
Overview of Ferroptosis and Synthetic Lethality Strategies

 ,
,
Abstract
1. Introduction
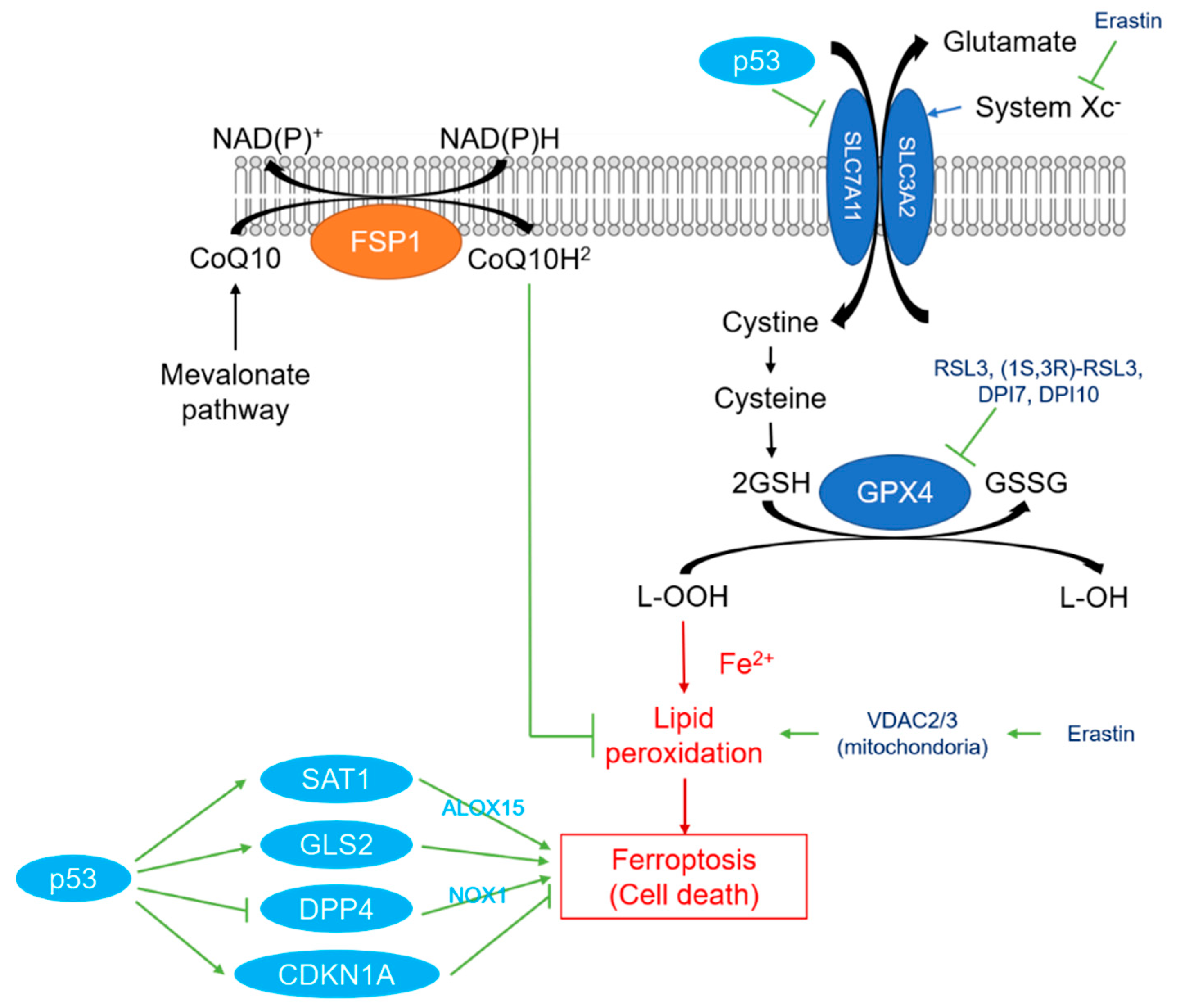
2. Overview of Ferroptosis
3. Overview of Synthetic Lethality
4. Anticancer Drug Discovery Using Synthetic Lethality
5. Synthetic Lethality and Ferroptosis
6. Synthetic Lethality and Ferroptosis—Related to RAS Mutation
7. Synthetic Lethality and Ferroptosis—Unrelated to RAS Mutation
7.1. Related to Ferroptosis Inhibitors
7.2. Related to Ferroptosis Inducers
8. Challenges and Problems
9. Prospects for Synthetic Lethal Strategies Using Ferroptosis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Nijman, S.M. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Garama, D.J.; Harris, T.J.; White, C.L.; Rossello, F.J.; Abdul-Hay, M.; Gough, D.J.; Levy, D.E. A Synthetic Lethal Interaction between Glutathione Synthesis and Mitochondrial Reactive Oxygen Species Provides a Tumor-Specific Vulnerability Dependent on STAT3. Mol. Cell Biol. 2015, 35, 3646–3656. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Friedmann-Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Jiang, L.; Hickman, J.H.; Wang, S.J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–e6812. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Mullard, A. Synthetic lethality screens point the way to new cancer drug targets. Nat. Rev. Drug Discov. 2017, 16, 589–591. [Google Scholar] [CrossRef]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Kobayashi, H.; Ohno, S.; Sasaki, Y.; Matsuura, M. Hereditary breast and ovarian cancer susceptibility genes (review). Oncol. Rep. 2013, 30, 1019–1029. [Google Scholar] [CrossRef]
- McDonald, E.R., 3rd; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef] [PubMed]
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 2016, 6, 914–929. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Dempster, J.M.; Pacini, C.; Pantel, S.; Behan, F.M.; Green, T.; Krill-Burger, J.; Beaver, C.M.; Younger, S.T.; Zhivich, V.; Najgebauer, H.; et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 2019, 10, 5817. [Google Scholar] [CrossRef]
- Chan, E.M.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019, 568, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, L.; Corti, G.; Picco, G.; Isella, C.; Montone, M.; Arcella, P.; Durinikova, E.; Zanella, E.R.; Novara, L.; Barbosa, F.; et al. Patient-Derived Xenografts and Matched Cell Lines Identify Pharmacogenomic Vulnerabilities in Colorectal Cancer. Clin. Cancer Res. 2019, 25, 6243–6259. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem 2017, 9, 1025–1033. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Kwon, E.J.; Kong, H.J.; Choi, J.Y.; Kim, Y.J.; Lee, E.W.; Kim, W.; Lee, H.; Cha, H.J. Systematic identification of a nuclear receptor-enriched predictive signature for erastin-induced ferroptosis. Redox Biol. 2020, 37, 101719. [Google Scholar] [CrossRef]
- Sugiyama, A.; Ohta, T.; Obata, M.; Takahashi, K.; Seino, M.; Nagase, S. xCT inhibitor sulfasalazine depletes paclitaxel-resistant tumor cells through ferroptosis in uterine serous carcinoma. Oncol. Lett 2020, 20, 2689–2700. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, R.Z.; Saez, B.; Sharda, A.; van Gastel, N.; Yu, V.W.C.; Baryawno, N.; Scadden, E.W.; Acharya, S.; Chattophadhyay, S.; Huang, C.; et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood 2020, 136, 1303–1316. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Milligan, J.; Wohlschlegel, J.A.; Ooi, A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018, 109, 2757–2766. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Shintani, S.; Hirata, Y.; Suina, K.; Semba, T.; Yamasaki, J.; Umene, K.; Ishikawa, M.; Saya, H.; Nagano, O. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency-resistant cancer cells. Oncotarget 2018, 9, 33832–33843. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [PubMed]
- To, T.L.; Cuadros, A.M.; Shah, H.; Hung, W.H.W.; Li, Y.; Kim, S.H.; Rubin, D.H.F.; Boe, R.H.; Rath, S.; Eaton, J.K.; et al. A Compendium of Genetic Modifiers of Mitochondrial Dysfunction Reveals Intra-organelle Buffering. Cell 2019, 179, 1222–1238.e17. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Suzuki, T.; Takahashi, Y.; Hishinuma, E.; Saigusa, D.; Yamamoto, M. Geldanamycin-Derived HSP90 Inhibitors Are Synthetic Lethal with NRF2. Mol. Cell. Biol. 2020, 40, 22. [Google Scholar] [CrossRef]
- Joly, J.H.; Delfarah, A.; Phung, P.S.; Parrish, S.; Graham, N.A. A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose. J. Biol. Chem. 2020, 295, 1350–1365. [Google Scholar] [CrossRef]
- Lorenzato, A.; Magrì, A.; Matafora, V.; Audrito, V.; Arcella, P.; Lazzari, L.; Montone, M.; Lamba, S.; Deaglio, S.; Siena, S.; et al. Vitamin C Restricts the Emergence of Acquired Resistance to EGFR-Targeted Therapies in Colorectal Cancer. Cancers 2020, 12, 3. [Google Scholar] [CrossRef]
- Verma, N.; Vinik, Y.; Saroha, A.; Nair, N.U.; Ruppin, E.; Mills, G.; Karn, T.; Dubey, V.; Khera, L.; Raj, H.; et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci. Adv. 2020, 6, 34. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. NRF2-Dependent Bioactivation of Mitomycin C as a Novel Strategy To Target KEAP1-NRF2 Pathway Activation in Human Cancer. Mol. Cell Biol. 2021, 41, 2. [Google Scholar] [CrossRef]
- Chen, S.; Bu, D.; Zhu, J.; Yue, T.; Guo, S.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. Endogenous hydrogen sulfide regulates xCT stability through persulfidation of OTUB1 at cysteine 91 in colon cancer cells. Neoplasia 2021, 23, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Invest. 2021, 131, 12. [Google Scholar] [CrossRef] [PubMed]
- Weïwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Stockwell, B.R. A powerful cell-protection system prevents cell death by ferroptosis. Nature 2019, 575, 597–598. [Google Scholar] [CrossRef]
- Miura, G. A back-up plan. Nat. Chem. Biol. 2019, 15, 1131. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Gambardella, V.; Gimeno-Valiente, F.; Tarazona, N.; Martinez-Ciarpaglini, C.; Roda, D.; Fleitas, T.; Tolosa, P.; Cejalvo, J.M.; Huerta, M.; Roselló, S.; et al. NRF2 through RPS6 Activation Is Related to Anti-HER2 Drug Resistance in HER2-Amplified Gastric Cancer. Clin. Cancer Res. 2019, 25, 1639–1649. [Google Scholar] [CrossRef]
- Hu, X.F.; Yao, J.; Gao, S.G.; Wang, X.S.; Peng, X.Q.; Yang, Y.T.; Feng, X.S. Nrf2 overexpression predicts prognosis and 5-FU resistance in gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5231–5235. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Ishigami, S.; Arigami, T.; Uenosono, Y.; Yanagita, S.; Uchikado, Y.; Kita, Y.; Nishizono, Y.; Okumura, H.; Nakajo, A.; et al. Clinicopathological significance of nuclear factor (erythroid-2)-related factor 2 (Nrf2) expression in gastric cancer. BMC Cancer 2015, 15, 5. [Google Scholar] [CrossRef]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Badros, A.Z.; Jagannath, S.; Tarantolo, S.; Wolf, J.L.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin with bortezomib: Activity in relapsed/refractory patients with multiple myeloma. Br. J. Haematol. 2010, 150, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Chanan-Khan, A.A.; Lonial, S.; Krishnan, A.Y.; Carroll, M.P.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: Results of a phase 1/2 study. Br. J. Haematol. 2011, 153, 729–740. [Google Scholar] [CrossRef]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Miller, K.; Rosen, L.; Schneider, B.; Chap, L.; Hannah, A.; Zhong, Z.; Ma, W.; Hudis, C.; Modi, S. A phase I dose-escalation trial of trastuzumab and alvespimycin hydrochloride (KOS-1022; 17 DMAG) in the treatment of advanced solid tumors. Clin. Cancer Res. 2012, 18, 5090–5098. [Google Scholar] [CrossRef]
- Sequist, L.V.; Gettinger, S.; Senzer, N.N.; Martins, R.G.; Jänne, P.A.; Lilenbaum, R.; Gray, J.E.; Iafrate, A.J.; Katayama, R.; Hafeez, N.; et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4953–4960. [Google Scholar] [CrossRef]
- Wagner, A.J.; Chugh, R.; Rosen, L.S.; Morgan, J.A.; George, S.; Gordon, M.; Dunbar, J.; Normant, E.; Grayzel, D.; Demetri, G.D. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin. Cancer Res. 2013, 19, 6020–6029. [Google Scholar] [CrossRef]
- Bradner, W.T. Mitomycin C: A clinical update. Cancer Treat. Rev. 2001, 27, 35–50. [Google Scholar] [CrossRef]
- Sculier, J.P.; Ghisdal, L.; Berghmans, T.; Branle, F.; Lafitte, J.J.; Vallot, F.; Meert, A.P.; Lemaitre, F.; Steels, E.; Burniat, A.; et al. The role of mitomycin in the treatment of non-small cell lung cancer: A systematic review with meta-analysis of the literature. Br. J. Cancer 2001, 84, 1150–1155. [Google Scholar] [CrossRef][Green Version]
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.L.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320.e10. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- Jung, S.A.; Lee, D.H.; Moon, J.H.; Hong, S.W.; Shin, J.S.; Hwang, I.Y.; Shin, Y.J.; Kim, J.H.; Gong, E.Y.; Kim, S.M.; et al. Corrigendum to ‘L-Ascorbic acid can abrogate SVCT-2-dependent cetuximab resistance mediated by mutant KRAS in human colon cancer cells’: [Free Radic. Biol. Med. 95 (2016) 200–208]. Free Radic Biol Med. 2016, 97, 620. [Google Scholar] [CrossRef]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, F.; Sun, F.; Gu, K.; Dong, S.; He, D. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst. Rev. 2015, 4, Cd011076. [Google Scholar] [CrossRef]
- Stangel, M.; Linker, R.A. Dimethyl fumarate (BG-12) for the treatment of multiple sclerosis. Expert Rev. Clin. Pharm. 2013, 6, 355–362. [Google Scholar] [CrossRef]
- Neubauer, A.; Maharry, K.; Mrózek, K.; Thiede, C.; Marcucci, G.; Paschka, P.; Mayer, R.J.; Larson, R.A.; Liu, E.T.; Bloomfield, C.D. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2008, 26, 4603–4609. [Google Scholar] [CrossRef]
- McLornan, D.P.; List, A.; Mufti, G.J. Applying synthetic lethality for the selective targeting of cancer. N. Engl. J. Med. 2014, 371, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Rosen, P.J.; Wagner, A.J.; Sahasrabudhe, S.; Weiss, G.J.; Lee, P.; Fuerst, M.; Robbins, P.; Litka, P.; Hoff, D.D.V. A phase I pharmacodynamic and pharmacokinetic study of a Ras inhibitor, PRLX 93936, in patients with advanced solid tumors. J. Clin. Oncol. 2010, 28, e13042. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Schlossman, R.L.; Gasparetto, C.J.; Berdeja, J.G.; Morris, J.; Jacobstein, D.A.; Anderson, K.C.; Mitsiades, C.S.; Laubach, J.P.; Richardson, P.G. An Open-Label, Dose Escalation, Multi-Center Phase 1 Study of PRLX 93936, an Agent Synthetically Active Against the Activated Ras Pathway, in the Treatment of Relapsed or Relapsed and Refractory Multiple Myeloma. Blood 2014, 124, 2140. [Google Scholar] [CrossRef]
- Ohman, K.A.; Hashim, Y.M.; Vangveravong, S.; Nywening, T.M.; Cullinan, D.R.; Goedegebuure, S.P.; Liu, J.; Van Tine, B.A.; Tiriac, H.; Tuveson, D.A.; et al. Conjugation to the sigma-2 ligand SV119 overcomes uptake blockade and converts dm-Erastin into a potent pancreatic cancer therapeutic. Oncotarget 2016, 7, 33529–33541. [Google Scholar] [CrossRef]
- Larraufie, M.H.; Yang, W.S.; Jiang, E.; Thomas, A.G.; Slusher, B.S.; Stockwell, B.R. Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg Med. Chem. Lett. 2015, 25, 4787–4792. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Ferroptosis | Apoptosis | Necroptosis | Autophagy | ||
|---|---|---|---|---|---|
| Morphological features | Smaller mitochondria, reduced mitochondria crista, elevated mitochondrial membrane densities, increased rupture of mitochondrial membrane | Cell rounding, nuclear condensation, membrane blebbing, apoptotic body formation | Cell swelling, rupture of plasma membrane, moderate chromatin condensation | Formation of double membraned autolysosomes (Autophagic vacuolization) | |
| Biological features | Iron accumulation and lipid peroxidation | Activation of caspases and DNA fragmentation | Drop in ATP levels, cytosolic necrosome formation | increased autophagic flux and lysosomal activity | |
| Regulatory pathways | Xc- /GPX4, MVA, sulfur transfer pathway, P62-Keap1-NRF2 pathway, P53/SLC7A11, ATG5-ATG7-NCOA4 pathway, P53-SAT1-ALOX15 pathway, HSPB1-TRF1, FSP1-COQ10-NAD(P)H pathway | Death receptor pathway, mitochondrial pathway and endoplasmic reticulum pathway; Caspase, P53, Bcl-2 mediated signaling pathway | TNF-R1 and RIP1/RIP3-MLKL related signaling pathways; PKC-MAPK-AP-1 related signaling pathway; ROS-related metabolic regulation pathway | mTOR, Beclin-1, P53 signaling pathway | |
| Major regulators | Positive | TFR1, ACSL4, NCOA4, VDAC2/3 | Caspase, pro-apoptotic BCL2 family (e.g., BAX), TP53 | RIPK1, RIPK3, MLKL | ATG5, ATG7, Becilin-1, Other ATG famiy proteins |
| Negative | GPX4, SLC7A11, FSP1 NRF2(NFE2L2), HSPB1, HSPA5 | anti-apoptotic BCL2 family (e.g., BCL2) | ESCRT-III, cIAPs, LUBAC, PPM1B, and AURKA | mTOR | |
| Dual | TP53 | ||||
| Inducer | Class1: Inhibit system Xc- and prevent cystine import | Erastin, Sorafenib, Sulfasalazine |
| Class2: Inhibit GPX4 | RSL3, (1S,3R)-RSL3, DPI7, DPI10 | |
| Class3: Degrade GPX4, bind to SQS, and deplete antioxidant CoQ10 | FIN56 | |
| Class4: Oxidize ferrous iron and lipidome directly, and inactivate GPX4 indirectly | FINO2 | |
| Supplement: Target VDACs, degrade GPX4 | Erastin | |
| Inhibitor | Class1: Inhibit accumulation of iron | DFO, Deferoxamine mesylate, 2,2′-pyridine |
| Class2: Inhibit lipid peroxidation | Fer-1, SRS11–9, SRS16–86, Liproxststatin-1, Vitamin E |
| Author | Synthetic Lethal Factors (A) | Synthetic Lethal Factors (B) | RAS Mutation | Summary of Synthetic Lethality | Tissues and Cells | Verification of Ferroptosis | Other Type of Cell Death | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Increase in Intracellular Reactive Oxygen Species and Lipid Peroxides | Intracellular Iron Accumulation/Inhibition by Iron Chelators | Cell Death by Ferotosis Inducer | Morphological Changes Characteristic of Ferroptosis | Bioenergetic Changes: Intracellular ATP Depletion | Lack of Other PCD Features | Escape of Cell Death by Ferroptosis Inhibitors | |||||||
| Yang, W.S., et al. [13] | RSL3, ML162, anf DPI10 | oncogenic-HRAS | RSL3, ML162, and DPI10 induce ferroptosis in engineered oncogenic-RAS fibroblast-derived tumorigenic cell lines | oncogenic-HRAS cells, 117 human cancer cell lines | ○ | ○ | ○ | ||||||
| Yu, Y., et al. [51] | Erastin | Cytarabine and Doxorubicin | Erastin induces cell death in AML cells in a dose-dependent manner through a mixture of ferroptosis, apoptosis, necroptosis, and autophagy. | HL-60 cells (AML, NRAS_Q61L) | mixed types of cell death associated with ferroptosis, apoptosis, necroptosis, and autophagy | ||||||||
| Chio, I.I.C., et al. [46] | MK 2206 (pan-AKT inhibitor) | BSO | oncogenic-KRAS | Combined targeting of AKT and glutathione synthesis inhibits pancreatic cancer | human and mouse Kras mutant PDA cells, Suit2 xenograft model | ○ | |||||||
| Kerins, M.J., et al. [52] | Erastin, RSL3, ML162, glutamate | FH deficiency | Ferroptosis inducers are selectively toxic to FH−/− cell line UOK262, because C93 of GPX4 is post-translationally modified by fumarates that accumulate in conditions of FH−/−, and C93 modification represses GPX4 activity. | HK2 fumarate hydratase knockout cell lines | ○ | ○ | ○ | ||||||
| Okazaki, S., et al. [53] | Sulfasalazine (xc- inhibitor) | Dyclonine (oral anesthetics) | Sulfasalazine-resistant head and neck squamous cell carcinoma (HNSCC) cells highly express ALDH3A1 and knockdown of ALDH3A1 sensitized these cells to sulfasalazine. The combination of dyclonine and sulfasalazine cooperatively suppressed the growth of highly ALDH3A1-expressing HNSCC or gastric tumors that were resistant to sulfasalazine monotherapy. | Sulfasalazine-resistant human HNSCC cells | non-programmed cell death | ||||||||
| Bersuker, K., et al. [28] | RSL3 | FSP1 | In CRISPR-Cas9 screening, RSL3 induces synthetic lethality in U-2 OS FSP1 knockout cell lines. FSP1 acts parallel to GPX4 to inhibit ferroptosis. | U-2 OS FSP1 knockout cell lines | ○ | ||||||||
| Ogiwara, H., et al. [54] | GCL catalytic subunit (GCLC) | ARID1A deficiency | Cancer cells lacking ARID1A are specifically vulnerable to glutathione and inhibition of GCLC. | ARID1A-knockout HCT116 colon cancer cells | apoptosis | ||||||||
| To, T.L., et al. [55] | GPX4 | Mitochondrial Inhibitors (antimycin, oligomycin, ethidium bromide) | Genome-wide CRISPR screening using small molecule mitochondrial inhibitors showed that genes involved in the glycolytic system (PFKP), pentose phosphate pathway (G6PD), and defense against lipid peroxidation (GPX4) were closely associated with synthetic lethality. | K562, HAP1, HeLa | ○ | ○ | |||||||
| Hu, K., et al. [22] | SLC7A10 | oncogenic-KRAS | In KRAS-mutant lung adenocarcinoma transplanted mice, treatment with SLC7A11 inhibitor (HG106) resulted in tumor suppression and prolonged survival. | KRAS-mutant lung adenocarcinoma cells, preclinical lung cancer mouse model | apoptosis | ||||||||
| Kwon, O.S., et al. [47] | Erastin | oncogenic-RAS | Erastin is a synthetic lethal compound against cancer expressing an oncogenic RAS. The activity of transcription factors, including NRF2 and AhR, serve as important markers of erastin resistance. | mesenchymal lung cancer cell lines | ○ | ○ | ○ | ||||||
| Sugiyama, A., et al. [49] | Sulfasalazine (xc- inhibitor) | JNK (RAS effector) activation | Sulfasalazine is highly cytotoxic in paclitaxel-resistant uterine serous carcinoma. Interaction of ROS accumulation and JNK pathway activation increases susceptibility to SAS-induced ferroptosis. | human uterine serous carcinoma cell lines | ○ | ○ | |||||||
| Yusuf, R.Z., et al. [50] | RSL3, GPX4 knock down | Aldh3a2 inhibition | Aldh3a2 inhibition in combination with GPX4 inhibition leads to synthetic lethality in mouse and human AML cells. | human AML cells, Aldh-mut and Aldh-Ctrl mice were used to generate MLL-AF9 leukemia through retroviral transduction | ○ | ○ | ○ | ||||||
| Baird, L., et al. [56] | NRF2 | 17-AAG, 17-DMAG, IPI-504 | 17-AAG is synthetic lethal with NRF2 in human cancer cell lines | Keap1 knockout cells | No description of what type of cell death. | ||||||||
| Joly, J. H., et al. [57] | GSH | GLUT1 | Co-targeting GLUT1 and GSH synthesis induces synthetic lethal cell death in high xCT-expressing cell lines susceptible to glucose deprivation | xCT-high cell lines of glioblastoma and Ewing’s sarcoma | No description of what type of cell death. | ||||||||
| Lorenzato, A., et al. [58] | vitamine C | Cetuximab | The combination of vitamin C and cetuximab causes synthetic lethality triggered by ATP depletion and oxidative stress. This in turn suppresses the acquisition of resistance to anti-EGFR antibodies. | Advanced colorectal cancer patient-derived xenografts | ○ | ○ | ○ | ○ | |||||
| Verma, N., et al. [59] | Ferroptosis inducers (FIN56, erastin) | BET inhibitor (JQ1) + proteasome inhibitor (BTZ) | Co-inhibition of BET (bromodomain and extra-terminal) and proteasome induces ferotosis and synthetic lethality in triple-negative breast cancer (TNBC) cell lines. | TNBC cell lines | ○ | ○ | |||||||
| Baird, L. and M. Yamamoto. [60] | NRF2 | Mitomycin C | Aberrant NRF2 activation confers enhanced mitomycin C sensitivity in human cancer cell lines | NRF2-activated human cancer cells | No description of what type of cell death. | ||||||||
| Chen, S., et al. [61] | erastin | AOAA | AOAA and Erastin resulted in synthetic lethality both in vitro and in vivo, which was mediated through increased ferroptosis and apoptosis. | Tissue microarrays of colorectal cancer | apoptosis + ferroptosis | ||||||||
| Singhal, R., et al. [62] | Erastin, RSL3, DMF | HIF-2α | Ferroptosis inducer (Erastin, RSL3) and DMF led to selective synthetic lethality in HIF-2a expressing tumor enteroids. | colon cancer cells and colon tumors in mice | ○ | ○ | ○ | ○ | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinowaki, Y.; Taguchi, T.; Onishi, I.; Kirimura, S.; Kitagawa, M.; Yamamoto, K. Overview of Ferroptosis and Synthetic Lethality Strategies. Int. J. Mol. Sci. 2021, 22, 9271. https://doi.org/10.3390/ijms22179271
Kinowaki Y, Taguchi T, Onishi I, Kirimura S, Kitagawa M, Yamamoto K. Overview of Ferroptosis and Synthetic Lethality Strategies. International Journal of Molecular Sciences. 2021; 22(17):9271. https://doi.org/10.3390/ijms22179271
Chicago/Turabian StyleKinowaki, Yuko, Towako Taguchi, Iichiroh Onishi, Susumu Kirimura, Masanobu Kitagawa, and Kouhei Yamamoto. 2021. "Overview of Ferroptosis and Synthetic Lethality Strategies" International Journal of Molecular Sciences 22, no. 17: 9271. https://doi.org/10.3390/ijms22179271
APA StyleKinowaki, Y., Taguchi, T., Onishi, I., Kirimura, S., Kitagawa, M., & Yamamoto, K. (2021). Overview of Ferroptosis and Synthetic Lethality Strategies. International Journal of Molecular Sciences, 22(17), 9271. https://doi.org/10.3390/ijms22179271