
Effect of Angiotensin II on Chondrocyte Degeneration and Protection via Differential Usage of Angiotensin II Receptors


Abstract
:1. Introduction
2. Results
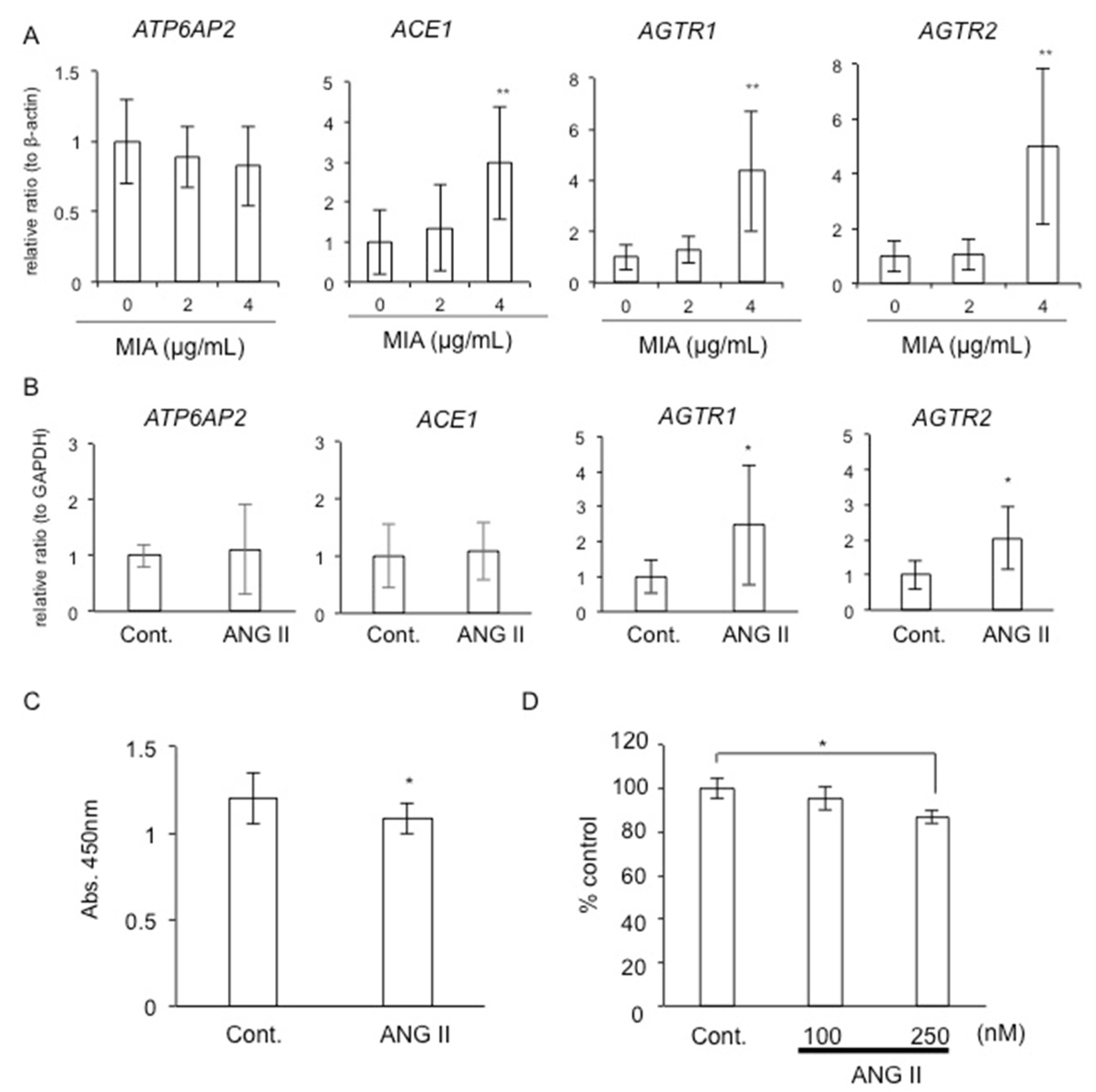
2.1. Gene Expression of Local RAS Components in HCS-2/8 Cells Treated with MIA
2.2. Gene Expression of Local RAS Components by ANG II
2.3. Effect of ANG II on Chondrocyte Proliferation and Differentiation
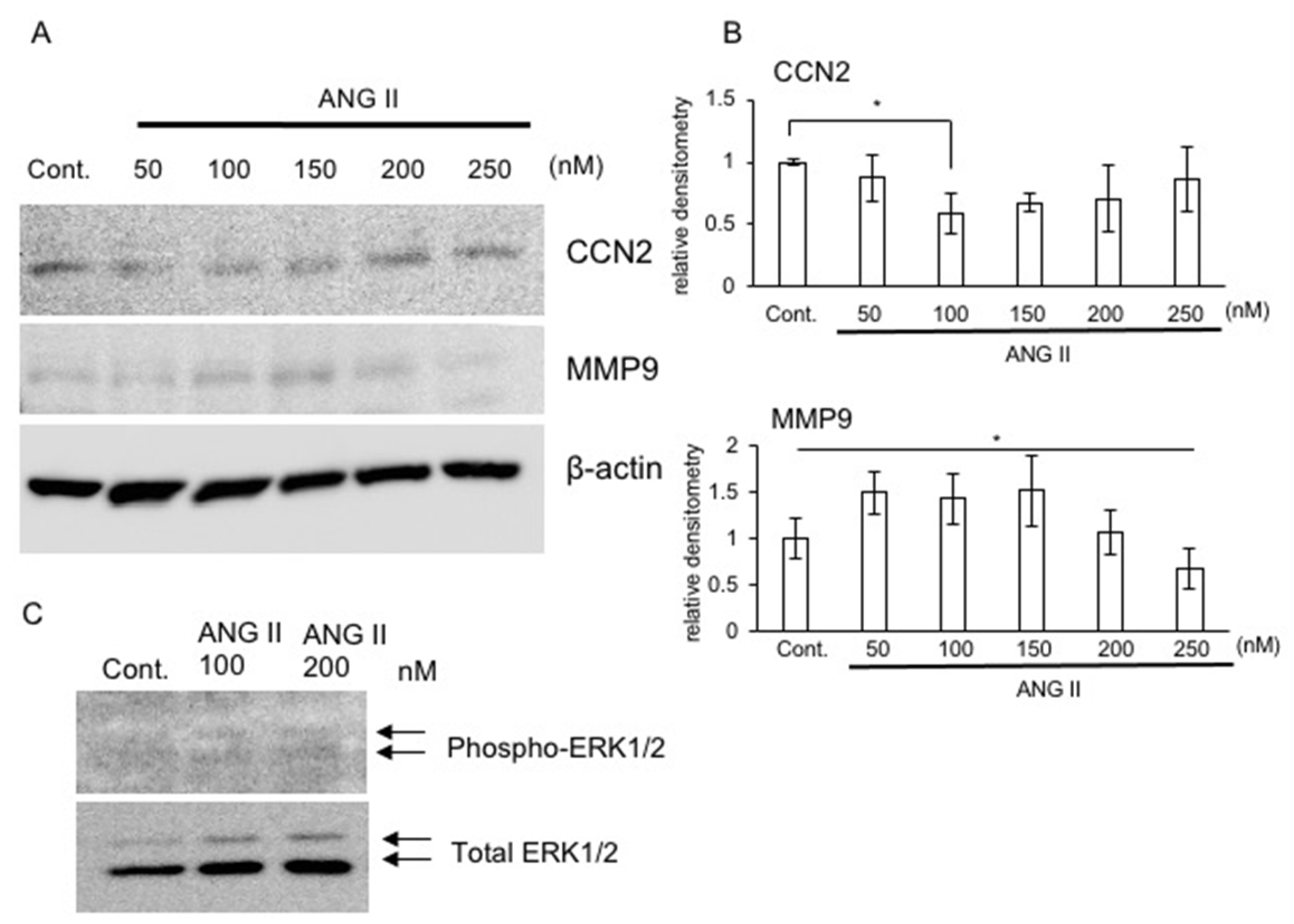
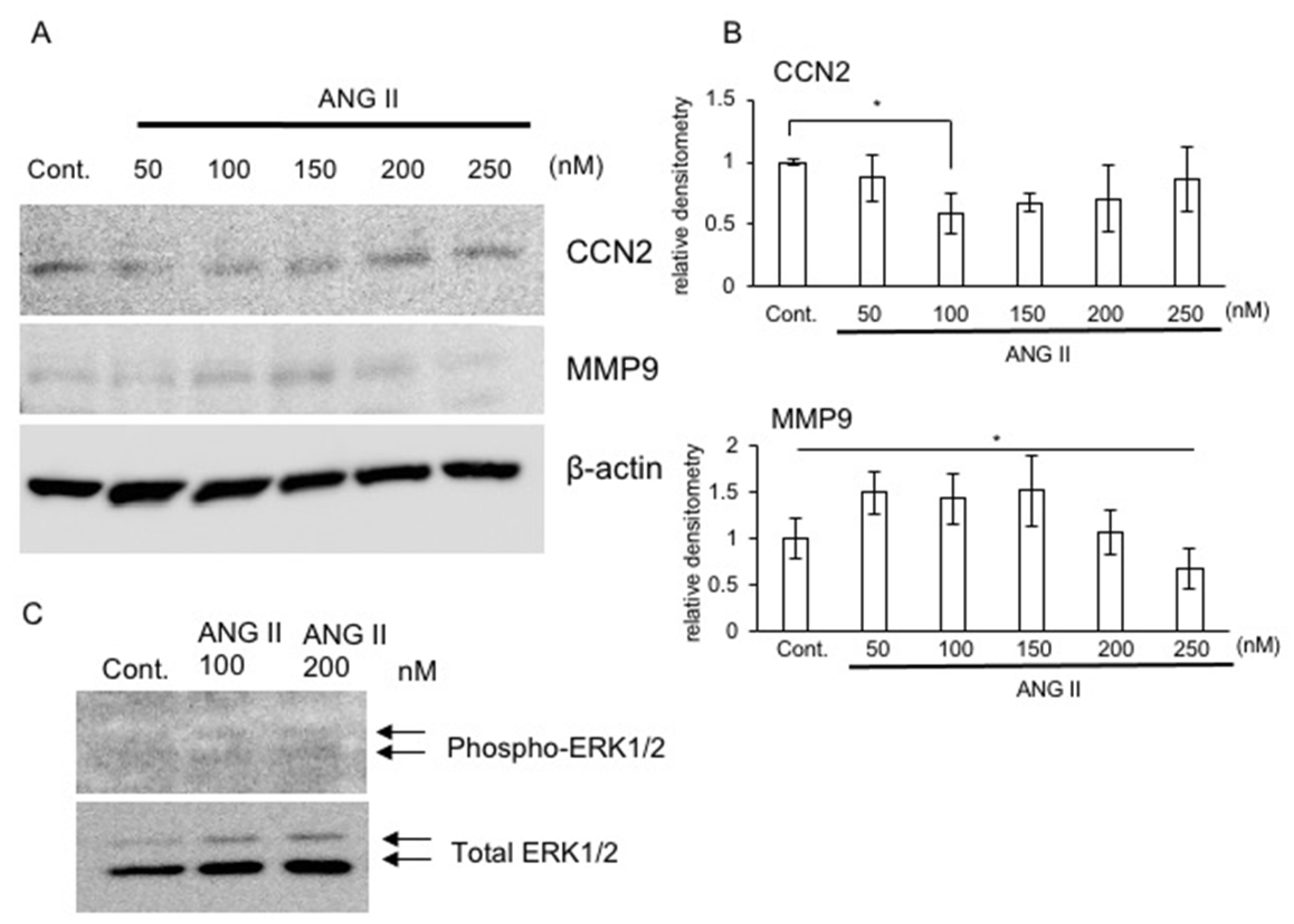
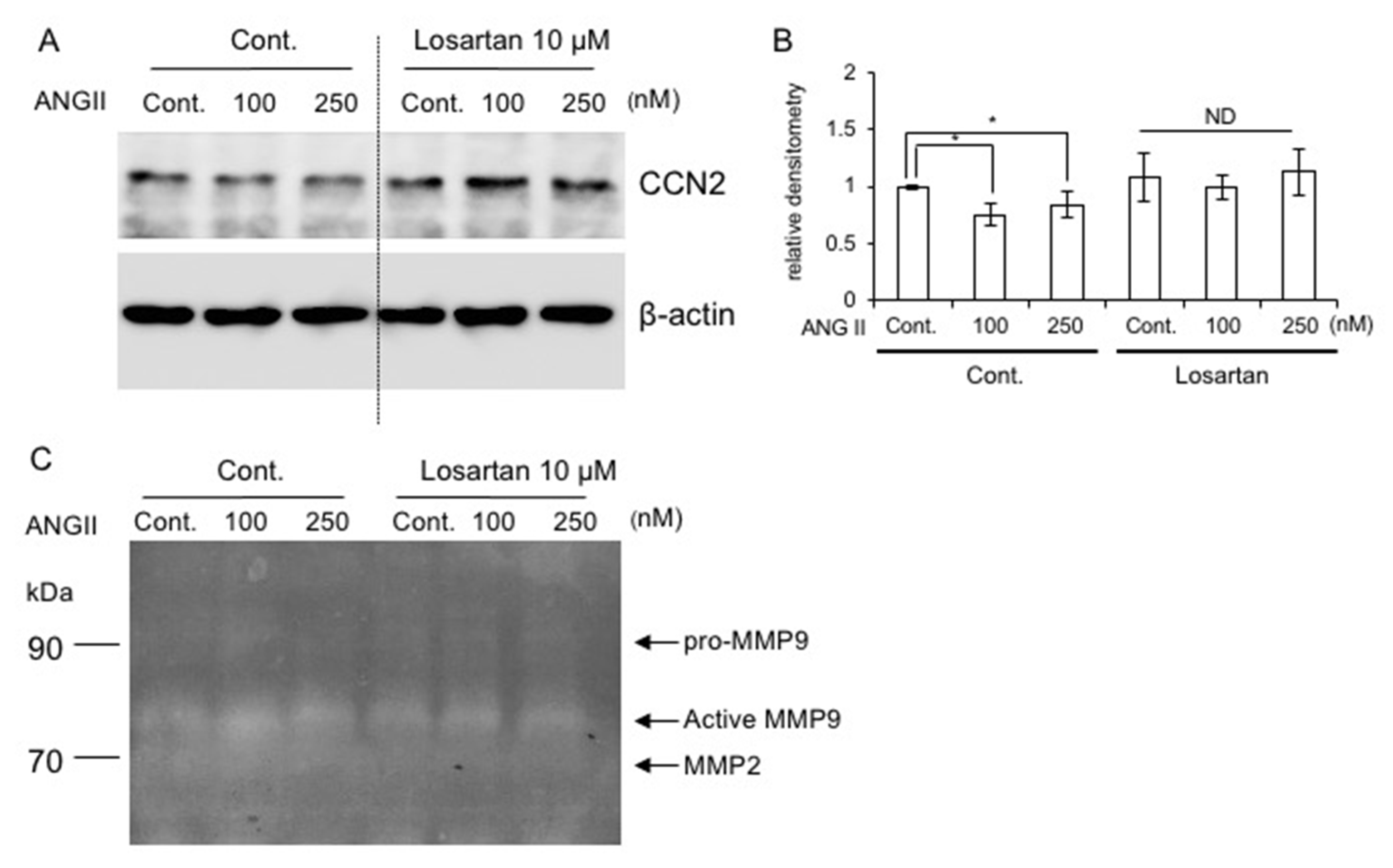
2.4. Production of CCN2 and MMP9 in the Presence of ANG II
2.5. Signaling Pathway of ANG II in RCS Cells
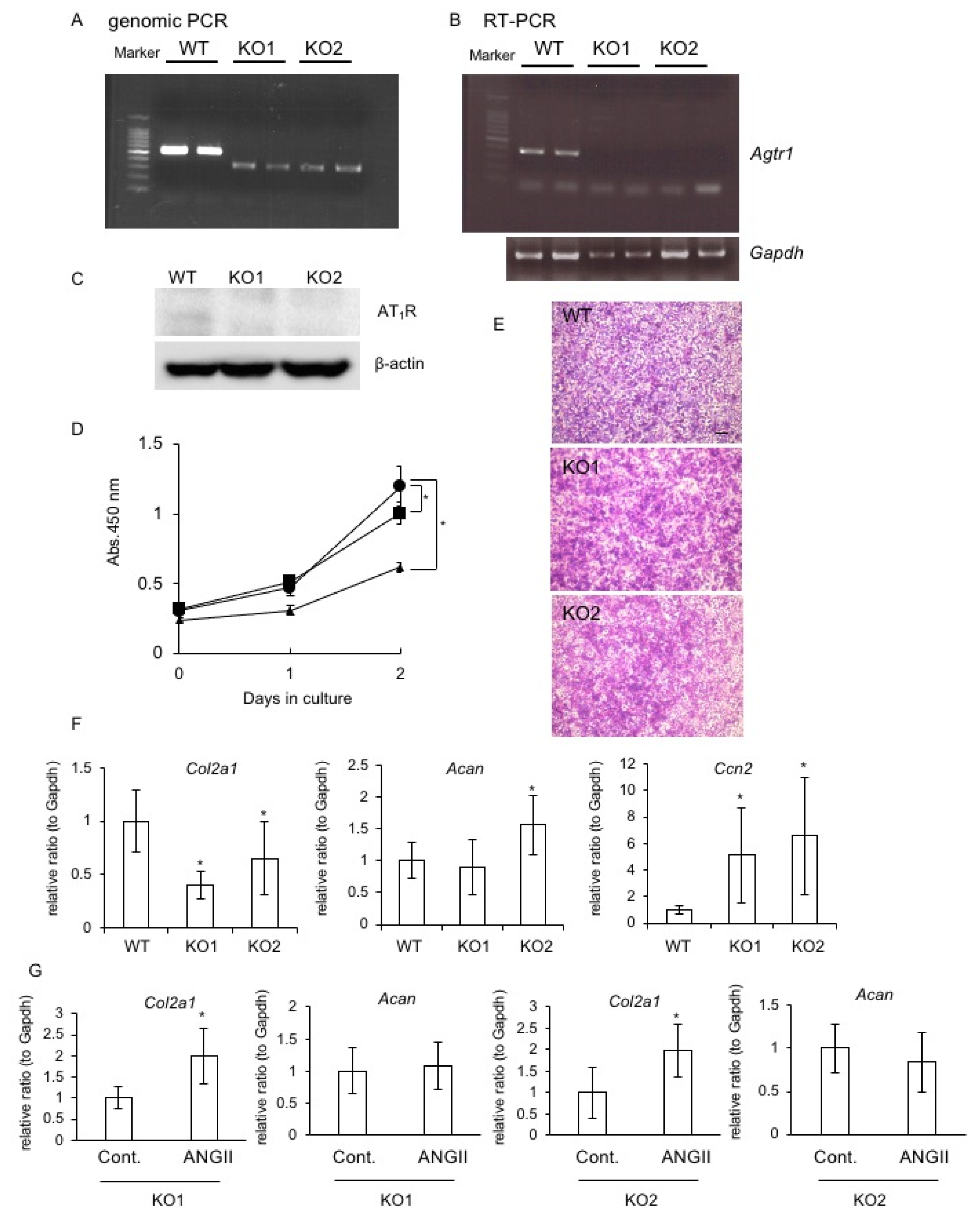
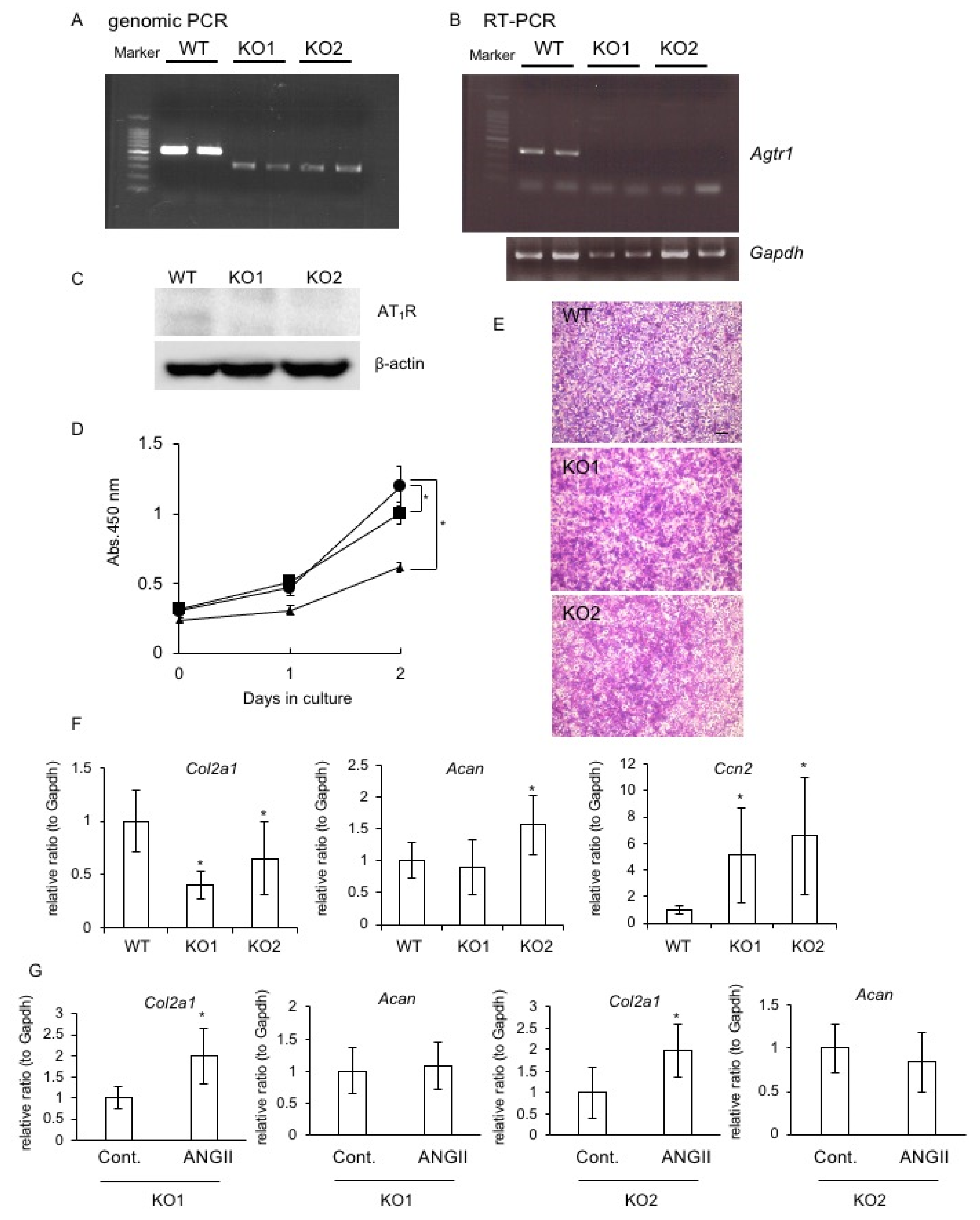
2.6. Generation of Agtr1a-Deficient RCS Cells and the Effect on Chondrocyte Proliferation and Differentiation
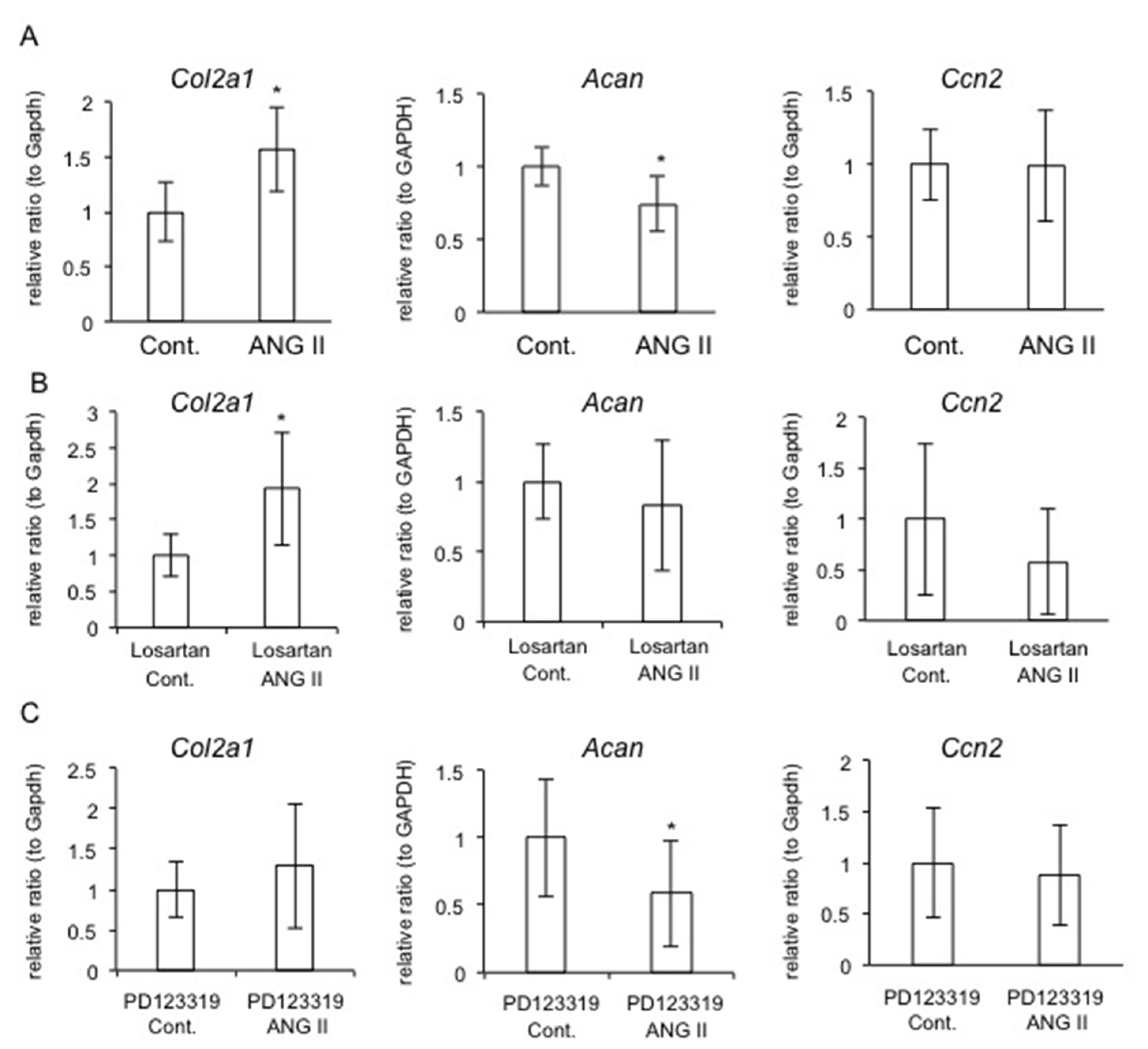
2.7. Effect of AT1R or AT2R Blockade on Col2a1, Acan, and Ccn2 Expression
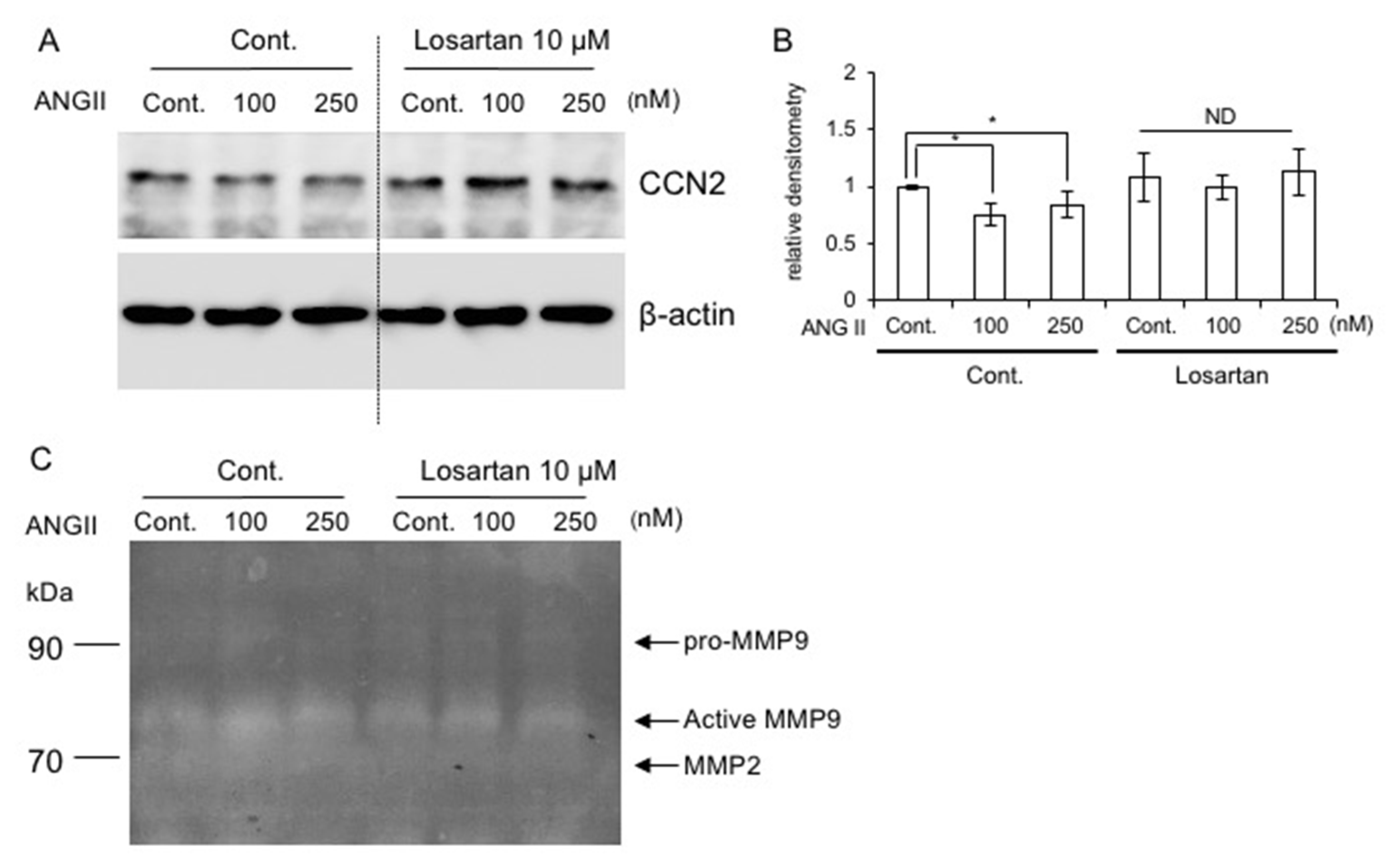
2.8. Effect of AT1R Blockade on CCN2 Production
2.9. Regulation of Gene Expression of Local RAS Components by Age
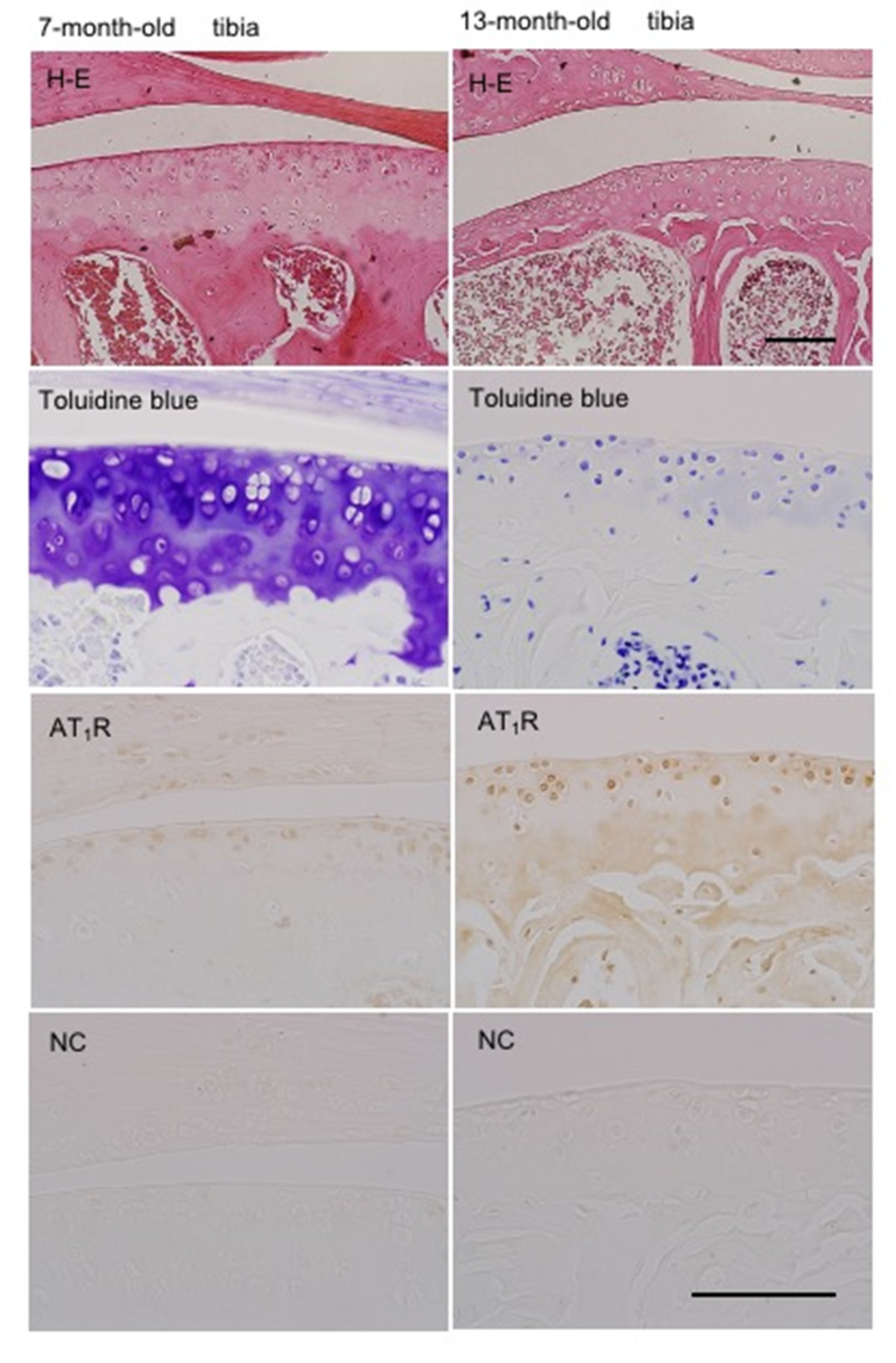
2.10. Localization of AT1R in Articular Cartilage According to Age
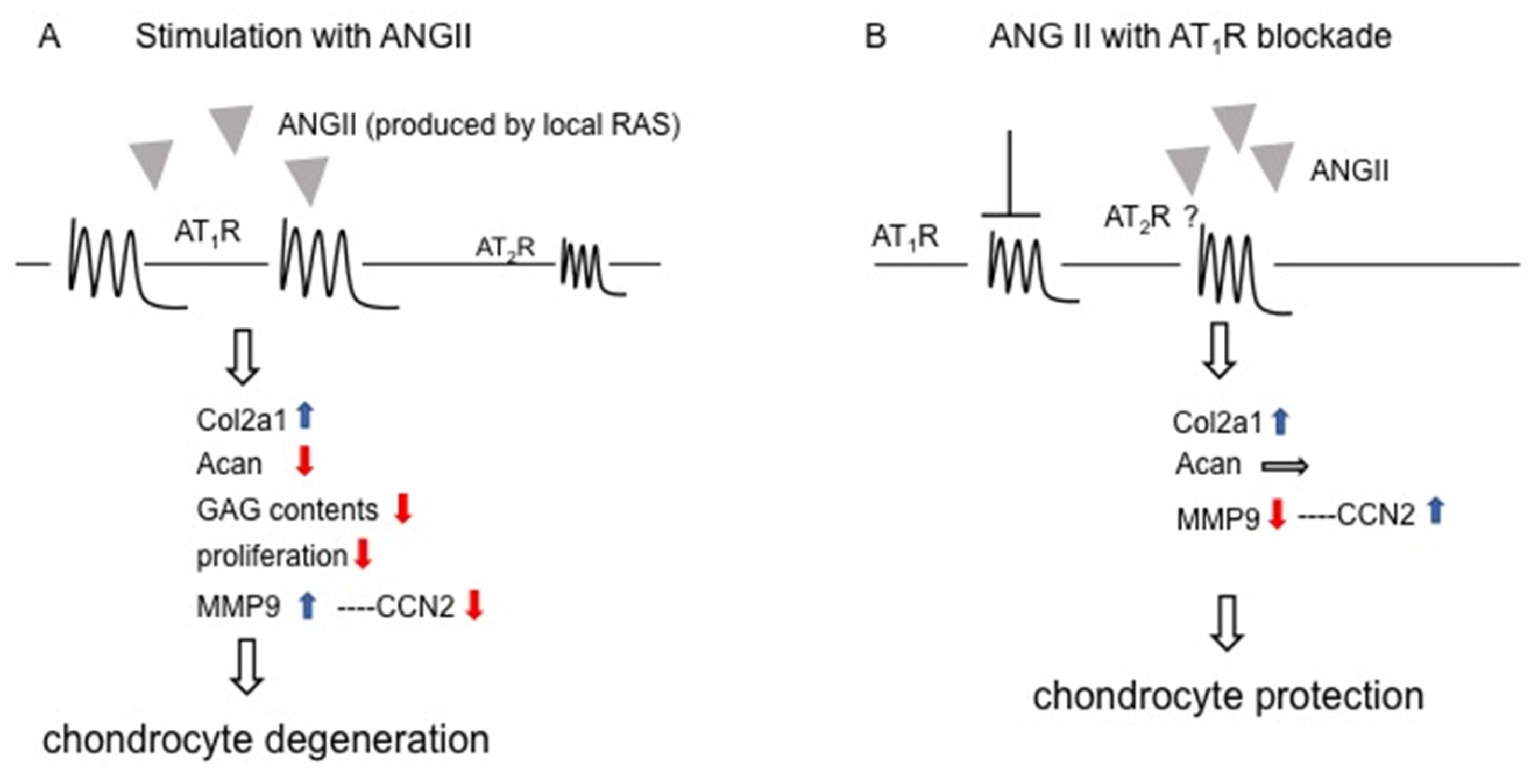
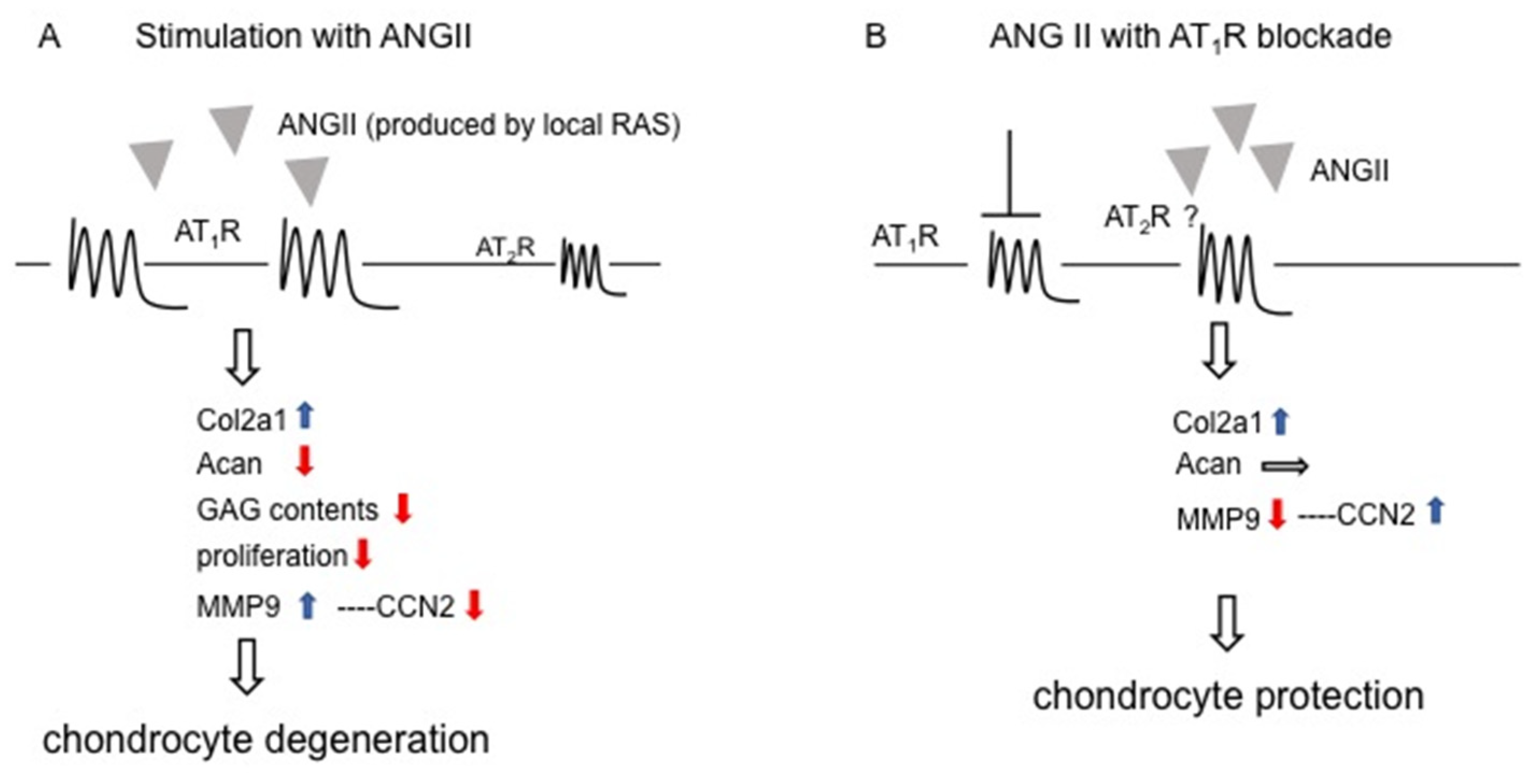
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Isolation of Chondrocytes from the Xiphoid Process
4.3. Cell Cultures
4.4. Generation of Agtr1a-Deficient RCS Cells by Using the CRISPER-Cas9 System
4.5. Quantitative Reverse Transcription (RT)-PCR Analysis
4.6. Western Blot Analysis
4.7. Cell Proliferation Assay
4.8. Measurement of Sulfated Glycosaminoglycan (GAG) Content
4.9. Gelatin Zymography
4.10. Immunohistochemistry
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Blagojevic, M.; Jinks, C.; Jeffery, A.; Jordan, K.P. Risk factors for onset of osteoarthritis of the knee in older adults: A systematic review and meta-analysis. Osteoarthr. Cartilage 2010, 18, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Chen, Q. Adipokines: New therapeutic target for osteoarthritis? Curr. Rheumatol. Rep. 2019, 21, 71. [Google Scholar] [CrossRef]
- Rasha, F.; Ramalingam, L.; Gollahon, L.; Layeequr Rahman, R.; Mizanoor Rahman, S.; Menikdiwela, K.; Moustaid-Moussa, N. Mechanisms linking the renin-angiotensin system, obesity, and breast cancer. Endocr. Relat. Cancer 2019, 26, R653–R672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, Y.; Matsuo, K.; Murata, M.; Yudoh, K.; Nakamura, H.; Shimizu, H.; Beppu, M.; Inaba, Y.; Saito, T.; Kato, T.; et al. Expression of angiotensin II receptor-1 in human articular chondrocytes. Arthritis 2012, 2021, 648537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, J.L.; Sigmund, C.D. Overview of the renin-angiotensin system-an endocrine and paracrine system. Endocrinology 2003, 144, 2179–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steckelings, U.M.; Kaschina, E.; Unger, T. The AT2 receptor--a matter of love and hate. Peptides 2005, 26, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Pahlavani, M.; Kalupahana, N.S.; Ramalingam, L.; Moustaid-Moussa, N. Regulation and functions of the renin-angiotensin system in white and brown adipose tissue. Comp. Physiol. 2017, 7, 1137–1150. [Google Scholar] [CrossRef]
- Mercure, C.; Thibault, G.; Lussier-Cacan, S.; Davignon, J.; Schiffrin, E.L.; Reudelhuber, T.L. Molecular analysis of human prorenin prosegment variants in vitro and in vivo. J. Biol. Chem. 1995, 270, 16355–16359. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.; Delarue, F.; Burcklé, C.A.; Bouzhir, L.; Giller, T.; Sraer, J.D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular response to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef]
- Nguyen, G.; Burcklé, C.A.; Sraer, J.D. Renin/prorenin-receptor biochemistry and functional significance. Cirr. Hypertens. Rep. 2004, 6, 129–132. [Google Scholar] [CrossRef]
- Li, W.; Peng, H.; Dale, M.; Seth, D.M.; Feng, Y. The prorenin and (pro)renin receptor: New players in the brain renin-angiotensin system? Int. J. Hypertens. 2012, 290635. [Google Scholar] [CrossRef] [PubMed]
- Quadri, S.S.; Cooper, C.; Ghaffar, D.; Vaishnav, H.; Nahar, L. The pathological role of pro(renin) receptor in renal inflammation. J. Exp. Pharmacol. 2021, 13, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Krop, M.; Lu, X.; Jan Danser, A.H.; Meima, M.E. The (pro)renin receptor. A decade of research: What have we learned? Pflug. Arch. 2013, 465, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin system. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Tsukamoto, I.; Akagi, M.; Inoue, S.; Yamagishi, K.; Mori, S.; Asada, S. Expressions of local renin-angiotensin system components in chondrocytes. Eur. J. Histochem. 2014, 58, 2387. [Google Scholar] [CrossRef]
- Tsukamoto, I.; Inoue, S.; Teramura, T.; Takehara, T.; Ohtani, K.; Akagi, M. Activating types 1 and 2 angiotensin II receptors modulate the hypertrophic differentiation of chondrocytes. FEBS Open Bio. 2013, 3, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakamatsu, T.; Iwasaki, Y.; Yamamoto, S.; Matsuo, K.; Goto, S.; Narita, I.; Kazama, J.J.; Tanaka, K.; Ito, A.; Ozasa, R.; et al. Type I angiotensin II receptor blockade reduces uremia-induced deterioration of bone material properties. J. Bone Min. Res. 2021, 36, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B.; Takigawa, M. CCN Proteins: A New Family of Cell Growth and Differentiation Regulators; Perbal, B., Takigawa, M., Eds.; Imperial College Press: London, UK, 2005; pp. 1–311. [Google Scholar]
- Takigawa, M. The CCN proteins: An overview. Methods Mol. Biol. 2017, 1489, 1–8. [Google Scholar] [CrossRef]
- Takigawa, M. CCN2: A master regulator of the genesis of bone and cartilage. J. Cell Commun. Signal. 2013, 7, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Nishida, T.; Shimo, T.; Kobayashi, K.; Kubo, T.; Tamatani, T.; Tezuka, K.; Takigawa, M. Effects of CTGF/Hcs24, a product of a hypertrophic chondrocyte-specific gene, on the proliferation and differentiation of chondrocytes in culture. Endocrinology 2000, 141, 264–273. [Google Scholar] [CrossRef]
- Nishida, T.; Nakanishi, T.; Asano, M.; Shimo, T.; Takigawa, M. Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J. Cell Physiol. 2000, 184, 197–206. [Google Scholar] [CrossRef]
- Nishida, T.; Emura, K.; Kubota, S.; Lyons, K.M.; Takigawa, M. CCN family 2/connective tissue growth factor (CCN2/CTGF) promotes osteoclastogenesis via induction of and interaction with dendritic cell-specific transmembrane protein (DC-STAMP). J. Bone Min. Res. 2011, 26, 351–363. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Kojima, S.; Kuboki, T.; Nakao, K.; Kushibiki, T.; Tabata, Y.; Takigawa, M. Regeneration of defects in articular cartilage in rat knee joints by CCN2 (connective tissue growth factor). J. Bone Min. Res. 2004, 19, 1308–1319. [Google Scholar] [CrossRef]
- Rüster, C.; Wolf, G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J. Am. Soc. Neprol. 2011, 22, 1189–1199. [Google Scholar] [CrossRef] [Green Version]
- Cabello-Verrugio, C.; Morales, M.G.; Cabrera, D.; Vio, C.P.; Brandan, E. Angiotnsin II receptor type I blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J. Cell Mol. Med. 2012, 16, 752–764. [Google Scholar] [CrossRef]
- Takigawa, M.; Tajima, K.; Pan, H.-O.; Enomoto, M.; Kinoshita, A.; Suzuki, F.; Takano, Y.; Mori, Y. Establishment of a clonal human chondrosarcoma cell line with cartilage phenotypes. Cancer Res. 1989, 49, 3996–4002. [Google Scholar] [PubMed]
- Mukhopadhyay, K.; Lefebvre, V.; Zhou, G.; Garofalo, S.; Kimura, J.H.; de Crombrugghe, B. Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-α1 (II) collagen gene. J. Biol. Chem. 1995, 270, 27711–27719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, S.; Cho, W.; Cho, H.; Lee, E.; Son, Y. Xiphoid process-derived chondrocytes: A novel cell source for elastic cartilage regeneration. Stem Cells Transl. Med. 2014, 3, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Gentili, C.; Cancedda, R. Cartilage and bone extracellular matrix. Curr. Pharm. Des. 2009, 15, 1334–1348. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Aoyama, E.; Yamanaka, N.; Lyons, K.M.; Takigawa, M. Low-intensity pulsed ultrasound (LIPUS) treatment of cultured chondrocytes stimulates production of CCN family protein 2 (CCN2), a protein involved in the regeneration of articular cartilage: Mechanism underlying this stimulation. Osteoarthr. Cartilage 2017, 25, 759–769. [Google Scholar] [CrossRef]
- Nishida, T.; Nagao, Y.; Hashitani, S.; Yamanaka, N.; Takigawa, M.; Kubota, S. Suppression of adipocyte differentiation by low-intensity pulsed ultrasound via inhibition of insulin signaling and promotion of CCN family protein 2. J. Cell Biochem. 2020, 29680. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Kubota, S.; Aoyama, E.; Takigawa, M. Impaired glycolytic metabolism causes chondrocyte hypertrophy-like changes via promotion of phospho-Smad1/5/8 translocation into nucleus. Osteoarthr. Cartilage 2013, 21, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Nakanishi, T.; Kimura, Y.; Hattori, T.; Sasaki, K.; Norimatsu, H.; Takahashi, K.; Takigawa, M. Nitric oxide mediates interleukin-1-induced matrix degradation and basic fibroblast growth factor release in cultured rabbit articular chondrocytes: A possible mechanism of pathological neovascularization in arthritis. Endocrinology 1996, 137, 3729–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, A.; Nishida, T.; Takashiba, S.; Kubota, S.; Takigawa, M. Regulatory mechanism of CCN2 production by serotonin (5-HT) via 5-HT2A and 5-HT2B receptors in chondrocytes. PLoS ONE 2017, 12, e0188014. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Primer Sequences | Expected Length (bp) | |
|---|---|---|---|
| WT | KO | ||
| Guide RNA1 | (F) 5′-TAATACGACTCACTATAGCGATGAAGTATACATTTCGG-3′ (R) 5′-TTCTAGCTCTAAAACCCACCGAAATGTATACTTCATCG-3′ | ||
| Guide RNA2 | (F) 5′-TAATACGACTCACTATAGTGGGCAGTCTATACCGCTA-3′ (R) 5′-TTCTAGCTCTAAAACTAGCGGTATAGACTGCCCA-3′ | ||
| Genomic PCR | (F) 5′-CAAGCGTCTTTCTTCTCAATCTC-3′ (R) 5′-GATGTCATCGTTTCTTGGTTTG-3′ | 526 | 290 |
| RT-PCR | (F) 5′-TGGGCAGTCTATACCGCTA-3′ (R) 5′-GCCCTATGGGGAGCGTCGAA-3′ | 334 | ND |
| Gene | Accession No. | Species | Primer Sequences | PCR Product Length (bp) |
|---|---|---|---|---|
| GAPDH | XM_011241214.1 | human /mouse | (F) 5′-GCCAAAAGGGTCATCATCTC-3′ (R) 5′-GTCTTCTGGGTGGCAGTGAT-3′ | 214 |
| ATP6AP2 | NM_005765.3 | human | (F) 5′-CCTCATTAGGAAGACAAGGACTATCC-3′ (R) 5′-GGGTTCTTCGCTTGTTTTGC-3′ | 50 |
| ACE1 | NM_000789.4 | human | (F) 5′-AAGCAGGACGGCTTCACAGA-3′ (R) 5′-GGGTCCCCTGAGGTTGATGTAT-3′ | 182 |
| AGTR1 | NM_009585.4 | human | (F) 5′-CTTCAGCCAGCGTCAGTT-3′ (R) 5′-TGCAGGTGACTTTGGCTA-3′ | 137 |
| AGTR2 | NM_000686.5 | human | (F) 5′-ACTTCGGGCTTGTGAACATC-3′ (R) 5′-TAAATCAGCCACAGCGAGGT-3′ | 217 |
| ACTB | NM_001101.3 | human | (F) 5′-GATCATTGCTCCTCCTGAGC-3′ (F) 5′-ACTCCTGCTTGCTGATCCAC-3′ | 100 |
| Acan | XM_039101035.1 | rat | (F) 5′-GATGTCCCCTGCAATTACCA-3′ (R) 5′-TCTGTGCAAGTGATTCGAGG-3′ | 229 |
| Col2a1 | NM_012929.1 | rat | (F) 5′-CCCAGAACATCACCTACCAC-3′ (R) 5′-GGTACTCGATGATGGTCTTG-3′ | 200 |
| Ccn2 | NM_022266.2 | rat | (F) 5′-ATCCCTGCGACCCACACAAG-3′ (R) 5′-CAACTGCTTTGGAAGGACTCGC-3′ | 144 |
| Gapdh | NM_017008.4 | rat | (F) 5′-GCCAAAAGGGTCATCATCTC-3′ (R) 5′-GTCTTCTGAGTGGCAGTGAT-3′ | 214 |
| CCN2 | NM_010217.2 | mouse | (F) 5′-CCACCCGAGTTACCAATGAC -3′ (R) 5′-GTGCAGCCAGAAAGCTCA-3′ | 151 |
| Atp6ap2 | NM_027439.4 | mouse | (F) 5′-CACATTGCGGCAGCTCCGTAA-3′ (R) 5′-CTCACAAGGGATGTGTCGAAT-3′ | 377 |
| Ace1 | NM_207624.6 | mouse | (F) 5′-AACAAACATGATGGCCACATCCCG-3′ (R) 5′-CGTGTAGCCATTGAGCTTGGCAAT-3′ | 147 |
| Agtr1 | NM_177322.3 | mouse | (F) 5′-CCATTGTCCACCCGATGAAG-3′ (R) 5′-TGCAGGTGACTTTGGCCAC-3′ | 62 |
| Agtr2 | NM_007429.5 | mouse | (F) 5′-CAGCAGCCGTCCTTTTGATAA-3′ (R) 5′-TTATCTGATGGTTTGTGTGAGCAA -3′ | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, T.; Akashi, S.; Takigawa, M.; Kubota, S. Effect of Angiotensin II on Chondrocyte Degeneration and Protection via Differential Usage of Angiotensin II Receptors. Int. J. Mol. Sci. 2021, 22, 9204. https://doi.org/10.3390/ijms22179204
Nishida T, Akashi S, Takigawa M, Kubota S. Effect of Angiotensin II on Chondrocyte Degeneration and Protection via Differential Usage of Angiotensin II Receptors. International Journal of Molecular Sciences. 2021; 22(17):9204. https://doi.org/10.3390/ijms22179204
Chicago/Turabian StyleNishida, Takashi, Sho Akashi, Masaharu Takigawa, and Satoshi Kubota. 2021. "Effect of Angiotensin II on Chondrocyte Degeneration and Protection via Differential Usage of Angiotensin II Receptors" International Journal of Molecular Sciences 22, no. 17: 9204. https://doi.org/10.3390/ijms22179204
APA StyleNishida, T., Akashi, S., Takigawa, M., & Kubota, S. (2021). Effect of Angiotensin II on Chondrocyte Degeneration and Protection via Differential Usage of Angiotensin II Receptors. International Journal of Molecular Sciences, 22(17), 9204. https://doi.org/10.3390/ijms22179204