan, T.; Guillemet, D.; Kennedy, D.O. The Acute and Chronic Cognitive Effects of a Sage Extract: A Randomized, Placebo Controlled Study in Healthy Humans. Nutrients 2021, 13, 218. [Google Scholar] [CrossRef]
- Dinel, A.L.; Lucas, C.; Guillemet, D.; Layé, S.; Pallet, V.; Joffre, C. Chronic Supplementation with a Mix of Salvia officinalis and Salvia Lavandulaefolia Improves Morris Water Maze Learning in Normal Adult C57BL/6J Mice. Nutrients 2020, 12, 1777. [Google Scholar] [CrossRef]
- Perry, N.S.L.; Menzies, R.; Hodgson, F.; Wedgewood, P.; Howes, M.J.R.; Brooker, H.J.; Wesnes, K.A.; Perry, E.K. A Randomised Double-Blind Placebo-Controlled Pilot Trial of a Combined Extract of Sage, Rosemary and Melissa, Traditional Herbal Medicines, on the Enhancement of Memory in Normal Healthy Subjects, Including Influence of Age. Phytomedicine 2018, 39, 42–48. [Google Scholar] [CrossRef]
- Kook, M.G.; Choi, S.W.; Seo, Y.; Kim, D.W.; Song, B.K.; Son, I.; Kim, S.; Kang, K.S. KCHO-1, a Novel Herbal Anti-Inflammatory Compound, Attenuates Oxidative Stress in an Animal Model of Amyotrophic Lateral Sclerosis. J. Vet. Sci. 2017, 18, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, J.K.; Son, M.J.; Kim, D.; Song, B.; Son, I.; Kang, H.W.; Lee, J.; Kim, S. Mecasin Treatment in Patients with Amyotrophic Lateral Sclerosis: Study Protocol for a Randomized Controlled Trial. Trials 2018, 19, 225. [Google Scholar] [CrossRef]
- Lin, C.L.; Wang, S.E.; Hsu, C.H.; Sheu, S.J.; Wu, C.H. Oral Treatment with Herbal Formula B307 Alleviates Cardiac Failure in Aging R6/2 Mice with Huntington’s Disease via Suppressing Oxidative Stress, Inflammation, and Apoptosis. Clin. Interv. Aging 2015, 10, 1173–1187. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Search of: Salvia officinalis—Results by Topic—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/results/browse?cond=Salvia+officinalis&brwse=cond_alpha_all (accessed on 23 August 2021).
- Bonesi, M.; Tenuta, M.C.; Loizzo, M.R.; Sicari, V.; Tundis, R. Potential Application of Prunus Armeniaca L. and P. Domestica L. Leaf Essential Oils as Antioxidant and of Cholinesterases Inhibitors. Antioxidants 2019, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Morabbi Najafabad, A.; Jamei, R. Free Radical Scavenging Capacity and Antioxidant Activity of Methanolic and Ethanolic Extracts of Plum (Prunus domestica L.) in Both Fresh and Dried Samples. Avicenna J. Phytomed. 2014, 4, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.U.; Amin, R.; Shahid, M.; Amin, M.; Zaib, S.; Iqbal, J. A Multi-Target Therapeutic Potential of Prunus Domestica Gum Stabilized Nanoparticles Exhibited Prospective Anticancer, Antibacterial, Urease-Inhibition, Anti-Inflammatory and Analgesic Properties. BMC Complementary Altern. Med. 2017, 17, s12906–s13017. [Google Scholar] [CrossRef] [PubMed]
- Bouayed, J.; Rammal, H.; Dicko, A.; Younos, C.; Soulimani, R. Chlorogenic Acid, a Polyphenol from Prunus Domestica (Mirabelle), with Coupled Anxiolytic and Antioxidant Effects. J. Neurol. Sci. 2007, 262, 77–84. [Google Scholar] [CrossRef]
- Shahidi, S.; Setareye, S.; Mahmoodi, M. Effect of Prunus domestica L. (Mirabelle) on Learning and Memory in Mice. Anc. Sci. Life 2013, 32, 139. [Google Scholar] [CrossRef]
- Vahedi-Mazdabadi, Y.; Karimpour-Razkenari, E.; Akbarzadeh, T.; Lotfian, H.; Toushih, M.; Roshanravan, N.; Saeedi, M.; Ostadrahimi, A. In Vitro Anti-Cholinesterase and Neuroprotective Activities of Sweet and Bitter Apricot Kernels (Prunus armeniaca L.). Iran. J. Pharm. Res. 2020, 19, 216–224. [Google Scholar] [CrossRef]
- Dauthal, P.; Mukhopadhyay, M. In-Vitro Free Radical Scavenging Activity of Biosynthesized Gold and Silver Nanoparticles Using Prunus Armeniaca (Apricot) Fruit Extract Nanomaterials in Energy, Health and Environment. Guest Editors: Puru Jena, Samy El Shall, Anil Kandalam. J. Nanoparticle Res. 2013, 15, 1–11. [Google Scholar] [CrossRef]
- Minaiyan, M.; Ghannadi, A.; Asadi, M.; Etemad, M.; Mahzouni, P. Anti-Inflammatory Effect of Prunus Armeniaca L. (Apricot) Extracts Ameliorates TNBS-Induced Ulcerative Colitis in Rats. Res. Pharm. Sci. 2014, 9, 225–231. [Google Scholar] [PubMed]
- Hwang, H.J.; Kim, P.; Kim, C.J.; Lee, H.J.; Shim, I.; Yin, C.S.; Yang, Y.; Hahm, D.H. Antinociceptive Effect of Amygdalin Isolated from Prunus Armeniaca on Formalin-Induced Pain in Rats. Biol. Pharm. Bull. 2008, 31, 1559–1564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kulkarni, K.S.; Kasture, S.B.; Mengi, S.A. Efficacy Study of Prunus Amygdalus (Almond) Nuts in Scopolamine-Induced Amnesia in Rats. Indian J. Pharmacol. 2010, 42, 168–173. [Google Scholar] [CrossRef]
- Pinelo, M.; Rubilar, M.; Sineiro, J.; Núñez, M.J. Extraction of Antioxidant Phenolics from Almond Hulls (Prunus Amygdalus) and Pine Sawdust (Pinus pinaster). Food Chem. 2004, 85, 267–273. [Google Scholar] [CrossRef]
- Kim, M.S.; Jeon, W.K.; Lee, K.W.; Park, Y.H.; Han, J.S. Ameliorating Effects of Ethanol Extract of Fructus Mume on Scopolamine-Induced Memory Impairment in Mice. Evid. Based Complementary Altern. Med. 2015, 2015, 102734. [Google Scholar] [CrossRef]
- Park, J.C.; Ma, J.; Jeon, W.K.; Han, J.S. Fructus Mume Extracts Alleviate Cognitive Impairments in 5XFAD Transgenic Mice. BMC Complementary Altern. Med. 2016, 16, s12906–s13016. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.K.; Ma, J.; Choi, B.R.; Han, S.H.; Jin, Q.; Hwang, B.Y.; Han, J.S. Effects of Fructus Mume Extract on MAPK and NF-ΚB Signaling and the Resultant Improvement in the Cognitive Deficits Induced by Chronic Cerebral Hypoperfusion. Evid.-Based Complementary Altern. Med. 2012, 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Bang, J.H.; Lee, J.; Han, J.S.; Kang, H.W.; Jeon, W.K. Fructus Mume Ethanol Extract Prevents Inflammation and Normalizes the Septohippocampal Cholinergic System in a Rat Model of Chronic Cerebral Hypoperfusion. J. Med. Food 2016, 19, 196–204. [Google Scholar] [CrossRef]
- Coppari, S.; Colomba, M.; Fraternale, D.; Brinkmann, V.; Romeo, M.; Rocchi, M.B.L.; Di Giacomo, B.; Mari, M.; Guidi, L.; Ramakrishna, S.; et al. Antioxidant and Anti-inflammaging Ability of Prune (Prunus spinosa L.) Extract Result in Improved Wound Healing Efficacy. Antioxidants 2021, 10, 374. [Google Scholar] [CrossRef]
- Pozzo, L.; Russo, R.; Frassinetti, S.; Vizzarri, F.; Árvay, J.; Vornoli, A.; Casamassima, D.; Palazzo, M.; Della Croce, C.M.; Longo, V. Wild Italian Prunus Spinosa L. Fruit Exerts in vitro Antimicrobial Activity and Protects against in Vitro and in vivo Oxidative Stress. Foods 2020, 9, 5. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Li, N.; Xu, B.; Duan, J.; Yuan, C.; Yuan, Q.; Yang, Q.; Qing, H.; Dai, Z.; et al. The Anti-Depression-Like Effects of Zhengtian Capsule via Induction of Neurogenesis and the Neurotrophic Signaling Pathway. Front. Pharmacol. 2020, 11, 01338. [Google Scholar] [CrossRef] [PubMed]
- Tohda, C.; Tamura, T.; Komatsu, K. Repair of Amyloid β(25-35)-Induced Memory Impairment and Synaptic Loss by a Kampo Formula, Zokumei-To. Brain Res. 2003, 990, 141–147. [Google Scholar] [CrossRef]
- Abou Baker, D.H.; Ibrahim, B.M.M.; Hassan, N.S.; Yousuf, A.F.; Gengaihi, S. El Exploiting Citrus Aurantium Seeds and Their Secondary Metabolites in the Management of Alzheimer Disease. Toxicol. Rep. 2020, 7, 723–729. [Google Scholar] [CrossRef]
- Tundis, R.; Loizzo, M.R.; Bonesi, M.; Menichini, F.; Mastellone, V.; Colica, C.; Menichini, F. Comparative Study on the Antioxidant Capacity and Cholinesterase Inhibitory Activity of Citrus Aurantifolia Swingle, C. aurantium, L., and C. bergamia Risso and Poit. Peel Essential Oils. J. Food Sci. 2012, 77, H40–H46. [Google Scholar] [CrossRef]
- Loizzo, M.R.; Tundis, R.; Bonesi, M.; Menichini, F.; De Luca, D.; Colica, C.; Menichini, F. Evaluation of Citrus Aurantifolia Peel and Leaves Extracts for Their Chemical Composition, Antioxidant and Anti-Cholinesterase Activities. J. Sci. Food Agric. 2012, 92, 2960–2967. [Google Scholar] [CrossRef] [PubMed]
- Heo, H.J.; Kim, M.J.; Lee, J.M.; Choi, S.J.; Cho, H.Y.; Hong, B.; Kim, H.K.; Kim, E.; Shin, D.H. Naringenin from Citrus Junos Has an Inhibitory Effect on Acetylcholinesterase and a Mitigating Effect on Amnesia. Dement. Geriatr. Cogn. Disord. 2004, 17, 151–157. [Google Scholar] [CrossRef]
- Zaki, H.F.; Abd-El-Fattah, M.A.; Attia, A.S. Naringenin Protects against Scopolamine-Induced Dementia in Rats. Bull. Fac. Pharm. Cairo Univ. 2014, 52, 15–25. [Google Scholar] [CrossRef]
- Zaidun, N.H.; Thent, Z.C.; Latiff, A.A. Combating Oxidative Stress Disorders with Citrus Flavonoid: Naringenin. Life Sci. 2018, 208, 111–122. [Google Scholar] [CrossRef]
- Ghofrani, S.; Joghataei, M.T.; Mohseni, S.; Baluchnejadmojarad, T.; Bagheri, M.; Khamse, S.; Roghani, M. Naringenin Improves Learning and Memory in an Alzheimer’s Disease Rat Model: Insights into the Underlying Mechanisms. Eur. J. Pharmacol. 2015, 764, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Liaquat, L.; Ahmad, S.; Batool, Z.; Siddiqui, R.A.; Tabassum, S.; Shahzad, S.; Rafiq, S.; Naz, N. Naringenin Protects AlCl3/D-Galactose Induced Neurotoxicity in Rat Model of AD via Attenuation of Acetylcholinesterase Levels and Inhibition of Oxidative Stress. PLoS ONE 2020, 15, e0227631. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.B.; Khan, M.M.; Khan, A.; Ahmed, M.E.; Ishrat, T.; Tabassum, R.; Vaibhav, K.; Ahmad, A.; Islam, F. Naringenin Ameliorates Alzheimer’s Disease (AD)-Type Neurodegeneration with Cognitive Impairment (AD-TNDCI) Caused by the Intracerebroventricular- Streptozotocin in Rat Model. Neurochem. Int. 2012, 61, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, W.Q.; Zha, H.; Yu, H.R. Effect of Naringenin on Learning and Memory Ability on Model Rats with Alzheimer Disease. J. Chin. Med. Mater. 2013, 36, 271–276. [Google Scholar]
- Khajevand-Khazaei, M.R.; Ziaee, P.; Motevalizadeh, S.A.; Rohani, M.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Roghani, M. Naringenin Ameliorates Learning and Memory Impairment Following Systemic Lipopolysaccharide Challenge in the Rat. Eur. J. Pharmacol. 2018, 826, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Md, S.; Gan, S.Y.; Haw, Y.H.; Ho, C.L.; Wong, S.; Choudhury, H. In Vitro Neuroprotective Effects of Naringenin Nanoemulsion against β-Amyloid Toxicity through the Regulation of Amyloidogenesis and Tau Phosphorylation. Int. J. Biol. Macromol. 2018, 118, 1211–1219. [Google Scholar] [CrossRef]
- Yang, Z.; Kuboyama, T.; Tohda, C. Naringenin Promotes Microglial M2 Polarization and Aβ Degradation Enzyme Expression. Phytother. Res. 2019, 33, 1114–1121. [Google Scholar] [CrossRef]
- Rahigude, A.; Bhutada, P.; Kaulaskar, S.; Aswar, M.; Otari, K. Participation of Antioxidant and Cholinergic System in Protective Effect of Naringenin against Type-2 Diabetes-Induced Memory Dysfunction in Rats. Neuroscience 2012, 226, 62–72. [Google Scholar] [CrossRef]
- Lou, H.; Jing, X.; Wei, X.; Shi, H.; Ren, D.; Zhang, X. Naringenin Protects against 6-OHDA-Induced Neurotoxicity via Activation of the Nrf2/ARE Signaling Pathway. Neuropharmacology 2014, 79, 380–388. [Google Scholar] [CrossRef]
- Sonia Angeline, M.; Sarkar, A.; Anand, K.; Ambasta, R.K.; Kumar, P. Sesamol and Naringenin Reverse the Effect of Rotenone-Induced PD Rat Model. Neuroscience 2013, 254, 379–394. [Google Scholar] [CrossRef]
- Mani, S.; Sekar, S.; Barathidasan, R.; Manivasagam, T.; Thenmozhi, A.J.; Sevanan, M.; Chidambaram, S.B.; Essa, M.M.; Guillemin, G.J.; Sakharkar, M.K. Naringenin Decreases α-Synuclein Expression and Neuroinflammation in MPTP-Induced Parkinson’s Disease Model in Mice. Neurotox. Res. 2018, 33, 656–670. [Google Scholar] [CrossRef]
- Kesh, S.; Kannan, R.R.; Balakrishnan, A. Naringenin Alleviates 6-Hydroxydopamine Induced Parkinsonism in SHSY5Y Cells and Zebrafish Model. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 239, 108893. [Google Scholar] [CrossRef]
- Chen, C.; Wei, Y.Z.; He, X.M.; Li, D.D.; Wang, G.Q.; Li, J.J.; Zhang, F. Naringenin Produces Neuroprotection against LPS-Induced Dopamine Neurotoxicity via the Inhibition of Microglial NLRP3 Inflammasome Activation. Front. Immunol. 2019, 10, 936. [Google Scholar] [CrossRef]
- Mani, M.; Balasubramanian, S.; Manikandan, K.R.; Kulandaivel, B. Neuroprotective Potential of Naringenin-Loaded Solid-Lipid Nanoparticles against Rotenone-Induced Parkinson’s Disease Model. J. Appl. Pharm. Sci. 2021, 11, 19–28. [Google Scholar] [CrossRef]
- Salman, M.; Sharma, P.; Alam, M.I.; Tabassum, H.; Parvez, S. Naringenin Mitigates Behavioral Alterations and Provides Neuroprotection against 3-Nitropropinoic Acid-Induced Huntington’s Disease like Symptoms in Rats. Nutr. Neurosci. 2021, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, Y.; Niu, X.; Tang, H.; Meydani, S.N.; Wu, D. Dietary Naringenin Supplementation Attenuates Experimental Autoimmune Encephalomyelitis by Modulating Autoimmune Inflammatory Responses in Mice. J. Nutr. Biochem. 2018, 54, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Sang, H.; Wang, J. Naringenin Attenuates Experimental Autoimmune Encephalomyelitis by Protecting the Intact of Blood-Brain Barrier and Controlling Inflammatory Cell Migration. J. Nutr. Biochem. 2021, 89, 108560. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Rajasekaran, R. Molecular Binding Response of Naringin and Naringenin to H46R Mutant SOD1 Protein in Combating Protein Aggregation Using Density Functional Theory and Discrete Molecular Dynamics. Prog. Biophys. Mol. Biol. 2019, 145, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Olugbemide, A.S.; Ben-Azu, B.; Bakre, A.G.; Ajayi, A.M.; Femi-Akinlosotu, O.; Umukoro, S. Naringenin Improves Depressive- and Anxiety-like Behaviors in Mice Exposed to Repeated Hypoxic Stress through Modulation of Oxido-Inflammatory Mediators and NF-KB/BDNF Expressions. Brain Res. Bull. 2021, 169, 214–227. [Google Scholar] [CrossRef]
- Xue, N.; Wu, X.; Wu, L.; Li, L.; Wang, F. Antinociceptive and Anti-Inflammatory Effect of Naringenin in Different Nociceptive and Inflammatory Mice Models. Life Sci. 2019, 217, 148–154. [Google Scholar] [CrossRef]
- Yi, L.T.; Li, C.F.; Zhan, X.; Cui, C.C.; Xiao, F.; Zhou, L.P.; Xie, Y. Involvement of Monoaminergic System in the Antidepressant-like Effect of the Flavonoid Naringenin in Mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 1223–1228. [Google Scholar] [CrossRef]
- Bansal, Y.; Singh, R.; Saroj, P.; Sodhi, R.K.; Kuhad, A. Naringenin Protects against Oxido-Inflammatory Aberrations and Altered Tryptophan Metabolism in Olfactory Bulbectomized-Mice Model of Depression. Toxicol. Appl. Pharmacol. 2018, 355, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.T.; Li, J.; Li, H.C.; Su, D.X.; Quan, X.B.; He, X.C.; Wang, X.H. Antidepressant-like Behavioral, Neurochemical and Neuroendocrine Effects of Naringenin in the Mouse Repeated Tail Suspension Test. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 39, 175–181. [Google Scholar] [CrossRef]
- Senol, F.S.; Ankli, A.; Reich, E.; Orhan, I.E. HPTLC Fingerprinting and Cholinesterase Inhibitory and Metal-Chelating Capacity of Various Citrus Cultivars and Olea Europaea. Food Technol. Biotechnol. 2016, 54, 275–281. [Google Scholar] [CrossRef]
- Lee, S.; Youn, K.; Lim, G.T.; Lee, J.; Jun, M. In Silico Docking and in Vitro Approaches towards BACE1 and Cholinesterases Inhibitory Effect of Citrus Flavanones. Molecules 2018, 23, 1509. [Google Scholar] [CrossRef]
- Li, C.; Zug, C.; Qu, H.; Schluesener, H.; Zhang, Z. Hesperidin Ameliorates Behavioral Impairments and Neuropathology of Transgenic APP/PS1 Mice. Behav. Brain Res. 2015, 281, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, L.; Zhu, X.; Wu, W.; Wang, Y. Hesperidin Alleviates Cognitive Impairment, Mitochondrial Dysfunction and Oxidative Stress in a Mouse Model of Alzheimer’s Disease. Cellular and Molecular Neurobiology 2014, 34, 1209–1221. [Google Scholar] [CrossRef]
- Justin-Thenmozhi, J.A.; Raja, W.T.R.; Manivasagam, T.; Janakiraman, U.; Essa, M.M. Hesperidin Ameliorates Cognitive Dysfunction, Oxidative Stress and Apoptosis against Aluminium Chloride Induced Rat Model of Alzheimer’s Disease. Nutr. Neurosci. 2017, 20, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Justin-Thenmozhi, A.; Dhivya Bharathi, M.; Kiruthika, R.; Manivasagam, T.; Borah, A.; Essa, M.M. Attenuation of Aluminum Chloride-Induced Neuroinflammation and Caspase Activation Through the AKT/GSK-3β Pathway by Hesperidin in Wistar Rats. Neurotox. Res. 2018, 34, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.M.; Li, S.Q.; Zhu, X.Y.; Wang, Y.; Wu, W.L.; Zhang, X.J. Protective Effects of Hesperidin against Amyloid-β (Aβ) Induced Neurotoxicity through the Voltage Dependent Anion Channel 1 (VDAC1)-Mediated Mitochondrial Apoptotic Pathway in PC12 Cells. Neurochem. Res. 2013, 38, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.; Diniz, L.P.; Buosi, A.; Neves, G.; Stipursky, J.; Gomes, F.C.A. Flavonoid Hesperidin Induces Synapse Formation and Improves Memory Performance through the Astrocytic TGF-Β1. Front. Aging Neurosci. 2017, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.S.; Goes, A.T.R.; Boeira, S.P.; Prigol, M.; Jesse, C.R. Protective Effect of Hesperidin in a Model of Parkinson’s Disease Induced by 6-Hydroxydopamine in Aged Mice. Nutrition 2014, 30, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.S.; Ladd, F.V.L.; Ladd, A.A.B.L.; Moreira, A.L.; Boeira, S.P.; Cattelan Souza, L. Hesperidin Protects against Behavioral Alterations and Loss of Dopaminergic Neurons in 6-OHDA-Lesioned Mice: The Role of Mitochondrial Dysfunction and Apoptosis. Metab. Brain Dis. 2021, 36, 153–167. [Google Scholar] [CrossRef]
- Antunes, M.S.; Cattelan Souza, L.; Ladd, F.V.L.; Ladd, A.A.B.L.; Moreira, A.L.; Bortolotto, V.C.; Silva, M.R.P.; Araújo, S.M.; Prigol, M.; Nogueira, C.W.; et al. Hesperidin Ameliorates Anxiety-Depressive-Like Behavior in 6-OHDA Model of Parkinson’s Disease by Regulating Striatal Cytokine and Neurotrophic Factors Levels and Dopaminergic Innervation Loss in the Striatum of Mice. Mol. Neurobiol. 2020, 57, 3027–3041. [Google Scholar] [CrossRef] [PubMed]
- Poetini, M.R.; Araujo, S.M.; Trindade de Paula, M.; Bortolotto, V.C.; Meichtry, L.B.; Polet de Almeida, F.; Jesse, C.R.; Kunz, S.N.; Prigol, M. Hesperidin Attenuates Iron-Induced Oxidative Damage and Dopamine Depletion in Drosophila Melanogaster Model of Parkinson’s Disease. Chem. Biol. Interact. 2018, 279, 177–186. [Google Scholar] [CrossRef]
- Kesh, S.; Kannan, R.R.; Sivaji, K.; Balakrishnan, A. Hesperidin Downregulates Kinases Lrrk2 and Gsk3β in a 6-OHDA Induced Parkinson’s Disease Model. Neurosci. Lett. 2021, 740, 135426. [Google Scholar] [CrossRef] [PubMed]
- Tamilselvam, K.; Braidy, N.; Manivasagam, T.; Essa, M.M.; Prasad, N.R.; Karthikeyan, S.; Thenmozhi, A.J.; Selvaraju, S.; Guillemin, G.J. Neuroprotective Effects of Hesperidin, a Plant Flavanone, on Rotenone-Induced Oxidative Stress and Apoptosis in a Cellular Model for Parkinson’s Disease. Oxidative Med. Cell. Longev. 2013, 2013, 102741. [Google Scholar] [CrossRef]
- Manivasagam, T.; Nataraj, J.; Tamilselvam, K.; Essa, M.; Janakiraman, U. Antioxidant and Anti-Inflammatory Potential of Hesperidin against 1-Methyl-4-Phenyl-1, 2, 3, 6-Tetrahydropyridine-Induced Experimental Parkinson′s Disease in Mice. Int. J. Nutr. Pharmacol. Neurol. Dis. 2013, 3, 294. [Google Scholar] [CrossRef]
- Jahanshahi, M.; Elyasi, L.; Ghazvini, H.; Nikmahzar, E.; Taziki, S. Investigation of the Antioxidant Activity of Hesperidin against 6-Hydroxydopamine-Induced Cell Damage in SH-SY5Y Cells. Natl. J. Physiol. Pharm. Pharmacol. 2018, 8, 834–839. [Google Scholar] [CrossRef]
- Menze, E.T.; Tadros, M.G.; Abdel-Tawab, A.M.; Khalifa, A.E. Potential Neuroprotective Effects of Hesperidin on 3-Nitropropionic Acid-Induced Neurotoxicity in Rats. NeuroToxicology 2012, 33, 1265–1275. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Protective Effect of Hesperidin and Naringin against 3-Nitropropionic Acid Induced Huntington’s like Symptoms in Rats: Possible Role of Nitric Oxide. Behav. Brain Res. 2010, 206, 38–46. [Google Scholar] [CrossRef]
- Haghmorad, D.; Mahmoudi, M.B.; Salehipour, Z.; Jalayer, Z.; Momtazi brojeni, A.A.; Rastin, M.; Kokhaei, P.; Mahmoudi, M. Hesperidin Ameliorates Immunological Outcome and Reduces Neuroinflammation in the Mouse Model of Multiple Sclerosis. J. Neuroimmunol. 2017, 302, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, O.; Ozcan, C.; Kamisli, O.; Cetin, A.; Basak, N.; Aytac, B. Hesperidin, a Citrus Flavonoid, Has the Ameliorative Effects Against Experimental Autoimmune Encephalomyelitis (EAE) in a C57BL/J6 Mouse Model. Neurochem. Res. 2015, 40, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Haghmorad, D.; Amini, A.A.; Mahmoudi, M.B.; Soltani, S.; Tabasi, N.; Jalayer, Z.; Rastin, M.; Mahmoudi, M. Immune Response Profile in Hesperidin Treated Mice Suffering from Experimental Autoimmune Encephalomyelitis. Clin. Biochem. 2011, 44, S166–S167. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, H.; Liu, Y.; Chen, Y.; Liu, Y.; Yin, X. The Antidepressant-Like Effects of Hesperidin in Streptozotocin-Induced Diabetic Rats by Activating Nrf2/ARE/Glyoxalase 1 Pathway. Front. Pharmacol. 2020, 11, 01325. [Google Scholar] [CrossRef]
- Souza, L.C.; de Gomes, M.G.; Goes, A.T.R.; Del Fabbro, L.; Filho, C.B.; Boeira, S.P.; Jesse, C.R. Evidence for the Involvement of the Serotonergic 5-HT1A Receptors in the Antidepressant-like Effect Caused by Hesperidin in Mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 40, 103–109. [Google Scholar] [CrossRef]
- Viswanatha, G.L.; Shylaja, H.; Sandeep Rao, K.S.; Santhosh Kumar, V.R.; Jagadeesh, M. Hesperidin Ameliorates Immobilization-Stress-Induced Behavioral and Biochemical Alterations and Mitochondrial Dysfunction in Mice by Modulating Nitrergic Pathway. ISRN Pharmacol. 2012, 2012, 479570. [Google Scholar] [CrossRef]
- Kosari-Nasab, M.; Shokouhi, G.; Ghorbanihaghjo, A.; Abbasi, M.M.; Salari, A.A. Hesperidin Attenuates Depression-Related Symptoms in Mice with Mild Traumatic Brain Injury. Life Sci. 2018, 213, 198–205. [Google Scholar] [CrossRef]
- Lee, B.; Choi, G.M.; Sur, B. Antidepressant-Like Effects of Hesperidin in Animal Model of Post-Traumatic Stress Disorder. Chin. J. Integr. Med. 2021, 27, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.S.; Jesse, C.R.; Ruff, J.R.; de Oliveira Espinosa, D.; Gomes, N.S.; Altvater, E.E.T.; Donato, F.; Giacomeli, R.; Boeira, S.P. Hesperidin Reverses Cognitive and Depressive Disturbances Induced by Olfactory Bulbectomy in Mice by Modulating Hippocampal Neurotrophins and Cytokine Levels and Acetylcholinesterase Activity. Eur. J. Pharmacol. 2016, 789, 411–420. [Google Scholar] [CrossRef]
- Donato, F.; de Gomes, M.G.; Goes, A.T.R.; Filho, C.B.; Del Fabbro, L.; Antunes, M.S.; Souza, L.C.; Boeira, S.P.; Jesse, C.R. Hesperidin Exerts Antidepressant-like Effects in Acute and Chronic Treatments in Mice: Possible Role of l-Arginine-NO-CGMP Pathway and BDNF Levels. Brain Res. Bull. 2014, 104, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Tsai, S.Y.; Lin, J.A.; Wu, C.H.; Yen, G.C. Cytoprotective Effects of Hesperetin and Hesperidin against Amyloid β-Induced Impairment of Glucose Transport through Downregulation of Neuronal Autophagy. Mol. Nutr. Food Res. 2012, 56, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-ΚB Signaling in an Aβ Mouse Model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.H.; Kim, M.E.; Cho, J.H.; Lee, Y.; Lee, J.; Park, Y.D.; Lee, J.S. Hesperetin Inhibits Neuroinflammation on Microglia by Suppressing Inflammatory Cytokines and MAPK Pathways. Arch. Pharmacal Res. 2019, 42, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Kiasalari, Z.; Khalili, M.; Baluchnejadmojarad, T.; Roghani, M. Protective Effect of Oral Hesperetin Against Unilateral Striatal 6-Hydroxydopamine Damage in the Rat. Neurochem. Res. 2016, 41, 1065–1072. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Wang, L.; Gu, Z.; Huan, Z.; Fu, H.; Liu, Q. Hesperetin Protects SH-SY5Y Cells against 6- Hydroxydopamine-Induced Neurotoxicity via Activation of NRF2/ARE Signaling Pathways. Trop. J. Pharm. Res. 2020, 19, 1197–1201. [Google Scholar] [CrossRef]
- Alizadeh, S.; Moghaddam, A.H.; Zare, M. The Antioxidant Effect of Hesperetin and Nano-Hesperetin on Activity of Catalase and Superoxide Dismutase Enzymes in the Hippocampus of Animal Model of Parkinson’s Disease. Res. J. Pharmacogn. 2017, 4, 29–30. [Google Scholar]
- Dobhal, Y.; Kandpal, E. Evaluation of Antiparkinson Activity of Hesperetin & Pyridoxine against Olanzepine & Manganese Induced Parkinsonism. J. Pharm. Pharmacol. Sci. Res. 2017, 2, 109. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-ΚB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef]
- Baradaran, S.; Hajizadeh Moghaddam, A.; Ghasemi-Kasman, M. Hesperetin Reduces Myelin Damage and Ameliorates Glial Activation in Lysolecithin-Induced Focal Demyelination Model of Rat Optic Chiasm. Life Sci. 2018, 207, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Khalili, M.; Alizadehmakvandi, A.; Roghani, M.; Amirimoghadam, S. The Effect of Hesperetin on Depression and Anxiety Induced by Reserpine Injection in Male Rats. J. Basic Clin. Pathophysiol. 2020, 8, 37–43. [Google Scholar] [CrossRef]
- Li, B.; Huang, A.L.; Zhang, Y.L.; Li, Z.; Ding, H.W.; Huang, C.; Meng, X.M.; Li, J. Design, Synthesis and Evaluation of Hesperetin Derivatives as Potential Multifunctional Anti-Alzheimer Agents. Molecules 2017, 22, 1067. [Google Scholar] [CrossRef]
- Chakraborty, S.; Basu, S. Multi-Functional Activities of Citrus Flavonoid Narirutin in Alzheimer’s Disease Therapeutics: An Integrated Screening Approach and in Vitro Validation. Int. J. Biol. Macromol. 2017, 103, 733–743. [Google Scholar] [CrossRef]
- Li, Y.; Du, Y.; Yang, J.; Xiu, Z.; Yang, N.; Zhang, J.; Gao, Y.; Li, B.; Shi, H. Narirutin Produces Antidepressant-like Effects in a Chronic Unpredictable Mild Stress Mouse Model. NeuroReport 2018, 29, 1264–1268. [Google Scholar] [CrossRef]
- Pinto, M.M.M.; Palmeira, A.; Fernandes, C.; Resende, D.I.S.P.; Sousa, E.; Cidade, H.; Tiritan, M.E.; Correia-da-Silva, M.; Cravo, S. From Natural Products to New Synthetic Small Molecules: A Journey through the World of Xanthones. Molecules 2021, 26, 431. [Google Scholar] [CrossRef] [PubMed]
- Borzdziłowska, P.; Bednarek, I. Xanthones as Natural Compounds with a Wide Spectrum of Biological Activity. Postepy Hig. I Med. Dosw. 2018, 72, 767–780. [Google Scholar] [CrossRef]
- Kou, X.; Song, L.; Wang, Y.; Yu, Q.; Ju, H.; Yang, A.; Shen, R. Design, Synthesis and Anti-Alzheimer’s Disease Activity Study of Xanthone Derivatives Based on Multi-Target Strategy. Bioorganic Med. Chem. Lett. 2020, 30, 126927. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Lan, W.; Liu, Z.; Huang, J.; Tang, H.; Wang, H. Synthesis and Biological Evaluation of 1, 3-Dihydroxyxanthone Mannich Base Derivatives as Anticholinesterase Agents. Chem. Cent. J. 2013, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, Y.; Qiang, X.; Cao, Z.; Xu, R.; Yang, X.; Xiao, G.; Song, Q.; Tan, Z.; Deng, Y. Multifunctional Thioxanthone Derivatives with Acetylcholinesterase, Monoamine Oxidases and β-Amyloid Aggregation Inhibitory Activities as Potential Agents against Alzheimer’s Disease. Bioorganic Med. Chem. 2017, 25, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Cruz, I.; Puthongking, P.; Cravo, S.; Palmeira, A.; Cidade, H.; Pinto, M.; Sousa, E. Xanthone and Flavone Derivatives as Dual Agents with Acetylcholinesterase Inhibition and Antioxidant Activity as Potential Anti-Alzheimer Agents. J. Chem. 2017, 2017, 8587260. [Google Scholar] [CrossRef]
- Alawi, M.S.; Awad, T.A.; Mohamed, M.A.; Khalid, A.; Ismail, E.M.O.; Alfatih, F.; Naz, S.; UL-Haq, Z. Insights into the Molecular Basis of Acetylcholinesterase Inhibition by Xanthones: An Integrative in Silico and in Vitro Approach. Mol. Simul. 2020, 46, 253–261. [Google Scholar] [CrossRef]
- Chi, X.Q.; Hou, B.; Yang, L.; Zi, C.T.; Lv, Y.F.; Li, J.Y.; Ren, F.C.; Yuan, M.Y.; Hu, J.M.; Zhou, J. Design, Synthesis and Cholinesterase Inhibitory Activity of α-Mangostin Derivatives. Nat. Prod. Res. 2020, 34, 1380–1388. [Google Scholar] [CrossRef]
- Remya, C.; Dileep, K.V.; Tintu, I.; Variyar, E.J.; Sadasivan, C. Design of Potent Inhibitors of Acetylcholinesterase Using Morin as the Starting Compound. Front. Life Sci. 2012, 6, 107–117. [Google Scholar] [CrossRef]
- Frandsen, J.; Choi, S.R.; Narayanasamy, P. Neural Glyoxalase Pathway Enhancement by Morin Derivatives in an Alzheimer’s Disease Model. ACS Chem. Neurosci. 2020, 11, 356–366. [Google Scholar] [CrossRef]
- Zhang, S.S.; Ma, Q.Y.; Zou, X.S.; Dai, H.F.; Huang, S.Z.; Luo, Y.; Yu, Z.F.; Luo, H.R.; Zhao, Y.X. Chemical Constituents from the Fungus Amauroderma Amoiensis and Their in Vitro Acetylcholinesterase Inhibitory Activities. Planta Med. 2013, 79, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.; Reis, P.B.P.S.; Machuqueiro, M.; Ressaissi, A.; Pacheco, R.; Serralheiro, M.L. Bioactivities of Centaurium Erythraea (Gentianaceae) Decoctions: Antioxidant Activity, Enzyme Inhibition and Docking Studies. Molecules 2019, 24, 3795. [Google Scholar] [CrossRef] [PubMed]
- Neagu, E.; Radu, G.L.; Albu, C.; Paun, G. Antioxidant Activity, Acetylcholinesterase and Tyrosinase Inhibitory Potential of Pulmonaria Officinalis and Centarium Umbellatum Extracts. Saudi J. Biol. Sci. 2018, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Grauzdytė, D.; Raudoniūtė, J.; Kulvinskienė, I.; Bagdonas, E.; Stasiulaitienė, I.; Martuzevičius, D.; Bironaitė, D.; Aldonytė, R.; Venskutonis, P.R. Cytoprotective Effects of Mangiferin and Z-Ligustilide in PAH-Exposed Human Airway Epithelium in Vitro. Nutrients 2019, 11, 218. [Google Scholar] [CrossRef]
- Sethiya, N.K.; Mishra, S.H. Investigation of Mangiferin, as a Promising Natural Polyphenol Xanthone on Multiple Targets of Alzheimer’s Disease. J. Biol. Act. Prod. Nat. 2014, 4, 111–119. [Google Scholar] [CrossRef]
- Biradar, S.M.; Joshi, H.; Chheda, T.K. Neuropharmacological Effect of Mangiferin on Brain Cholinesterase and Brain Biogenic Amines in the Management of Alzheimer’s Disease. Eur. J. Pharmacol. 2012, 683, 140–147. [Google Scholar] [CrossRef]
- Infante-Garcia, C.; Ramos-Rodriguez, J.J.; Delgado-Olmos, I.; Gamero-Carrasco, C.; Fernandez-Ponce, M.T.; Casas, L.; Mantell, C.; Garcia-Alloza, M. Long-Term Mangiferin Extract Treatment Improves Central Pathology and Cognitive Deficits in APP/PS1 Mice. Mol. Neurobiol. 2017, 54, 4696–4704. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, M.; Nataraj, J.; Essa, M.M.; Memon, M.A.; Manivasagam, T. Mangiferin Attenuates MPTP Induced Dopaminergic Neurodegeneration and Improves Motor Impairment, Redox Balance and Bcl-2/Bax Expression in Experimental Parkinson’s Disease Mice. Chem. Biol. Interact. 2013, 206, 239–247. [Google Scholar] [CrossRef]
- Lei, L.Y.; Wang, R.C.; Pan, Y.L.; Yue, Z.G.; Zhou, R.; Xie, P.; Tang, Z.S. Mangiferin Inhibited Neuroinflammation through Regulating Microglial Polarization and Suppressing NF-ΚB, NLRP3 Pathway. Chin. J. Nat. Med. 2021, 19, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.K.; Kisku, A.; Jangra, A. Mangiferin Ameliorates Intracerebroventricular-Quinolinic Acid-Induced Cognitive Deficits, Oxidative Stress, and Neuroinflammation in Wistar Rats. Indian J. Pharmacol. 2020, 52, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Jangra, A.; Lukhi, M.M.; Sulakhiya, K.; Baruah, C.C.; Lahkar, M. Protective Effect of Mangiferin against Lipopolysaccharide-Induced Depressive and Anxiety-like Behaviour in Mice. Eur. J. Pharmacol. 2014, 740, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Sánchez-Gómez, M.V.; Ruiz, A.; Cavaliere, F.; Ortiz-Sanz, C.; Quintela-López, T.; Capetillo-Zarate, E.; Solé-Domènech, S.; Matute, C. Mangiferin and Morin Attenuate Oxidative Stress, Mitochondrial Dysfunction, and Neurocytotoxicity, Induced by Amyloid Beta Oligomers. Oxidative Med. Cell. Longev. 2018, 2018, 2856063. [Google Scholar] [CrossRef]
- Du, Y.; Qu, J.; Zhang, W.; Bai, M.; Zhou, Q.; Zhang, Z.; Li, Z.; Miao, J. Morin Reverses Neuropathological and Cognitive Impairments in APPswe/PS1dE9 Mice by Targeting Multiple Pathogenic Mechanisms. Neuropharmacology 2016, 108, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gong, E.J.; Park, H.R.; Kim, M.E.; Piao, S.; Lee, E.; Jo, D.G.; Chung, H.Y.; Ha, N.C.; Mattson, M.P.; Lee, J. Morin Attenuates Tau Hyperphosphorylation by Inhibiting GSK3β. Neurobiol. Dis. 2011, 44, 223–230. [Google Scholar] [CrossRef]
- Kim, J.M.; Lee, E.K.; Park, G.; Kim, M.K.; Yokozawa, T.; Yu, B.P.; Chung, H.Y. Morin Modulates the Oxidative Stress-Induced NF-ΚB Pathway through Its Anti-Oxidant Activity. Free Radic. Res. 2010, 44, 454–461. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Cao, X.B.; Xiong, N.; Wang, H.C.; Huang, J.S.; Sun, S.G.; Wang, T. Morin Exerts Neuroprotective Actions in Parkinson Disease Models in Vitro and in Vivo. Acta Pharmacol. Sin. 2010, 31, 900–906. [Google Scholar] [CrossRef]
- Lee, K.M.; Lee, Y.; Chun, H.J.; Kim, A.H.; Kim, J.Y.; Lee, J.Y.; Ishigami, A.; Lee, J. Neuroprotective and Anti-Inflammatory Effects of Morin in a Murine Model of Parkinson’s Disease. J. Neurosci. Res. 2016, 94, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.A.M.; Gad, A.M.; Menze, E.T.; Badary, O.A.; El-Naga, R.N. Protective Effects of Morin against Depressive-like Behavior Prompted by Chronic Unpredictable Mild Stress in Rats: Possible Role of Inflammasome-Related Pathways. Biochem. Pharmacol. 2020, 180, 114140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.X.; Wang, Y.; Liu, T.; Wang, Y.X.; Chen, H.Z.; Xu, J.R.; Qiu, Y. α-Mangostin Decreases β-Amyloid Peptides Production via Modulation of Amyloidogenic Pathway. CNS Neurosci. Ther. 2017, 23, 526–534. [Google Scholar] [CrossRef]
- Chen, Y.; Bian, Y.; Wang, J.W.; Gong, T.T.; Ying, Y.M.; Ma, L.F.; Shan, W.G.; Xie, X.Q.; Zhan, Z.J. Effects of α-Mangostin Derivatives on the Alzheimer’s Disease Model of Rats and Their Mechanism: A Combination of Experimental Study and Computational Systems Pharmacology Analysis. ACS Omega 2020, 5, 9846–9863. [Google Scholar] [CrossRef] [PubMed]
- Sattayasai, J.; Chaonapan, P.; Arkaravichie, T.; Soi-ampornkul, R.; Junnu, S.; Charoensilp, P.; Samer, J.; Jantaravinid, J.; Masaratana, P.; Suktitipat, B.; et al. Protective Effects of Mangosteen Extract on H2O2-Induced Cytotoxicity in SK-N-SH Cells and Scopolamine-Induced Memory Impairment in Mice. PLoS ONE 2013, 8, 85053. [Google Scholar] [CrossRef]
- Oh, Y.; Do, H.T.T.; Kim, S.; Kim, Y.M.; Chin, Y.W.; Cho, J. Memory-Enhancing Effects of Mangosteen Pericarp Water Extract through Antioxidative Neuroprotection and Anti-Apoptotic Action. Antioxidants 2021, 10, 34. [Google Scholar] [CrossRef]
- Huang, H.J.; Chen, W.L.; Hsieh, R.H.; Hsieh-Li, H.M. Multifunctional Effects of Mangosteen Pericarp on Cognition in C57BL/6J and Triple Transgenic Alzheimer’s Mice. Evid. Based Complementary Altern. Med. 2014, 2014, 813672. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Li, J.; Tan, X.; Luo, S.; Liu, Y.; Meng, Y.; Wu, B.; Zhou, Y.; Yang, Y.; Chen, H.; et al. Natural Xanthone α-Mangostin Inhibits LPS-Induced Microglial Inflammatory Responses and Memory Impairment by Blocking the TAK1/NF-ΚB Signaling Pathway. Mol. Nutr. Food Res. 2020, 64, 2000096. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, W.; Ling, J.; Jiang, C. α-Mangostin Inhibits α-Synuclein-Induced Microglial Neuroinflammation and Neurotoxicity. Cell. Mol. Neurobiol. 2016, 36, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.M.; Li, L.D.; Duan, C.L.; Li, Y.J. Neuroprotective Effect of α-Mangostin on Mitochondrial Dysfunction and α-Synuclein Aggregation in Rotenone-Induced Model of Parkinson’s Disease in Differentiated SH-SY5Y Cells. J. Asian Nat. Prod. Res. 2017, 19, 833–845. [Google Scholar] [CrossRef]
- Parkhe, A.; Parekh, P.; Nalla, L.V.; Sharma, N.; Sharma, M.; Gadepalli, A.; Kate, A.; Khairnar, A. Protective Effect of Alpha Mangostin on Rotenone Induced Toxicity in Rat Model of Parkinson’s Disease. Neurosci. Lett. 2020, 716, 134652. [Google Scholar] [CrossRef]
- Nava Catorce, M.; Acero, G.; Pedraza-Chaverri, J.; Fragoso, G.; Govezensky, T.; Gevorkian, G. Alpha-Mangostin Attenuates Brain Inflammation Induced by Peripheral Lipopolysaccharide Administration in C57BL/6J Mice. J. Neuroimmunol. 2016, 297, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Pedraza-Chaverrí, J.; Reyes-Fermín, L.M.; Nolasco-Amaya, E.G.; Orozco-Ibarra, M.; Medina-Campos, O.N.; González-Cuahutencos, O.; Rivero-Cruz, I.; Mata, R. ROS Scavenging Capacity and Neuroprotective Effect of α-Mangostin against 3-Nitropropionic Acid in Cerebellar Granule Neurons. Exp. Toxicol. Pathol. 2009, 61, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Liu, X.; Liu, J.; Cai, E.; Zhao, Y.; Li, H.; Zhang, L.; Li, P.; Gao, Y. α-Mangostin Exhibits Antidepressant-like Effects Mediated by the Modification of GABAergic, Serotonergic and Dopaminergic Systems. Nat. Prod. Res. 2020, 34, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, L.; Zhu, S.; Xu, L.; Liu, S.; Yuan, C.; Guo, Y.; Wang, X. Neuroprotection Against Parkinson’s Disease Through the Activation of Akt/GSK3β Signaling Pathway by Tovophyllin A. Front. Neurosci. 2020, 14, 00723. [Google Scholar] [CrossRef] [PubMed]




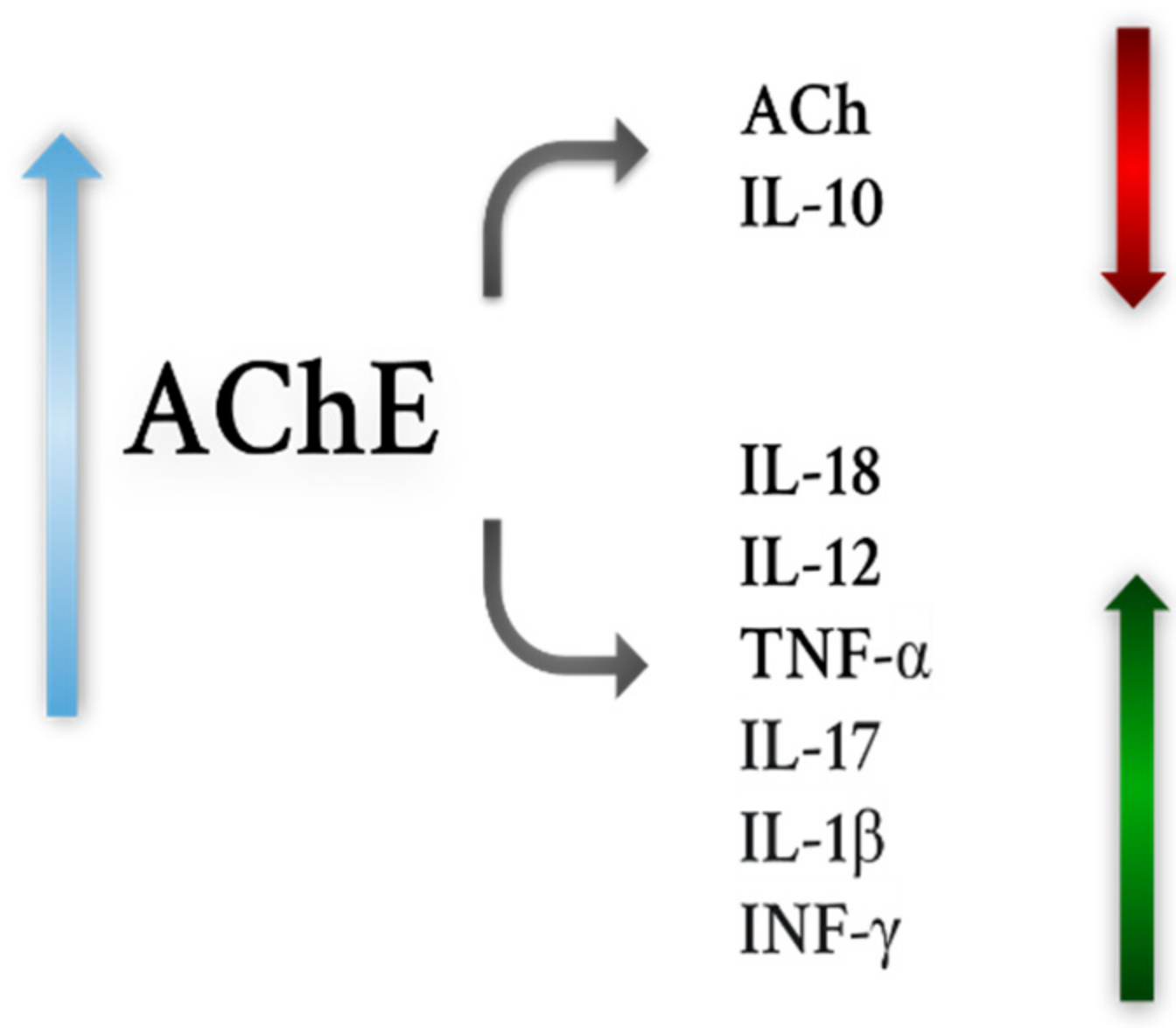{kind=link}
{kind=link}
{kind=link}
| Drug | Characteristics | Disadvantages |
|---|---|---|
| Well-known inhibitors of AChE | ||
| Rivastigmine | Inhibitor of AChE and BChE Drug registered for the symptomatic treatment of mild to intermediate dementia in AD and idiopathic PD Administered as an oral or transdermal patch Effects on βA levels Beneficial effects in AD and PD Anti-inflammatory effects | Side effects (mainly gastrointestinal) Oral administration requires dose control Discrepant study results regarding use in HD, MDD, BPAD, PSP No proven efficacy in SM No efficacy data in ALS, OPCA |
| Donepezil | Drug registered for the symptomatic treatment of mild, moderate and severe dementia in AD AChE inhibitor Has almost no drug interactions May be a matrix-type drug Improves the condition of patients with AD, PD Alleviates cholinergic deficits in some part Exhibits effects on βA, p-tau | Side effects (similar to rivastigmine) Gene polymorphisms affect response to the drug Divergent findings regarding use of the drug in SM, HD, BPAD, MDD, PSP No data available for ALS, OPCA |
| Galantamine | AChE inhibitor Registered as a drug in patients with mild to moderate forms of AD Large volume of distribution May be a prolonged-acting or immediate-acting drug Improves cognitive abilities in AD Anti-inflammatory, antioxidant activity -Effects on βA Beneficial effects on symptoms associated with HD | Side effects as above Discrepant findings regarding efficacy in PD, BPAD, MDD No data available for ALS, SM, PSP, OPCA |
| Huperzine A | It is not yet registered as a drug in any neurodegenerative disease (Ex. China) Mild to moderate side effects Neuroprotective effects Beneficial effects in patients with AD Effects on βA | No data available for other neurodegenerative diseases |
| Phenserine Posiphen | AChE inhibitor Effects:
Promising results in PD and AD in animals | Phenserine failed in clinical trials No data available for other neurodegenerative diseases |
| New compounds with potential use in the treatment of neurodegenerative diseases and depression | ||
| Thunberginol C | Inhibitors of AChE and BChE Anti-inflammatory activity (?) | A small amount of scientific research |
| Hydrangenol 8-O-glucoside pentaacetate | ||
| Hydrangenol | Effects:
| No data available on effects on AChE A small amount of scientific research |
| Skimmin | Effects:
| |
| Extracts and essential oils of Salvia spp. | Most promising compounds:
Effects on mood Effects on p-tau, βA, HTT, α-synuclein Effects on microglia Inhibitors of AChE Effects
| The chemical profile of an oil/extract depends on various climatic, soil or latitude and longitude factors |
| Extracts and essential oils of Prunus spp. | Most promising compounds:
AChE inhibitors | |
| Hesperidin | Effects on βA, p-tau Inhibitor of AChE and BChE Effects on cognitive abilities (better than donepezil) Promising results in AD, PD, HD, SM, ALS, MDD, BPAD Effects:
| |
| Limonoids | Effects on βA, AChE Effects on cognitive abilities (better than donepezil) Neuroprotective effect | |
| Naringenin | AChE Inhbitiror Effects on βA, Beneficial effects on cognitive abilities Promising results in PD, AD, MDD, HD, BPAD, ALS, SM Effects:
| |
| Hesperetin | Effect
| |
| Narirutin | Effects on βA Low toxicity Effects:
| |
| Mangiferin | AChE inhibitor Effects on p-tau, βA (α-Mangostin) Potential use: AD, PD, MDD Effects:
| |
| Morin | ||
| α-Mangostin | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walczak-Nowicka, Ł.J.; Herbet, M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9290. https://doi.org/10.3390/ijms22179290
Walczak-Nowicka ŁJ, Herbet M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis. International Journal of Molecular Sciences. 2021; 22(17):9290. https://doi.org/10.3390/ijms22179290
Chicago/Turabian StyleWalczak-Nowicka, Łucja Justyna, and Mariola Herbet. 2021. "Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis" International Journal of Molecular Sciences 22, no. 17: 9290. https://doi.org/10.3390/ijms22179290
APA StyleWalczak-Nowicka, Ł. J., & Herbet, M. (2021). Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis. International Journal of Molecular Sciences, 22(17), 9290. https://doi.org/10.3390/ijms22179290