
Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-κB Pathway in BV2 Microglial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
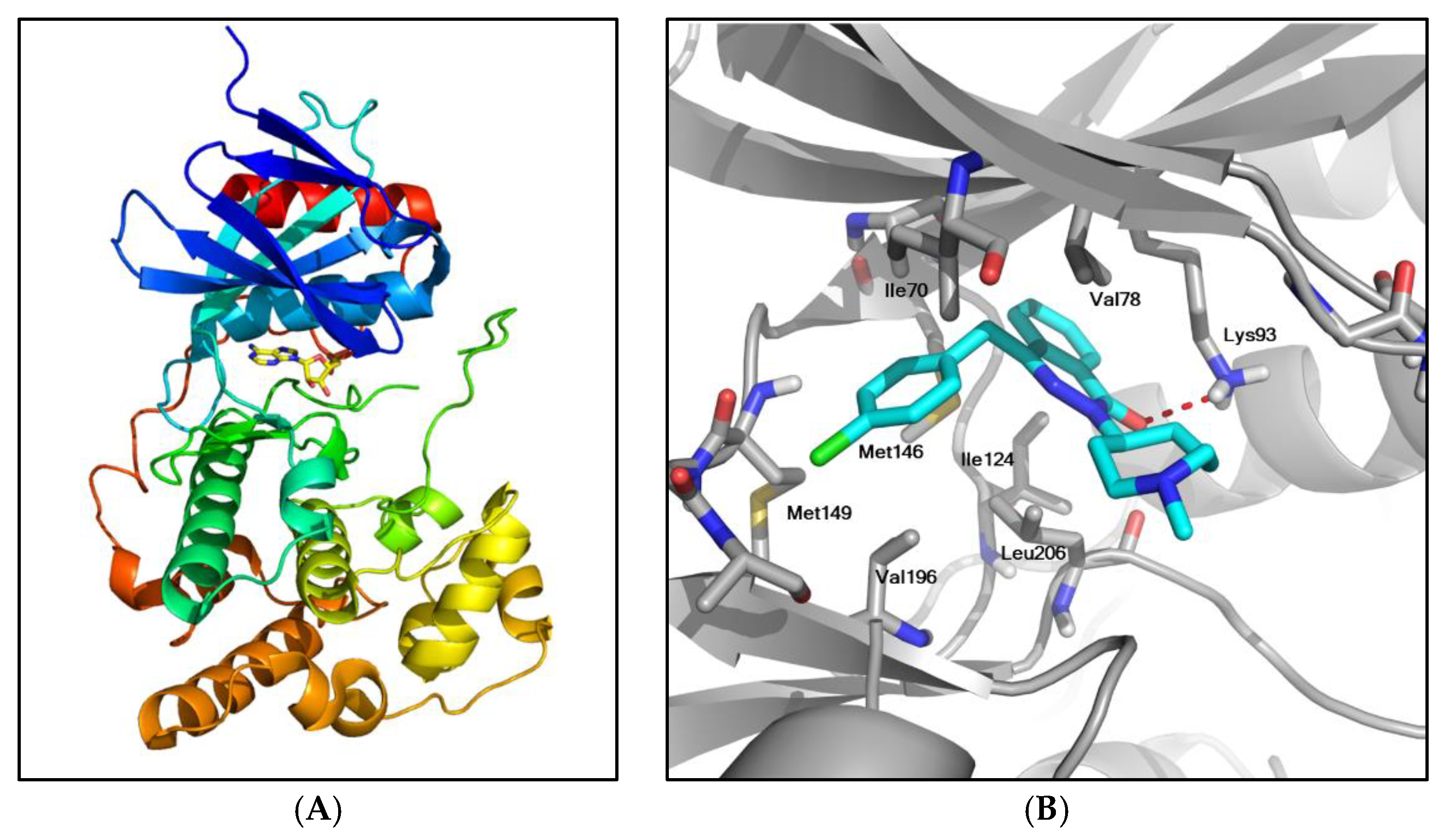
2.1. Structure-Based Virtual Screening
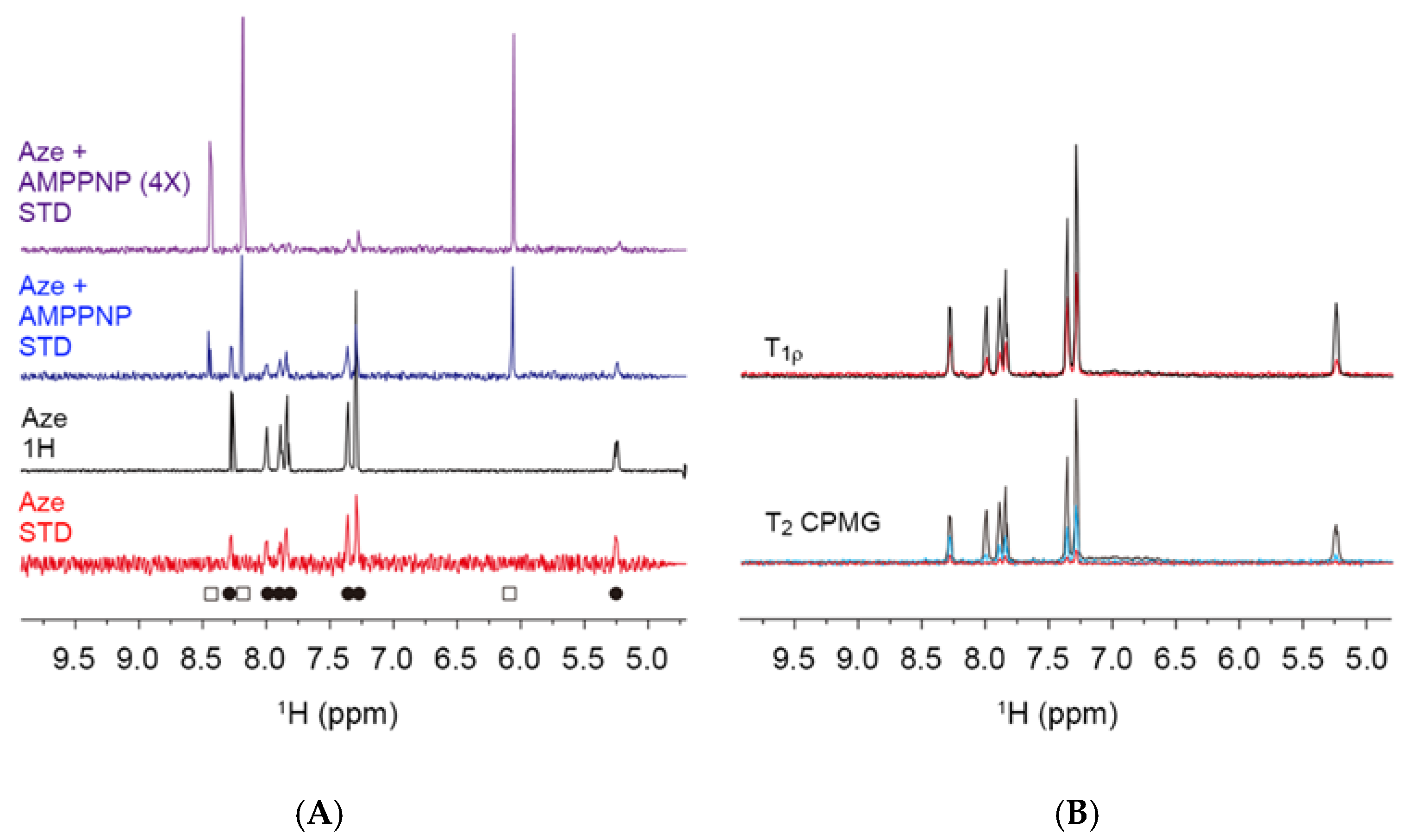
2.2. Nuclear Magnetic Resonance Spectroscopy
2.3. Effect of Aze on JNK3 Activity
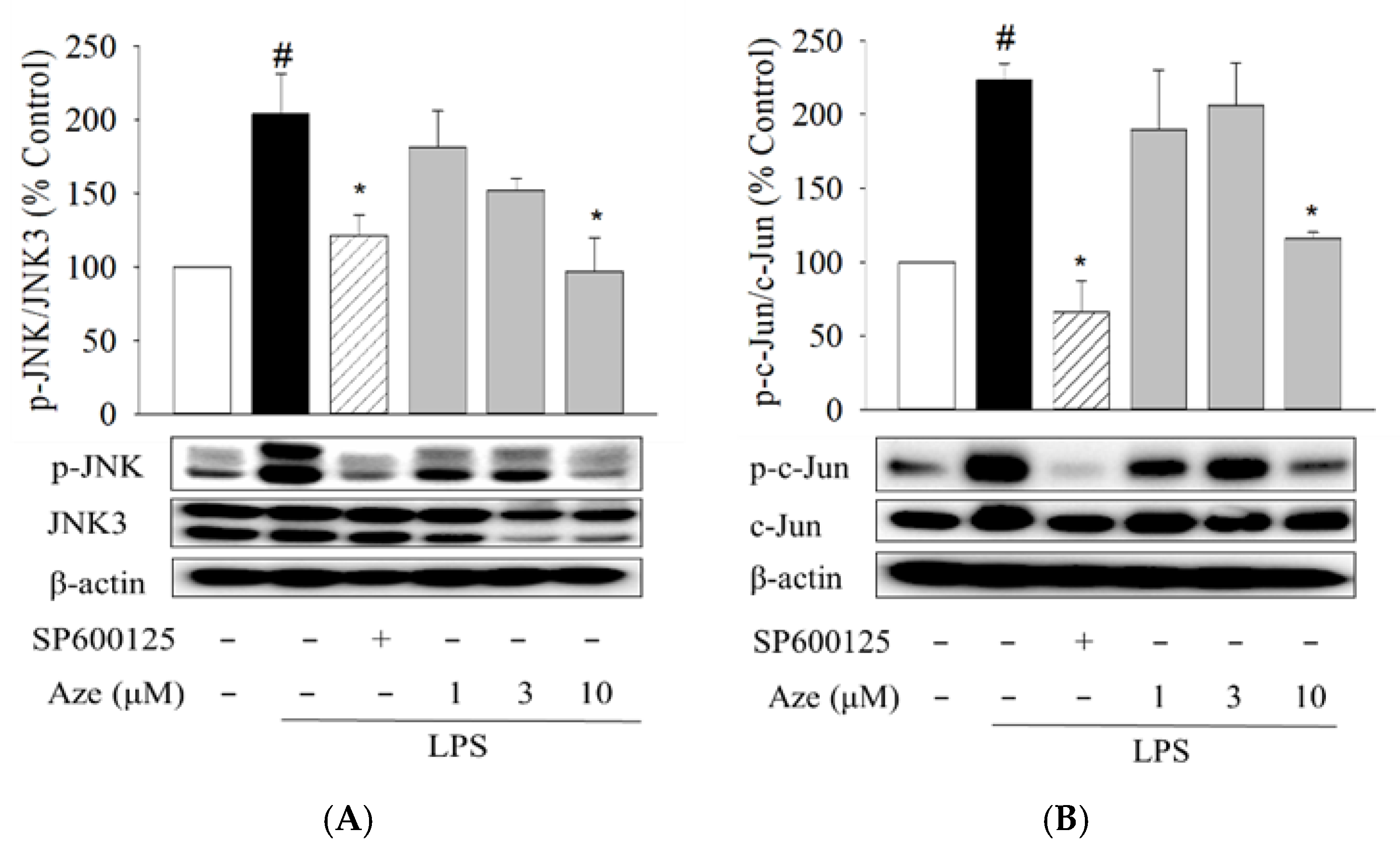
2.4. Effect of Aze on the LPS-Induced Phosphorylation of JNK and c-Jun in BV2 Cells
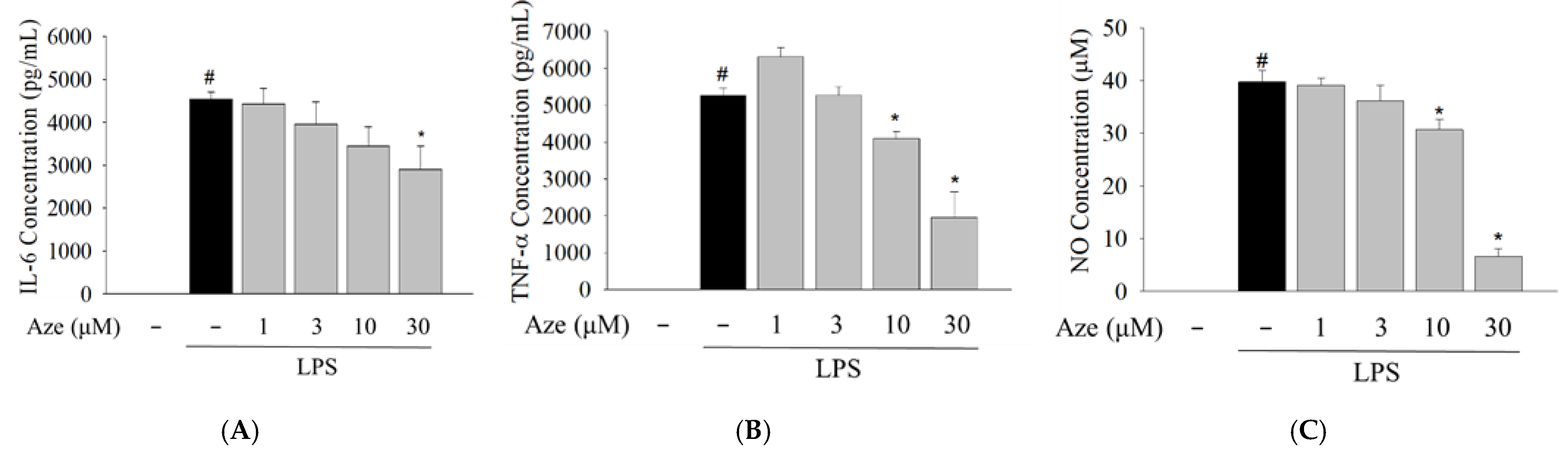
2.5. Effect of Aze on the LPS-Induced Production of Pro-Inflammatory Mediators in BV2 Cells
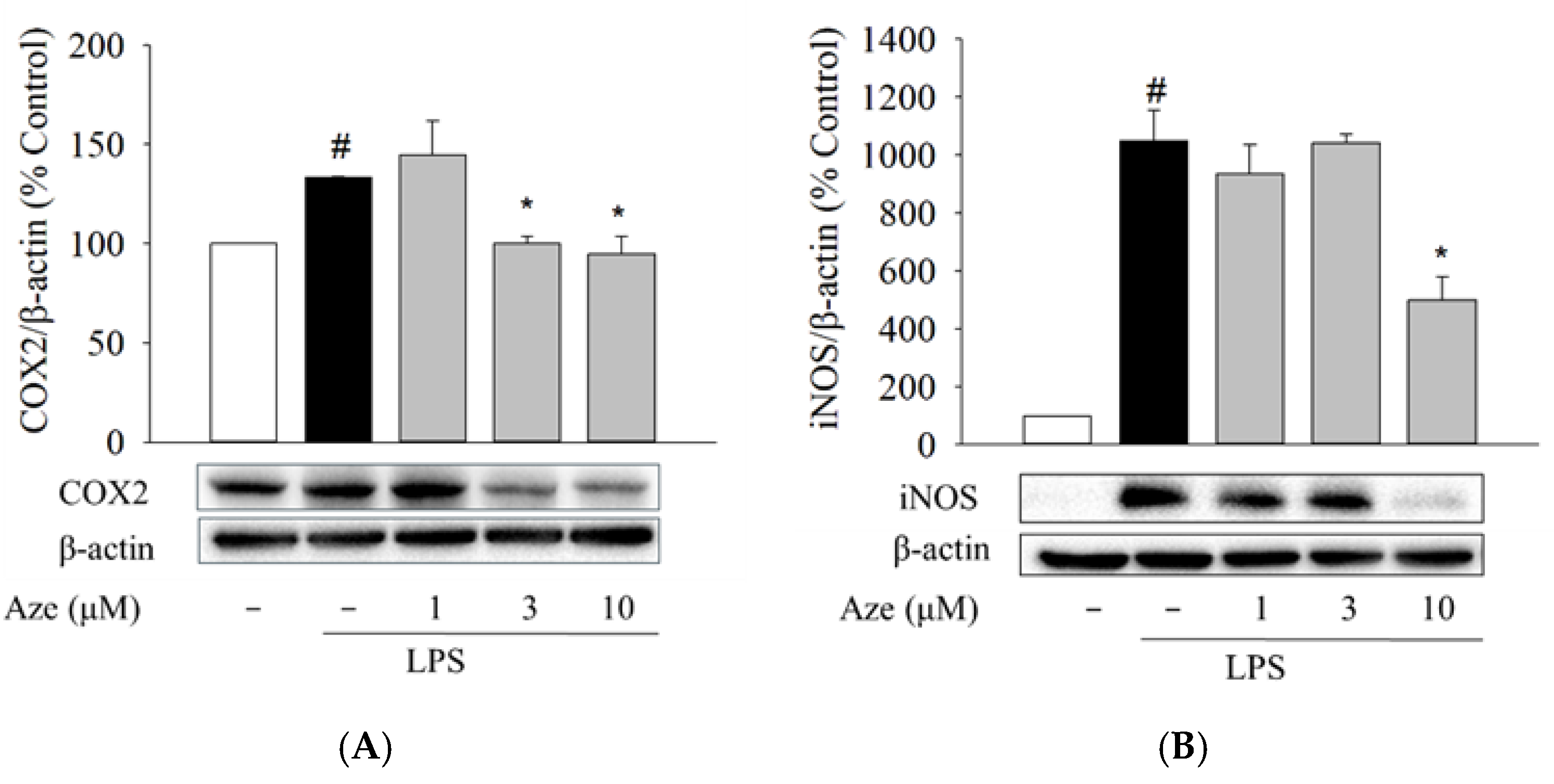
2.6. Effect of Aze on the LPS-Induced Expression of COX2 and iNOS in BV2 Cells
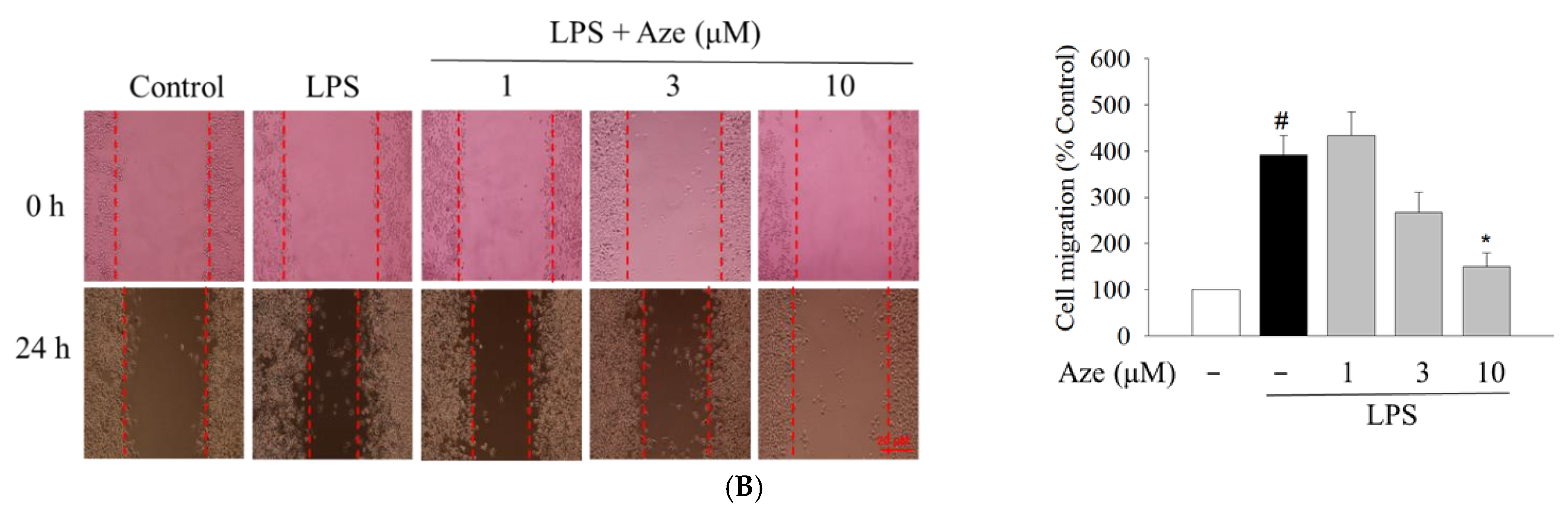
2.7. Effect of Aze on the LPS-Induced Cell Migration in BV2 Cells
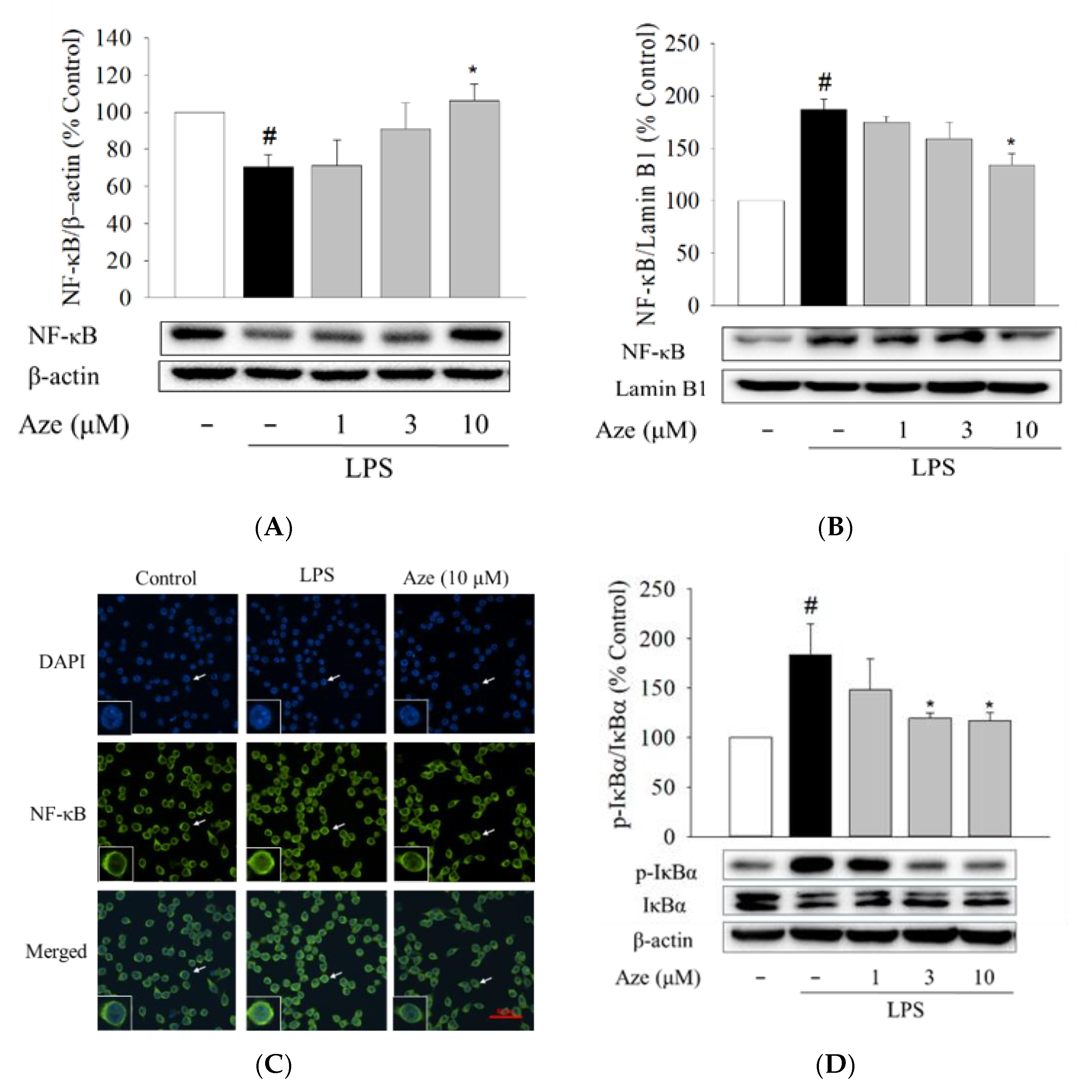
2.8. Effect of Aze on the LPS-Induced Nuclear Translocation of NF-κB in BV2 Cells
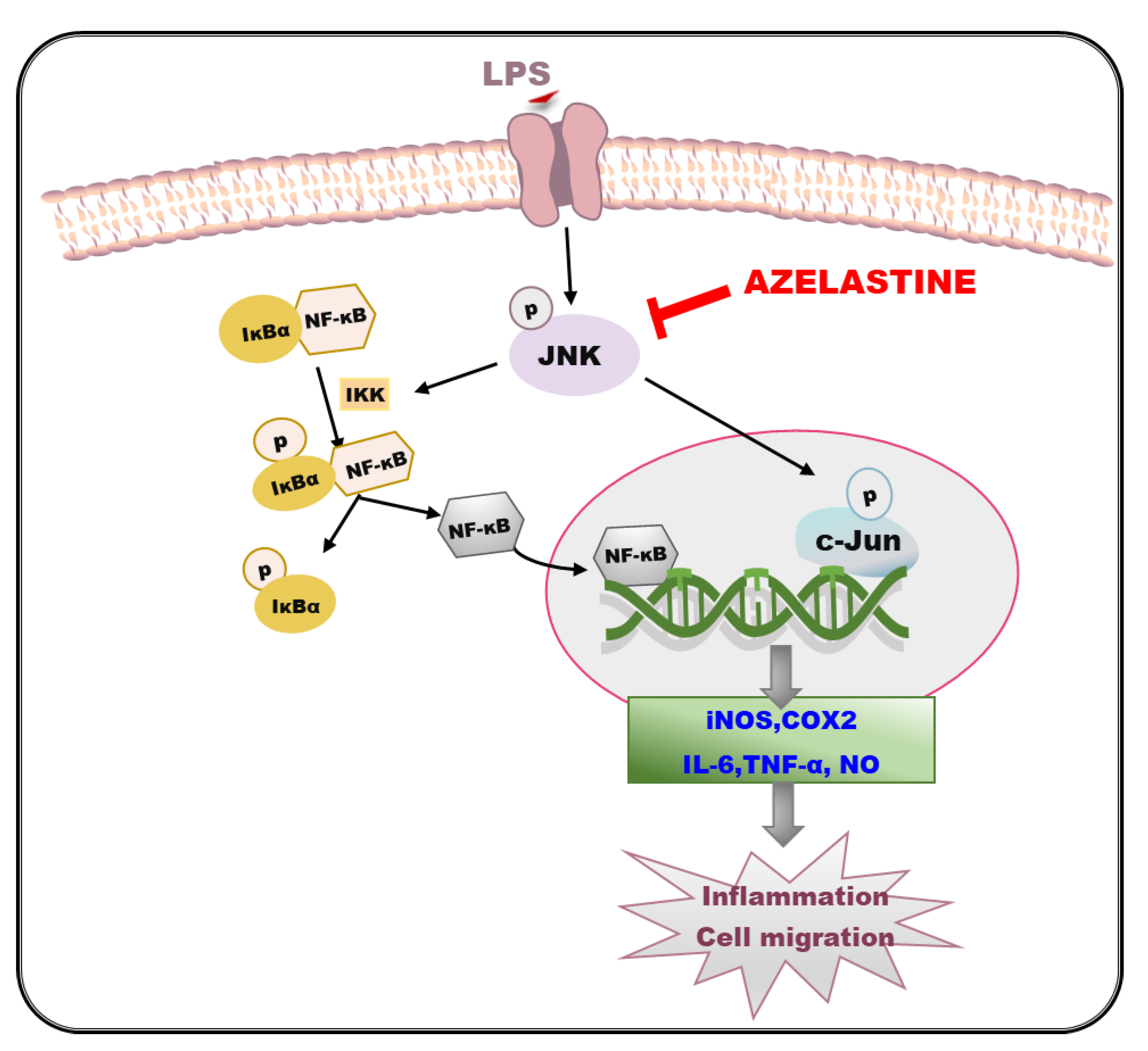
3. Discussion
4. Materials and Methods
4.1. Materials and Chemicals
4.2. Virtual Screening
4.3. NMR Study for Binding of Aze to JNK3
4.4. JNK3 Activity Assay
4.5. BV2 Cell Culture and Treatment of Cells
4.6. Western Blotting
4.7. Determinations of IL-6, TNF-α, and NO
4.8. Cell Migration Assays
4.8.1. Transwell Migration Assay
4.8.2. Wound Healing Assay
4.9. Immunocytochemistry
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPPNP | Adenylyl-imidodiphosphate |
| Aze | Azelastine |
| COX2 | Cyclooxygenase-2 |
| FDA | U.S. Food and Drug Administration |
| JNK | c-Jun N-terminal kinases |
| IκBs | Inhibitors of kappa |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| LPS | Lipopolysaccharide |
| NF-κB | Nuclear factor-kappa B |
| NMR | Nuclear magnetic resonance |
| NO | Nitric oxide |
| PDB | Protein data bank |
| SBVS | Structure-based virtual screening |
| SDF | Structure data file |
| STD | Saturation transfer difference |
| TNF-α | Tumor necrosis factor-alpha |
References
- Olajide, O.A.; Sarker, S.D. Chapter five—Anti-inflammatory natural products. In Annual Reports in Medicinal Chemistry; Sarker, S.D., Nahar, L., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 55, pp. 153–177. [Google Scholar]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; He, D.; Bai, Y. Microglia-mediated inflammation and neurodegenerative disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Wu, H.J.; Li, H.Q.; Qin, S.; Wang, Y.E.; Li, J.; Lou, H.F.; Chen, Z.; Li, X.M.; Luo, Q.M.; et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhara, D.; Oh, U.; Neigh, G.N. Neuroinflammation; Elsevier: Amsterdam, The Netherlands, 2020; Volume 175, pp. 235–259. [Google Scholar]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Mohit, A.A.; Martin, J.H.; Miller, C.A. p493F12 kinase: A novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 1995, 14, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, X.; Borsello, T. The JNK signalling transduction pathway in the brain. Front. Biosci. 2012, 4, 2110–2120. [Google Scholar] [CrossRef]
- Waetzig, V.; Czeloth, K.; Hidding, U.; Mielke, K.; Kanzow, M.; Brecht, S.; Goetz, M.; Lucius, R.; Herdegen, T.; Hanisch, U.K. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 2005, 50, 235–246. [Google Scholar] [CrossRef]
- Haeusgen, W.; Boehm, R.; Zhao, Y.; Herdegen, T.; Waetzig, V. Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience 2009, 161, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Kelley, K.W.; Johnson, R.W. Luteolin reduces IL-6 production in microglia by inhibiting JNK phosphorylation and activation of AP-1. Proc. Natl. Acad. Sci. USA 2008, 105, 7534–7539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocivavsek, A.; Burns, M.P.; Rebeck, G.W. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia 2009, 57, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, J.P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, P.L.; Settipane, R.A. Azelastine nasal spray: A review of pharmacology and clinical efficacy in allergic and nonallergic rhinitis. Allergy Asthma Proc. 2003, 24, 95–105. [Google Scholar]
- Bernstein, J.A. Azelastine hydrochloride: A review of pharmacology, pharmacokinetics, clinical efficacy and tolerability. Curr. Med. Res. Opin. 2007, 23, 2441–2452. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Mika, J. Targeting the microglial signaling pathways: New insights in the modulation of neuropathic pain. Curr. Med. Chem. 2016, 23, 2908–2928. [Google Scholar] [CrossRef] [Green Version]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Park, S.E.; Sapkota, K.; Kim, S.; Kim, H.; Kim, S.J. Kaempferol acts through mitogen-activated protein kinases and protein kinase B/AKT to elicit protection in a model of neuroinflammation in BV2 microglial cells. Br. J. Pharmacol. 2011, 164, 1008–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zheng, Y.; Luo, Y.; Du, Y.; Zhang, X.; Fu, J. Curcumin inhibits LPS-induced neuroinflammation by promoting microglial M2 polarization via TREM2/ TLR4/ NF-κB pathways in BV2 cells. Mol. Immunol. 2019, 116, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, R.; Tong, Y.; Chen, P.; Shen, Y.; Miao, S.; Liu, X. Neuroprotection by dihydrotestosterone in LPS-induced neuroinflammation. Neurobiol. Dis. 2020, 140, 104814. [Google Scholar] [CrossRef]
- Nijboer, C.H.; van der Kooij, M.A.; van Bel, F.; Ohl, F.; Heijnen, C.J.; Kavelaars, A. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav. Immun. 2010, 24, 812–821. [Google Scholar] [CrossRef]
- Xie, X.; Gu, Y.; Fox, T.; Coll, J.T.; Fleming, M.A.; Markland, W.; Caron, P.R.; Wilson, K.P.; Su, M.S. Crystal structure of JNK3: A kinase implicated in neuronal apoptosis. Structure 1998, 6, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Viegas, A.; Manso, J.; Nobrega, F.L.; Cabrita, E.J. Saturation-transfer difference (STD) NMR: A simple and fast method for ligand screening and characterization of protein binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Olejniczak, E.T.; Fesik, S.W. One-dimensional relaxation- and diffusion-edited NMR methods for screening compounds that bind to macromolecules. J. Am. Chem. Soc. 1997, 119, 12257–12261. [Google Scholar] [CrossRef]
- Salvi, N.; Buratto, R.; Bornet, A.; Ulzega, S.; Rentero Rebollo, I.; Angelini, A.; Heinis, C.; Bodenhausen, G. Boosting the sensitivity of ligand–protein screening by NMR of long-lived states. J. Am. Chem. Soc. 2012, 134, 11076–11079. [Google Scholar] [CrossRef] [Green Version]
- Mehan, S.; Meena, H.; Sharma, D.; Sankhla, R. JNK: A stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neurodegenerative abnormalities. J. Mol. Neurosci. 2011, 43, 376–390. [Google Scholar] [CrossRef]
- Zhang, J.; Lin, W.; Tang, M.; Zhao, Y.; Zhang, K.; Wang, X.; Li, Y. Inhibition of JNK ameliorates depressive-like behaviors and reduces the activation of pro-inflammatory cytokines and the phosphorylation of glucocorticoid receptors at serine 246 induced by neuroinflammation. Psychoneuroendocrinology 2020, 113, 104580. [Google Scholar] [CrossRef]
- Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.K.; Lee, H.; Cho, J. Anti-inflammatory and anti-migratory activities of isoquinoline-1-carboxamide derivatives in LPS-treated BV2 microglial cells via inhibition of MAPKs/NF-κB pathway. Int. J. Mol. Sci. 2020, 21, 2319. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.L.; Bui, B.P.; Lee, H.; Cho, J. A novel 1,8-naphthyridine-2-carboxamide derivative attenuates inflammatory responses and cell migration in LPS-treated BV2 cells via the suppression of ROS generation and TLR4/Myd88/NF-κB signaling pathway. Int. J. Mol. Sci. 2021, 22, 2527. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.N.; Zong, Y.; Zhong, L.M.; Li, Y.M.; Zhang, W.; Bian, L.G.; Ai, Q.L.; Liu, Y.D.; Sun, J.; Lu, D. Gastrodin inhibits expression of inducible NO synthase, cyclooxygenase-2 and proinflammatory cytokines in cultured LPS-stimulated microglia via MAPK pathways. PLoS ONE 2011, 6, e21891. [Google Scholar] [CrossRef]
- Miller, A.M.; Stella, N. Microglial cell migration stimulated by ATP and C5a involve distinct molecular mechanisms: Quantification of migration by a novel near-infrared method. Glia 2009, 57, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, M.L.; Ramani, K.; Ryoo, M. Ubiquitin-conjugating enzyme 9 phosphorylation as a novel mechanism for potentiation of the inflammatory response. Am. J. Pathol. 2016, 186, 2326–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabapathy, K. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 145–169. [Google Scholar] [CrossRef]
- De los Reyes Corrales, T.; Losada-Pérez, M.; Casas-Tintó, S. JNK pathway in CNS pathologies. Int. J. Mol. Sci. 2021, 22, 3883. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): Differences revealed by gene targeting. Bioessays 2006, 28, 923–934. [Google Scholar] [CrossRef]
- Deng, Z.; Yuan, C.; Yang, J.; Peng, Y.; Wang, W.; Wang, Y.; Gao, W. Behavioral defects induced by chronic social defeat stress are protected by Momordica charantia polysaccharides via attenuation of JNK3/PI3K/AKT neuroinflammatory pathway. Ann. Transl. Med. 2019, 7, 6. [Google Scholar] [CrossRef]
- Zulfiqar, Z.; Shah, F.A.; Shafique, S.; Alattar, A.; Ali, T.; Alvi, A.M.; Rashid, S.; Li, S. Repurposing FDA approved drugs as JNK3 inhibitor for prevention of neuroinflammation induced by MCAO in rats. J. Inflamm. Res. 2020, 13, 1185–1205. [Google Scholar] [CrossRef]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. A review of computational drug repositioning: Strategies, approaches, opportunities, challenges, and directions. J. Cheminform. 2020, 12, 46. [Google Scholar] [CrossRef]
- Govindaraj, R.G.; Naderi, M.; Singha, M.; Lemoine, J.; Brylinski, M. Large-scale computational drug repositioning to find treatments for rare diseases. NPJ Syst. Biol. Appl. 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, W.; Bhattiprolu, K.C.; Gubensäk, N.; Zangger, K. Investigating protein-ligand interactions by solution nuclear magnetic resonance spectroscopy. ChemPhysChem 2018, 19, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampron, A.; Elali, A.; Rivest, S. Innate immunity in the CNS: Redefining the relationship between the CNS and its environment. Neuron 2013, 78, 214–232. [Google Scholar] [CrossRef] [Green Version]
- Moniruzzaman, M.; Lee, G.; Bose, S.; Choi, M.; Jung, J.K.; Lee, H.; Cho, J. Antioxidant and anti-inflammatory activities of N-((3,4-dihydro-2H-benzo[h]chromene-2-yl)methyl)-4-methoxyaniline in LPS-induced BV2 microglial cells. Biol. Pharm. Bull. 2015, 38, 1831–1835. [Google Scholar] [CrossRef] [Green Version]
- Hidding, U.; Mielke, K.; Waetzig, V.; Brecht, S.; Hanisch, U.; Behrens, A.; Wagner, E.; Herdegen, T. The c-Jun N-terminal kinases in cerebral microglia: Immunological functions in the brain. Biochem. Pharmacol. 2002, 64, 781–788. [Google Scholar] [CrossRef]
- Yan, C.; Deng, C.; Liu, X.; Chen, Y.; Ye, J.; Cai, R.; Shen, Y.; Tang, H. TNF-α induction of IL-6 in alveolar type II epithelial cells: Contributions of JNK/c-Jun/AP-1 element, C/EBPδ/C/EBP binding site and IKK/NF-κB p65/κB site. Mol. Immunol. 2018, 101, 585–596. [Google Scholar] [CrossRef]
- McTavish, D.; Sorkin, E.M. Azelastine: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs 1989, 38, 778–800. [Google Scholar] [CrossRef]
- Aïd, S.; Bosetti, F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Roloff, F.; Singh, V.; Stangel, M.; Stern, M.; Bicker, G. Nitric oxide/cyclic GMP signaling regulates motility of a microglial cell line and primary microglia in vitro. Brain Res. 2014, 1564, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Dibaj, P.; Nadrigny, F.; Steffens, H.; Scheller, A.; Hirrlinger, J.; Schomburg, E.D.; Neusch, C.; Kirchhoff, F. NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia 2010, 58, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Sahley, C.L.; Muller, K.J. ATP and NO dually control migration of microglia to nerve lesions. Dev. Neurobiol. 2009, 69, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Wu, Y.; Ren, X.; Liu, Q.; Wang, J.; Liu, X. Isoorientin attenuates lipopolysaccharide-induced pro-inflammatory responses through down-regulation of ROS-related MAPK/NF-κB signaling pathway in BV-2 microglia. Mol. Cell Biochem. 2014, 386, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eo, Y.; Han, B.G.; Shim, M.; Lim, J.S.; Phuong, T.N.T.; Hoa, P.P.; Ahn, H.-C. Crystal structures of Apo- and AMP-bound human c-Jun N-terminal kinase 3. Bull. Korean Chem. Soc. 2016, 37, 1360–1363. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, P.L.; Bui, B.P.; Duong, M.T.H.; Lee, K.; Ahn, H.-C.; Cho, J. Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-κB Pathway in BV2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 9061. https://doi.org/10.3390/ijms22169061
Nguyen PL, Bui BP, Duong MTH, Lee K, Ahn H-C, Cho J. Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-κB Pathway in BV2 Microglial Cells. International Journal of Molecular Sciences. 2021; 22(16):9061. https://doi.org/10.3390/ijms22169061
Chicago/Turabian StyleNguyen, Phuong Linh, Bich Phuong Bui, Men Thi Hoai Duong, Kyeong Lee, Hee-Chul Ahn, and Jungsook Cho. 2021. "Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-κB Pathway in BV2 Microglial Cells" International Journal of Molecular Sciences 22, no. 16: 9061. https://doi.org/10.3390/ijms22169061
APA StyleNguyen, P. L., Bui, B. P., Duong, M. T. H., Lee, K., Ahn, H.-C., & Cho, J. (2021). Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-κB Pathway in BV2 Microglial Cells. International Journal of Molecular Sciences, 22(16), 9061. https://doi.org/10.3390/ijms22169061