
Involvement of Alarmins in the Pathogenesis and Progression of Multiple Myeloma

 ,
,  ,
,
Abstract
1. Introduction
2. Alarmins
2.1. High-Mobility Group Box-1 (HMGB1)
2.2. Heat Shock Proteins (HSPs)
2.3. S100 Proteins
3. Search Strategy
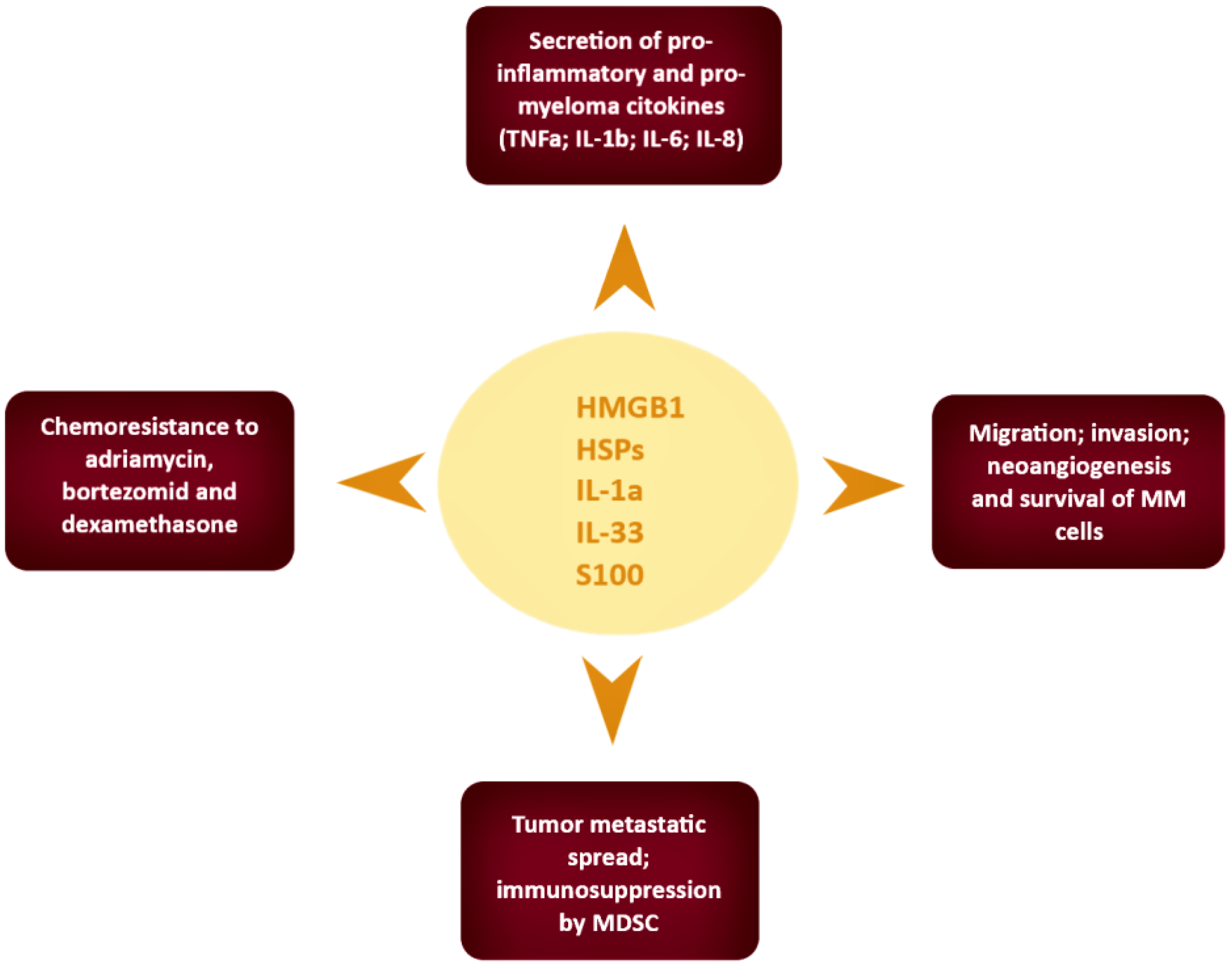
4. Results
4.1. HMGB1-Induced Chemoresistance
4.2. IL-1β as a Progression Factor of MM
4.3. Role of Proinflammatory Cytokines in MM Progression
4.4. S100 Protein as Responsible for Disease Progression
5. Discussion
5.1. Role of the Immune System in MM Progression
5.2. Neoangiogenesis in MM
5.3. Drug Resistance in MM
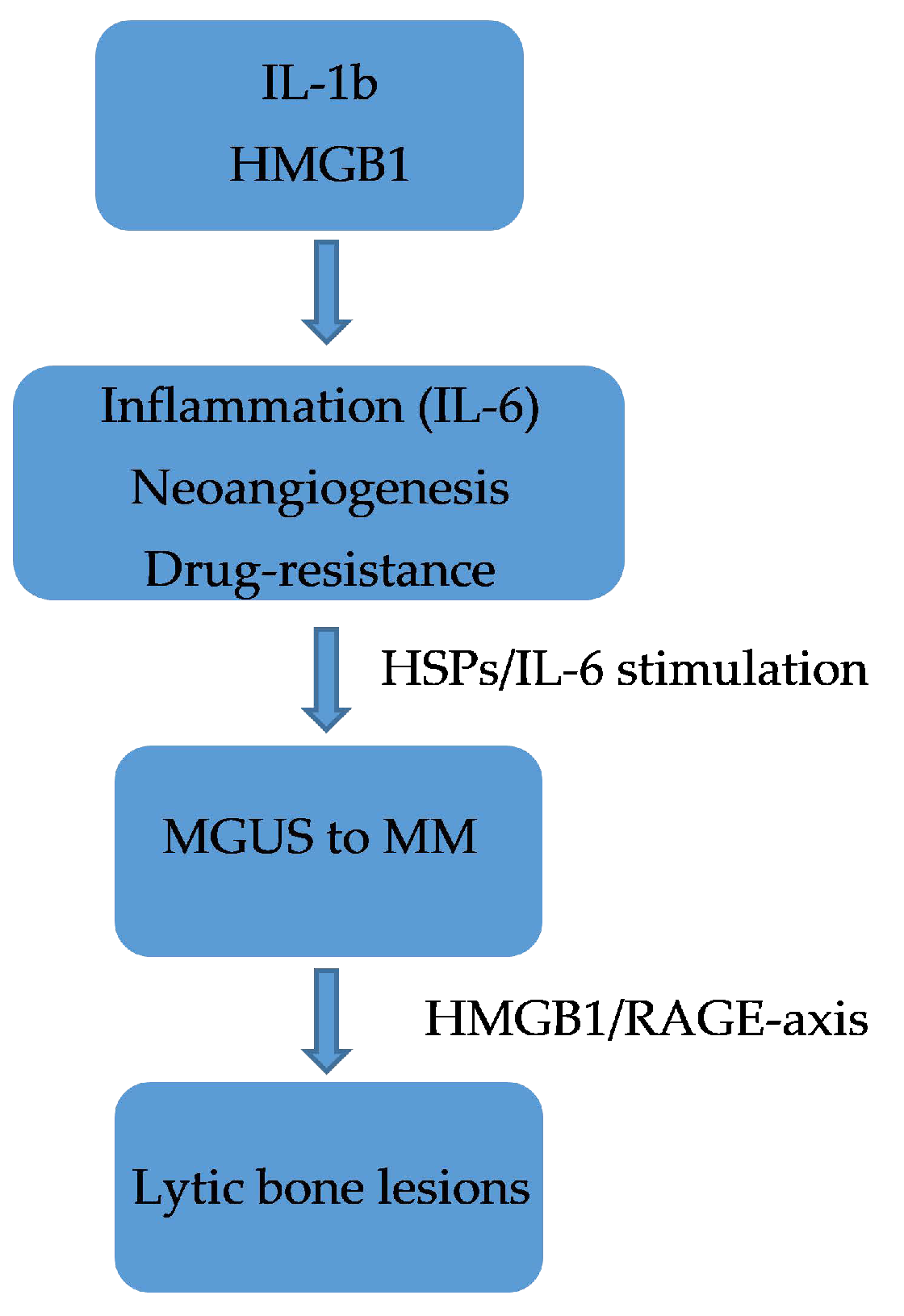
5.4. MGUS to MM: Role of Immunoglobulin Chains
5.5. HMGB1/RAGE Axis as Crosstalking between Immune Cells and Bone Tissue
6. Mutational Status, Inflammation, and MM Progression
7. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rollig, C.; Knop, S.; Bornhauser, M. Multiple myeloma. Lancet 2015, 385, 2197–2208. [Google Scholar] [CrossRef]
- Kunacheewa, C.; Orlowski, R.Z. New drugs in multiple myeloma. Annu. Rev. Med. 2019, 70, 521–547. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; Van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Legarda, M.A.; Cejalvo, M.J.; De La Rubia, J. Recent Advances in the Treatment of Patients with Multiple Myeloma. Cancers 2020, 12, 3576. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Benson, J.; Larson, D.; Therneau, T.; Dispenzieri, A.; Melton Iii, L.J.; Rajkumar, S.V. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin. Lymphoma Myeloma 2009, 9, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Melton, L.J., 3rd; Colby, C.L.; Therneau, T.M.; Clark, R.; Kumar, S.K.; Bradwell, A.; et al. Prevalence and risk of progression of light-chain monoclonal gammo-pathy of undetermined significance: A retrospective population-based cohort study. Lancet 2010, 375, 1721–1728. [Google Scholar] [CrossRef]
- Marinac, C.R.; Ghobrial, I.M.; Birmann, B.; Soiffer, J.; Rebbeck, T.R. Dissecting racial disparities in multiple myeloma. Blood Cancer J. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, W.M. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009, 113, 5418–5422. [Google Scholar] [CrossRef]
- Kumar, S.K.; Callander, N.S.; Alsina, M.; Atanackovic, D.; Biermann, J.S.; Castillo, J.; Chandler, J.C.; Costello, C.; Faiman, M.; Fung, H.C. NCCN Guidelines Insights: Multiple Myeloma, Version 3. J. Natl. Compr. Cancer Netw. 2018, 16, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H. Immunopathogenesis and immunotherapy of multiple myeloma. Int. J. Hematol. 2018, 107, 278–285. [Google Scholar] [CrossRef]
- Nikesitch, N.; Ling, S.C.W. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2015, 69, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Jahangir, A.; Chen, G.; Hong, C. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mech-anisms. Oncotarget 2013, 4, 2186. [Google Scholar]
- González, D.; Van Der Burg, M.; Garcia-Sanz, R.; Fenton, J.A.; Langerak, A.W.; González, M.; Van Dongen, J.; Miguel, J.S.; Morgan, G. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood 2007, 110, 3112–3121. [Google Scholar] [CrossRef] [PubMed]
- Kiertscher, S.M.; Luo, J.; Dubinett, S.M.; Roth, M.D. Tumors Promote Altered Maturation and Early Apoptosis of Monocyte-Derived Dendritic Cells. J. Immunol. 2000, 164, 1269–1276. [Google Scholar] [CrossRef]
- Greil, R.; Egle, A.; Villunger, A. On the Role and Significance of Fas (Apo-1/CD95) Ligand (FasL) Expression in Immune Privileged Tissues and Cancer Cells Using Multiple Myeloma as a Model. Leuk. Lymphoma 1998, 31, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Murray, A.; Pope, B.; Sze, D.M.; Gibson, J.; Ho, P.J.; Hart, D.N.; Joshua, D. Either interleukin-12 or interferon-γ can correct the dendritic cell defect induced by transforming growth factor β 1 in patients with myeloma. Br. J. Haematol. 2004, 125, 743–748. [Google Scholar] [CrossRef]
- Villunger, A.; Egle, A.; Marschitz, I.; Kos, M.; Böck, G.; Ludwig, H.; Geley, S.; Kofler, R.; Greil, R. Constitutive expression of Fas (Apo-1/CD95) ligand on multiple myeloma cells: A potential mechanism of tumor-induced suppression of immune sur-veillance. Blood 1997, 90, 12–20. [Google Scholar] [CrossRef]
- Yoneda, K.-I.; Morii, T.; Nieda, M.; Tsukaguchi, N.; Amano, I.; Tanaka, H.; Yagi, H.; Narita, N.; Kimura, H. The peripheral blood Vα24+NKT cell numbers decrease in patients with haematopoietic malignancy. Leuk. Res. 2005, 29, 147–152. [Google Scholar] [CrossRef]
- Koike, M.; Sekigawa, I.; Okada, M.; Matsumoto, M.; Iida, N.; Hashimoto, H.; Oshimi, K. Relationship between CD4+/CD8+ T cell ratio and T cell activation in multiple myeloma: Reference to IL-16. Leuk. Res. 2002, 26, 705–711. [Google Scholar] [CrossRef]
- Ogawara, H.; Handa, H.; Yamazaki, T.; Toda, T.; Yoshida, K.; Nishimoto, N.; Al-Ma’Quol, W.H.S.; Kaneko, Y.; Matsushima, T.; Tsukamoto, N.; et al. High Th1/Th2 ratio in patients with multiple myeloma. Leuk. Res. 2005, 29, 135–140. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, R.R.; Negendank, W.G.; Schreiber, A.D. Impaired granulocyte adherence in multiple myeloma: Relationship to complement system, granulocyte delivery, and infection. Blood 1978, 51, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H.G.; Cawley, J.C. Abnormal monoclonal antibody-defined helper/suppressor T-cell subpopulations in multiple mye-loma: Relationship to treatment and clinical stage. Br. J. Haematol. 1983, 53, 271–275. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Davies, F.E.; Owen, R.G.; English, A.; Pratt, G.; Child, J.A.; Jack, A.S.; Morgan, G.J. B-lymphocyte suppres-sion in multiple myeloma is a reversible phenomenon specific to normal B-cell progenitors and plasma cell precursors. Br. J. Haematol. 1998, 100, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Hájek, R.; Butch, A.W. Dendritic cell biology and the application of dendritic cells to immunotherapy of multiple myeloma. Med Oncol. 2000, 17, 2–15. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: Interplay of growth factors, their receptors and stromal interactions. Eur. J. Cancer 2006, 42, 1564–1573. [Google Scholar] [CrossRef]
- Caers, J.; Van Valckenborgh, E.; Menu, E.; Van Camp, B.; Vanderkerken, K. Unraveling the biology of multiple myeloma disease: Cancer stem cells, acquired intracellular changes and interactions with the surrounding micro-environment. Bull Cancer 2008, 95. [Google Scholar] [CrossRef]
- Fernando, R.C.; Mazzotti, D.R.; Azevedo, H.; Sandes, A.F.; Gil Rizzatti, E.; De Oliveira, M.B.; Alves, V.L.F.; Eugênio, A.I.P.; De Carvalho, F.; Dalboni, M.A.; et al. Transcriptome Analysis of Mesenchymal Stem Cells from Multiple Myeloma Patients Reveals Downregulation of Genes Involved in Cell Cycle Progression, Immune Response, and Bone Metabolism. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Spisek, R.; Kukreja, A.; Chen, L.-C.; Matthews, P.; Mazumder, A.; Vesole, D.; Jagannath, S.; Zebroski, H.A.; Simpson, A.J.; Ritter, G.; et al. Frequent and specific immunity to the embryonal stem cell-associated antigen SOX2 in patients with monoclonal gammopathy. J. Exp. Med. 2007, 204, 831–840. [Google Scholar] [CrossRef]
- Song, G.; Darr, D.B.; Santos, C.M.; Ross, M.; Valdivia, A.; Jordan, J.L.; Midkiff, B.R.; Cohen, S.; Nikolaishvili-Feinberg, N.; Miller, C.; et al. Effects of Tumor Microenvironment Heterogeneity on Nanoparticle Disposition and Efficacy in Breast Cancer Tumor Models. Clin. Cancer Res. 2014, 20, 6083–6095. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone Marrow Microenvironment in Multiple Myeloma Progression. J. Biomed. Biotechnol. 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Lee, S.J.; Borrello, I. Role of the Immune Response in Disease Progression and Therapy in Multiple Myeloma. Immunotoxins 2016, 169, 207–225. [Google Scholar] [CrossRef]
- Valkovic, T.; Gacic, V.; Nacinovic-Duletic, A. Multiple Myeloma Index for Risk of Infection. Cancer 2018, 9, 2211–2214. [Google Scholar]
- Allegra, A.; Innao, V.; Allegra, A.G.; Pugliese, M.; Di Salvo, E.; Ventura-Spagnolo, E.; Musolino, C.; Gangemi, S. Lymphocyte Subsets and Inflammatory Cytokines of Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma. Int. J. Mol. Sci. 2019, 20, 2822. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Gallucci, S.; Matzinger, P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2006, 81, 1–5. [Google Scholar] [CrossRef]
- Yang, D.; Wei, F.; Tewary, P.; Howard, O.M.; Oppenheim, J.J. Alarmin-induced cell migration. Eur. J. Immunol. 2013, 43, 1412–1418. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; Erlandsson-Harris, H.; Andersson, U. High-mobility group box protein 1 (HMGB1): An alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res. Ther. 2008, 10, 209. [Google Scholar] [CrossRef]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2016, 14, 43–64. [Google Scholar] [CrossRef]
- Yang, D.; Biragyn, A.; Hoover, D.M.; Lubkowski, J.; Oppenheim, J.J. Multiple Roles of Antimicrobial Defensins, Cathelicidins, and Eosinophil-Derived Neurotoxin in Host Defense. Annu. Rev. Immunol. 2004, 22, 181–215. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jin, Y.; Zhang, K.; Li, H.; Chen, W.; Meng, G.; Fang, X. Alarmin HNP-1 promotes pyroptosis and IL-1beta release through different roles of NLRP3 inflammasome via P2 × 7 in LPS-primed macrophages. Innate Immun. 2014, 20, 290–300. [Google Scholar] [CrossRef]
- Su, Z.; Zhang, P.; Yu, Y.; Lu, H.; Liu, Y.; Ni, P.; Su, X.; Wang, D.; Liu, Y.; Wang, J.; et al. HMGB1 Facilitated Macrophage Reprogramming towards a Proinflammatory M1-like Pheno-type in Experimental Autoimmune Myocarditis Development. Sci. Rep. 2016, 6, 21884. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malig-nancies. J. Hematol. Oncol. 2020, 13, 91. [Google Scholar] [CrossRef]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A New Group of Chromatin-Associated Proteins with a High Content of Acidic and Basic Amino Acids. J. Biol. Inorg. Chem. 1973, 38, 14–19. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef]
- Jube, S.; Rivera, Z.S.; Bianchi, M.E.; Powers, A.; Wang, E.; Pagano, I.; Pass, H.; Gaudino, G.; Carbone, M.; Yang, H. Cancer Cell Secretion of the DAMP Protein HMGB1 Supports Progression in Malignant Mesothelioma. Cancer Res. 2012, 72, 3290–3301. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef]
- Lotze, M.T.; Zeh, H.J.; Rubartelli, A.; Sparvero, L.J.; Amoscato, A.; Washburn, N.R.; Devera, M.E.; Liang, X.; Tor, M.; Billiar, T. The grateful dead: Damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol. Rev. 2007, 220, 60–81. [Google Scholar] [CrossRef]
- Kazama, H.; Ricci, J.E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of Immunological Tolerance by Apoptotic Cells Requires Caspase-Dependent Oxidation of High-Mobility Group Box-1 Protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef]
- Li, G.; Tang, D.; Lotze, M.T. Ménage à Trois in stress: DAMPs, redox and autophagy. Semin. Cancer Biol. 2013, 23, 380–390. [Google Scholar] [CrossRef]
- Li, G.; Liang, X.; Lotze, M.T. HMGB1: The central cytokine for all lymphoid cells. Front. Immunol. 2013, 4, 68. [Google Scholar] [CrossRef]
- Zhang, L.; Fok, J.H.L.; Davies, F.E. Heat shock proteins in multiple myeloma. Oncotarget 2014, 5, 1132–1148. [Google Scholar] [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Gen. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Endemann, M.; Bergmeister, H.; Bidmon, B.; Boehm, M.; Csaicsich, D.; Malaga-Dieguez, L.; Arbeiter, K.; Regele, H.; Herkner, K.; Aufricht, C. Evidence for HSP-mediated cytoskeletal stabilization in mesothelial cells during acute experimental peritoneal dialysis. Am. J. Physiol. Physiol. 2007, 292, F47–F56. [Google Scholar] [CrossRef]
- Slavin, M.A.; Grigg, A.P.; Schwarer, A.P.; Szer, J.; Spencer, A.; Sainani, A.; Thursky, K.; Roberts, A. A randomized comparison of empiric or pre-emptive antibiotic therapy after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007, 40, 157–163. [Google Scholar] [CrossRef]
- Joshi, A.D.; Oak, S.R.; Hartigan, A.J.; Finn, W.G.; Kunkel, S.L.; Duffy, K.E.; Das, A.; Hogaboam, C.M. Interleukin-33 contrib-utes to both M1 and M2 chemokine marker expression in human macrophages. BioMed Cent. Immunol. 2010, 11, 52–60. [Google Scholar] [CrossRef]
- Cevikbas, F.; Steinhoff, M. IL-33: A Novel Danger Signal System in Atopic Dermatitis. J. Investig. Dermatol. 2012, 132, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef] [PubMed]
- Di Salvo, E.; Ventura-Spagnolo, E.; Casciaro, M.; Navarra, M.; Gangemi, S. IL-33/IL-31 Axis: A Potential Inflammatory Pathway. Mediat. Inflamm. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Temajo, N.O.; Howard, N. The virus-induced HSPs regulate the apoptosis of operatus APCs that results in autoimmunity, not in homeostasis. Autoimmun. Rev. 2014, 13, 1013–1019. [Google Scholar] [CrossRef]
- Kalvakolanu, D.V.; Roy, S.K. CCAAT/Enhancer Binding Proteins and Interferon Signaling Pathways. J. Interf. Cytokine Res. 2005, 25, 757–769. [Google Scholar] [CrossRef]
- Imai, Y.; Maru, Y.; Tanaka, J. Action mechanisms of histone deacetylase inhibitors in the treatment of hematological malig-nancies. Cancer Sci. 2016, 107, 1543–1549. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Innao, V.; Allegra, A.G.; Pioggia, G.; Gangemi, S. Inflammatory and Anti-Inflammatory Equilibrium, Proliferative and Antiproliferative Balance: The Role of Cytokines in Multiple Myeloma. Mediat. Inflamm. 2017, 2017, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Guo, X.; He, D.; Zhang, E.; Chen, J.; Chen, Q.; Li, Y.; Yang, L.; Yang, Y.; Zhao, Y.; Wang, G.; et al. HMGB1 knock-down increases MM cell vulnerability by regulating autophagy and DNA damage repair. J. Exp. Clin. Cancer Res. 2018, 37, 205. [Google Scholar] [CrossRef]
- Ning, J.; Yang, R.; Wang, H.; Cui, L. HMGB1 enhances chemotherapy resistance in multiple myeloma cells by activating the nuclear factor-κB pathway. Exp. Ther. Med. 2021, 22, 705. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Musolino, C.; Pace, E.; Innao, V.; Di Salvo, E.; Ferraro, M.; Casciaro, M.; Spatari, G.; Tartarisco, G.; Allegra, A.G.; et al. Evaluation of the AGE/sRAGE Axis in Patients with Multiple Myeloma. Antioxidants 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Galliera, E.; Marazzi, M.G.; Vianello, E.; Drago, L.; Luzzati, A.; Bendinelli, P.; Maroni, P.; Tacchini, L.; Desiderio, M.A.; Romanelli, M.M.C. Circulating sRAGE in the diagnosis of osteolytic bone metastasis. J. Biol. Regul. Homeost. Agents 2016, 30, 1203–1208. [Google Scholar] [PubMed]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 synergistically enhances bortezomib-induced cytotoxicity in multiple myeloma. Cell Stress Chaperon 2020, 25, 357–367. [Google Scholar] [CrossRef]
- Jones, R.J.; Singh, R.K.; Shirazi, F.; Wan, J.; Wang, H.; Wang, X.; Ha, M.J.; Baljevic, M.; Kuiatse, I.; Davis, R.E.; et al. Intravenous Immunoglobulin G Suppresses Heat Shock Protein (HSP)-70 Expression and Enhances the Activity of HSP90 and Proteasome Inhibitors. Front. Immunol. 2020, 11, 1816. [Google Scholar] [CrossRef]
- Takagi, S.; Tsukamoto, S.; Park, J.; Johnson, K.E.; Kawano, Y.; Moschetta, M.; Liu, C.J.; Mishima, Y.; Kokubun, K.; Manier, S.; et al. Platelets Enhance Multiple My-eloma Progression via IL-1β Upregulation. Clin. Cancer Res. 2018, 24, 2430–2439. [Google Scholar] [CrossRef]
- Tsirakis, G.; Pappa, C.A.; Kolovou, A.; Kokonozaki, M.; Neonakis, I.; Alexandrakis, M.G. Clinical significance of interleukin-22 in multiple myeloma. Hematology 2014, 20, 143–147. [Google Scholar] [CrossRef]
- Murdaca, G.; Colombo, B.M.; Puppo, F. The role of Th17 lymphocytes in the autoimmune and chronic inflammatory diseases. Intern. Emerg. Med. 2011, 6, 487–495. [Google Scholar] [CrossRef]
- Ciprandi, G.; Amici, M.D.E.; Murdaca, G.; Fenoglio, D.; Ricciardolo, F.; Marseglia, G.L.; Tosca, M. Serum interleukin-17 levels are related to clinical severity in allergic rhinitis. Allergy 2009, 64, 1375–1378. [Google Scholar] [CrossRef]
- Ben Hmid, A.; Selmi, O.; Rekik, R.; Lamari, H.; Zamali, I.; Ladeb, S.; Safra, I.; Ben Othman, T.; Ben Romdhane, N.; Ben Ahmed, M. RORC overexpression as a sign of Th17 lymphocytes accumulation in multiple myeloma bone marrow. Cytokine 2020, 134, 155210. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Profita, M.; Alonci, A.; Saitta, S.; Russo, S.; Bonanno, A.; Innao, V.; Gangemi, S. Reduced IL-33 plasma levels in multiple myeloma patients are associated with more advanced stage of disease. Br. J. Haematol. 2013, 160, 709–710. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, Y.; Jiang, Y.; Qin, T.; Chen, J.; Chu, X.; Yi, Q.; Gao, S.; Wang, S. Dectin-1 signaling inhibits osteoclastogenesis via IL-33-induced inhibition of NFATc1. Oncotarget 2017, 8, 53366–53374. [Google Scholar] [CrossRef] [PubMed]
- Bosseboeuf, A.; Allain-Maillet, S.; Mennesson, N.; Tallet, A.; Rossi, C.; Garderet, L.; Caillot, D.; Moreau, P.; Piver, E.; Girodon, F.; et al. Pro-inflammatory State in Monoclonal Gam-mopathy of Undetermined Significance and in Multiple Myeloma Is Characterized by Low Sialylation of Pathogen-Specific and Other Monoclonal Immunoglobulins. Front. Immunol. 2017, 8, 1347. [Google Scholar] [CrossRef] [PubMed]
- Binsfeld, M.; Muller, J.; Lamour, V.; De Veirman, K.; De Raeve, H.; Bellahcène, A.; Van Valckenborgh, E.; Baron, F.; Beguin, Y.; Binsfeld, M.; et al. Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget. 2016, 7, 37931–37943. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.Y.; Wang, Y.; Wang, J.N.; Song, M.; Meng, Q.Q.; Han, X. Clinical significance of S100A6 and Notch1 in multiple myeloma patients. Zhonghua Xue Ye Xue Za Zhi 2017, 38, 285–289. [Google Scholar] [PubMed]
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Podar, K.; Akiyama, M.; Gupta, D.; Richardson, P.; Munshi, N.; Anderson, K.C. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol. Cancer Ther. 2002, 1, 539–544. [Google Scholar] [PubMed]
- Dezorella, N.; Pevsner-Fischer, M.; Deutsch, V.; Kay, S.; Baron, S.; Stern, R.; Tavor, S.; Nagler, A.; Naparstek, E.; Zipori, D.; et al. Mesenchymal stromal cells revert multiple myeloma cells to less differentiated phenotype by the combined activities of adhesive interactions and interleukin-6. Exp. Cell Res. 2009, 315, 1904–1913. [Google Scholar] [CrossRef]
- Cheung, W.-C.; Van Ness, B. Distinct IL-6 signal transduction leads to growth arrest and death in B cells or growth promotion and cell survival in myeloma cells. Leukemia 2002, 16, 1182–1188. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Yi, Q.; Osterborg, A. Idiotype-specific T cells in multiple myeloma: Targets for an immunotherapeutic intervention? Med. On-col. 1996, 13, 1–7. [Google Scholar]
- Yi, Q.; Osterborg, A.; Bergenbrant, S.; Mellstedt, H.; Holm, G.; Lefvert, A.K. Idiotype-reactive T-cell subsets and tumor load in monoclonal gammopathies. Blood 1995, 86, 3043–3049. [Google Scholar] [CrossRef]
- Cozzolino, F.; Torcia, M.; Aldinucci, D.; Rubartelli, A.; Miliani, A.; Shaw, A.R.; Lansdorp, P.M.; Di Guglielmo, R. Production of interleukin-1 by bone marrow myeloma cells. Blood 1989, 74, 380–387. [Google Scholar] [CrossRef]
- Donovan, K.; Lacy, M.; Kline, M.; Ahmann, G.; Heimbach, J.; Kyle, R.; Lust, J. Contrast in cytokine expression between patients with monoclonal gammopathy of undetermined significance or multiple myeloma. Leukemia 1998, 12, 593–600. [Google Scholar] [CrossRef][Green Version]
- Hawley, T.S.; Lach, B.; Burns, B.F.; May, L.T.; Sehgal, P.B. Hawley RG. Expression of retrovirally transduced IL-1 alpha in IL-6-dependent B cells: A murine model of aggressive multiple myeloma. Growth Factors 1991, 5, 327–338. [Google Scholar] [CrossRef]
- Hawley, R.G.; Wang, M.H.; Fong, A.Z.; Hawley, T.S. Association between ICAM-1 expression and metastatic capacity of murine B-cell hybridomas. Clin. Exp. Metastasis 1993, 11, 213–226. [Google Scholar] [CrossRef]
- Van Camp, B.; Durie, B.G.; Spier, C.; De Waele, M.; Van Riet, I.; Vela, E.; Frutiger, Y.; Richter, L.; Grogan, T.M. Plasma cells in multiple myeloma express a natural killer cell-associated antigen: CD56 (NKH-1; Leu-19). Blood 1990, 76, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, D.M.; Nagler, A.; Ginzton, N.; Greenberg, P.; Butcher, E.C. Hematopoietic progenitor cell expression of the H-CAM (CD44) homing-associated adhesion molecule. Blood 1990, 75, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Drach, J.; Gattringer, C.; Huber, H. Expression of the neural cell adhesion molecule (CD56) by human myeloma cells. Clin. Exp. Immunol. 1991, 83, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Corley, P. Induction of interleukin-1 and glucocorticoid hormones by HIV promotes viral replication and links human chromosome 2 to AIDS pathogenesis: Genetic mechanisms and therapeutic implications. Med. Hypotheses 1997, 48, 415–421. [Google Scholar] [CrossRef]
- Beaulieu, A.D.; Paquin, R.; Gosselin, J. Epstein-Barr virus modulates de novo protein synthesis in human neutrophils. Blood 1995, 86, 2789–2798. [Google Scholar] [CrossRef]
- Bitko, V.; Velazquez, A.; Yang, L.; Yang, Y.C.; Barik, S. Transcriptional induction of multiple cytokines by human respiratory syncytial virus requires activation of NF-kappa B and is inhibited by sodium salicylate and aspirin. Virology 1997, 232, 369–378. [Google Scholar] [CrossRef]
- Vacca, A.; Di Stefano, R.; Frassanito, A.; Iodice, G.; Dammacco, F. A disturbance of the IL-2/IL-2 receptor system parallels the activity of multiple myeloma. Clin. Exp. Immunol. 1991, 84, 429–434. [Google Scholar]
- Meyer, A.; Staratschek-Jox, A.; Springwald, A.; Wenk, H.; Wolf, J.; Wickenhauser, C.; Bullerdiek, J. Non-Hodgkin lymphoma expressing high levels of the danger-signalling protein HMGB1. Leuk. Lymphoma 2008, 49, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.; Li, Q.; Wu, P.; Ye, Y.; Tseng, H.Y.; Zhang, L.; Zhang, X.D. Autophagymediated HMGB1 release antagonizes apop-tosis of gastric cancer cells induced by vincristine via transcriptional regulation of Mcl-1. Autophagy 2012, 8, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Mansure, J.J.; Almajed, W.; Cury, F.; Ferbeyre, G.; Popovic, M.; Seuntjens, J.; Kassouf, W. The Role of HMGB1 in Radioresistance of Bladder Cancer. Mol. Cancer Ther. 2016, 15, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, Y.; Wang, Z.; Chen, L.; Dong, X.; Nie, X. Interference with HMGB1 increases the sensitivity to chemotherapy drugs by inhibiting HMGB1-mediated cell autophagy and inducing cell apoptosis. Tumor Biol. 2015, 36, 8585–8592. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Liu, J.; Xu, P.; Zhang, X.-M.; Tian, Y.-Y.; Xue, Y.-M.; Gao, X.-Y.; Liu, Y.; Wang, J.-H. Synergistic effect of HMGB1 knockdown and cordycepin in the K562 human chronic myeloid leukemia cell line. Mol. Med. Rep. 2015, 12, 4462–4468. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.-W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Liang, L.; Xiao, X.; Peng, Y.; Luo, Y.; Zhou, W.; Zhang, J.; Qiu, L.; Zhang, S.; Liu, F.; et al. Lycorine downregulates HMGB1 to inhibit autophagy and enhances bortezomib activity in multiple myeloma. Theranostics 2016, 6, 2209. [Google Scholar] [CrossRef]
- Cebrián, M.J.G.; Bauden, M.; Andersson, R.; Holdenrieder, S.; Ansari, D. Paradoxical Role of HMGB1 in Pancreatic Cancer: Tumor Suppressor or Tumor Promoter? Anticancer Res. 2016, 36, 4381–4390. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, H.; Sun, M.; Yin, Z.; Qian, J. High mobility group box 1-mediated autophagy promotes neuroblastoma cell chemoresistance. Oncol. Rep. 2015, 34, 2969–2976. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ding, Y.; Wang, S.; Zhang, Q.; Liu, L. Increased expression of high mobility group box 1 (HMGB1) is associated with progression and poor prognosis in human nasopharyngeal carcinoma. J. Pathol. 2008, 216, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Sezer, C.; Süren, D.; Yıldırım, M.; Demirpençe, O.; Kaya, V.; Alikanoğlu, A.S.; Bülbüller, N.; Yıldız, M. The role of High Mobility Group Box 1 (HMGB1) in colorectal cancer. Med. Sci. Monit. 2014, 20, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Flex, A.; Pecorini, G.; Angelini, F.; Arena, V.; Stigliano, E.; Gremese, E.; Tolusso, B.; Ferraccioli, G. The role of high-mobility group box protein 1 in collagen antibody-induced arthritis is dependent on vascular endothelial growth factor. Clin. Exp. Immunol. 2016, 184, 62–72. [Google Scholar] [CrossRef]
- Ciprandi, G.; Murdaca, G.; Colombo, B.M.; De Amici, M.; Marseglia, G.L. Serum vascular endothelial growth factor in aller-gic rhinitis and systemic lupus erythematosus. Hum. Immunol. 2008, 69, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Mulcrone, P.L.; Edwards, S.K.E.; Petrusca, D.N.; Haneline, L.S.; Delgado-Calle, J.; Roodman, G.D. Osteocyte Vegf-a contrib-utes to myeloma-associated angiogenesis and is regulated by Fgf23. Sci. Rep. 2020, 10, 17319. [Google Scholar] [CrossRef]
- Brito, A.B.; Lourenço, G.J.; Oliveira, G.B.; De Souza, C.A.; Vassallo, J.; Lima, C.S. Associations of VEGF and VEGFR2 poly-morphisms with increased risk and aggressiveness of multiple myeloma. Ann. Hematol. 2014, 93, 1363–1369. [Google Scholar]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spanò, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef]
- Solimando, A.G.; De Summa, S.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Effects of AntagomiRs on Different Lung Diseases in Human, Cellular, and Animal Models. Int. J. Mol. Sci. 2019, 20, 3938. [Google Scholar] [CrossRef]
- Zhang, C.; Ge, S.; Hu, C.; Yang, N.; Zhang, J. MiRNA-218, a new regulator of HMGB1, suppresses cell migration and invasion in non-small cell lung cancer. Acta Biochim. Biophys. Sin. 2013, 45, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, Y.; Long, J.; Guo, K.; Ge, C.; Du, R. microRNA-218 suppresses the proliferation, invasion and promotes apoptosis of pancreatic cancer cells by targeting HMGB1. Chin. J. Cancer Res. 2015, 27, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Yang, J.; Liu, C.; Zhou, P.; Xiao, L.; Zhang, K. MiR-218 inhibits HMGB1-mediated autophagy in endometrial carci-noma cells during chemotherapy. Int. J. Clin. Exp. Pathol. 2015, 8, 6617–6626. [Google Scholar] [PubMed]
- Pika, T.; Lochman, P.; Sandecka, V.; Maisnar, V.; Minarik, J.; Tichy, M.; Zapletalova, J.; Solcova, L.; Scudla, V.; Hajek, R. Immunoparesis in MGUS—Relationship of uninvolved immunoglobulin pair suppression and polyclonal immunoglobuline levels to MGUS risk categories. Neoplasma 2015, 62, 827–832. [Google Scholar] [CrossRef]
- Magnano, L.; Fernández de Larrea, C.; Elena, M.; Cibeira, M.T.; Tovar, N.; Aróstegui, J.I.; Pedrosa, F.; Rosiñol, L.; Filella, X.; Yagüe, J.; et al. Prognostic Impact of Serum Heavy/Light Chain Pairs in Patients with Monoclonal Gammopathy of Unde-termined Significance and Smoldering Myeloma: Long-Term Results from a Single Institution. Clin. Lymphoma Myeloma Leuk. 2016, 16, e71–e77. [Google Scholar] [CrossRef]
- Valković, T.; Gačić, V.; Ivandić, J.; Petrov, B.; Dobrila-Dintinjana, R.; Dadić-Hero, E.; Načinović-Duletić, A. Infections in Hospitalised Patients with Multiple Myeloma: Main Characteristics and Risk Factors. Turk J. Haematol. 2015, 32, 234–242. [Google Scholar] [CrossRef]
- Cherry, B.M.; Costello, R.; Zingone, A.; Burris, J.; Korde, N.; Manasanch, E.; Kwok, M.; Annunziata, C.; Roschewski, M.; Engels, E.A.; et al. Immunoparesis and monoclonal gammopathy of undetermined significance are disassociated in advanced age. Am. J. Hematol. 2012, 88, 89–92. [Google Scholar] [CrossRef]
- Fu, L.-L.; Cheng, Y.; Liu, B. Beclin-1: Autophagic regulator and therapeutic target in cancer. Int. J. Biochem. Cell Biol. 2013, 45, 921–924. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Tang, L.-J.; Shi, Y.-W.; Ren, W.; Hu, W.-X. Apoptosis induced by lycorine in KM3 cells is associated with the G0/G1 cell cycle arrest. Oncol. Rep. 2007, 17, 377–384. [Google Scholar] [CrossRef][Green Version]
- Takahashi, H.; Nishibori, M. Current status and future prospects in HMGB1 and receptor researches. Nihon Rinsho. Jpn. J. Clin. Med. 2016, 74, 703–711. [Google Scholar]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia-Pac. J. Clin. Oncol. 2020, 17, 193–208. [Google Scholar] [CrossRef]
- Chatterjee, S.; Azad, B.B.; Nimmagadda, S. The Intricate Role of CXCR4 in Cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef]
- Coniglio, S.J. Role of Tumor-Derived Chemokines in Osteolytic Bone Metastasis. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef]
- Paiva, B.; Corchete, L.A.; Vidriales, M.B.; Puig, N.; Maiso, P.; Rodriguez, I.; Alignani, D.; Burgos, L.; Sanchez, M.L.; Barcena, P.; et al. Spanish Myeloma Group/Program for the Study of Malignant Blood Diseases Therapeutics (GEM/PETHEMA) Cooperative Study Groups. Phenotypic and genomic analysis of multiple myeloma minimal residual disease tumor cells: A new model to under-stand chemoresistance. Blood 2016, 127, 1896–1906. [Google Scholar]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef] [PubMed]
- Charoonpatrapong, K.; Shah, R.; Robling, A.G.; Alvarez, M.; Clapp, D.W.; Chen, S.; Kopp, R.; Pavalko, F.M.; Yu, J.; Bidwell, J.P. HMGB1 expression and release by bone cells. J. Cell. Physiol. 2006, 207, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Metformin inhibits advanced glycation end products (AGEs)-induced growth and VEGF expression in MCF-7 breast cancer cells by suppressing AGEs receptor expression via AMP-activated pro-tein kinase. Horm. Metab. Res. 2013, 45, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Allegra, A.; Pioggia, G.; Tonacci, A.; Casciaro, M.; Musolino, C.; Gangemi, S. Synergic Crosstalk between Inflammation, Oxidative Stress, and Genomic Alterations in BCR-ABL-Negative Myeloproliferative Neoplasm. Antioxidants 2020, 9, 1037. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, Q.; Foltz, S.M.; Fowles, J.S.; Yao, L.; Wang, J.T.; Cao, S.; Sun, H.; Wendl, M.C.; Sethuraman, S.; et al. Co-evolution of tumor and immune cells during progression of multiple myeloma. Nat. Commun. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Walker, B.; Boyle, E.M.; Wardell, C.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Castaneda-Avila, M.A.; Ulbricht, C.M.; Epstein, M.M. Risk factors for monoclonal gammopathy of undetermined significance: A systematic review. Ann. Hematol. 2021, 100, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; Tibullo, D.; Camiolo, G.; Parrinello, N.L.; Romano, A.; Puglisi, F.; Barbato, A.; Conticello, C.; Lupo, G.; Anfuso, C.D.; et al. TLR4 signaling drives mesenchymal stromal cells commitment to promote tumor microenvironment transformation in multiple myeloma. Cell Death Dis. 2019, 10, 704–714. [Google Scholar] [CrossRef]
- Botta, C.; DI Martino, M.T.; Ciliberto, D.; Cucè, M.; Correale, P.; Rossi, M.; Tagliaferri, P.; Tassone, P. A gene expression inflammatory signature specifically predicts multiple myeloma evolution and patients survival. Blood Cancer J. 2016, 6, e511. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Tuchman, S.A.; Rosenblatt, J.M.; Mitsiades, C.S.; Colson, K.; Masone, K.; Warren, D.; Redd, R.A.; Grayson, D.; Richardson, P.G. Phase 1 open-label study of panobinostat, lenalidomide, bortezomib + dexamethasone in relapsed and re-lapsed/refractory multiple myeloma. Blood Cancer J. 2021, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers 2019, 11, 475. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Sant’Antonio, E.; Penna, G.; Alonci, A.; D’Angelo, A.; Russo, S.; Cannavò, A.; Gerace, D.; Musolino, C. Novel therapeutic strategies in multiple myeloma: Role of the heat shock protein inhibitors. Eur. J. Haematol. 2010, 86, 93–110. [Google Scholar] [CrossRef]
- Okawa, Y.; Hideshima, T.; Steed, P.; Vallet, S.; Hall, S.; Huang, K.; Rice, J.; Barabasz, A.; Foley, B.; Ikeda, H.; et al. SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and ERK. Blood 2009, 113, 846–855. [Google Scholar] [CrossRef]
- De Veirman, K.; De Beule, N.; Maes, K.; Menu, E.; De Bruyne, E.; De Raeve, H.; Fostier, K.; Moreaux, J.; Kassambara, A.; Hose, D.; et al. Extracellular S100A9 Protein in Bone Mar-row Supports Multiple Myeloma Survival by Stimulating Angiogenesis and Cytokine Secretion. Cancer Immunol. Res. 2017, 5, 839–846. [Google Scholar] [CrossRef]
- Zhao, Z.; Luo, Z.; Yang, Q.; Chang, H.; Liu, P.; Li, Z.; Guo, S.; Zhou, C.; Song, J.; Cao, W. Neutrophil-Derived MRP14 Supports Plasma Cell Commitment and Protects Myeloma Cells from Apoptosis. J. Immunol. Res. 2019, 2019, 9561350. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Geng, C.; Zhou, H.; Gao, W.; Chen, W. Serum exosomal microRNAs as novel biomarkers for multiple myeloma. Hematol. Oncol. 2019, 37, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Romano, R.; Bucci, C.; Marzetti, E. Extracellular Vesicles and Damage-Associated Molecular Patterns: A Pandora’s Box in Health and Disease. Front. Immunol. 2020, 11, 2993. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Author | Year | Type of Study | Objective | Outcome |
|---|---|---|---|---|
| Xing Gu, Donghua He, Enfan Zhang et al. | 2018 | Research paper | Evaluate the effect of HMGB1 and the mechanism involved in multiple myeloma drug resistance | HMGB1 may serve as a target for MM treatment |
| Jing Ning, Rui Yang, Hainan Wang, Lijuan Cui | 2021 | Research paper | Explore the exact molecular mechanism underlying HMGB1-mediated drug resistance in multiple myeloma | HMGB1 regulates drug resistance in MM cells by regulating NF-κB signalling pathway |
| Alessandro Allegra, Caterina Musolino, Elisabetta Pace et al. | 2019 | Research paper | Evaluate the advanced glycation end products/soluble receptor of advanced glycation end products (AGE/sRAGE) axis in patients with multiple myeloma (MM) | Serum concentrations of AGE and sRAGE could therefore become potential therapeutic targets in multiple myeloma |
| E Galliera, M G Marazzi, E Vianello et al. | 2016 | Research paper | To investigate the diagnostic potential of sRAGE to improve the detection and monitoring of bone metastasis | sRAGE might play a protective role in bone metastasis progression, and it may have diagnostic significance for detecting and monitoring osteolytic metastases |
| Lingjuan Huang, Yanmeng Wang, Ju Bai et al. | 2020 | Research paper | To investigate whether targeting HSP70 using a specific inhibitor VER-155008 combined with bortezomib could overcome the acquired resistance in multiple myeloma | Find of a strong synergistic interaction between VER-155008 and bortezomib may support for combination therapy in multiple myeloma patients |
| Richard J Jones, Ram K Singh, Fazal Shirazi et al. | 2020 | Research paper | To support the possibility that anti-HSP70-1 IgG contained in IVIgG can inhibit myeloma and MCL growth by interfering with a novel mechanism involving uptake of exogenous HSP70-1 which then induces its own promoter | IVIgG has a potential road map to identify new therapies that could be generated as monoclonal antibodies |
| Satoshi Takagi, Shokichi Tsukamoto, Jihye Park et al. | 2018 | Research paper | To investigate the association of platelet activation status with clinical stages in multiple myeloma (MM) patients and explored the role of platelets in MM progression | Platelets from MM patients were highly activated with disease progression. IL-1β is critical to platelet-mediated MM progression and might be a potential target for MM treatment |
| George Tsirakis, Constantina A Pappa, Anna Kolovouv et al. | 2015 | Research paper | To estimate serum levels of IL-22 in MM patients, both in activity and remission, in order to apprehend its possible participation in MM biology | Elevated levels of IL-22 in active MM patients, in parallel with disease activity, and in positive correlation with IL-1beta, may represent the inflammatory element of the disease |
| Murdaca G, Colombo BM, Puppo F | 2010 | review | Focus on recent information regarding IL-17 and its relevance in autoimmune and chronic inflammatory diseases | IL-17 plays a key role in various steps of RA, SLE, and other autoimmune and chronic inflammatory diseases development, and it is associated not only with T cell-mediated tissue injury but also with the production of pathogenic autoantibodies |
| G Ciprandi, M De Amici, G Murdaca, D Fenoglio, F Ricciardolo, G Marseglia, M Tosca | 2009 | Research paper | To investigate a possible relationship between serum IL-17 levels and clinical parameters in patients with allergic rhinitis studied during the pollen season | Serum IL-17 levels were significantly related to clinical symptoms, drug use, and peripheral eosinophil counts |
| Ahlem Ben Hmid, Oumayma Selmi, Raja Rekik et al. | 2020 | Research paper | Try to understand the role of Th17 lymphocytes in multiple myeloma | The involvement of Th17 cells in the pathophysiology of MM. Such data further support the use of anti-IL-17 antibodies as a therapeutic approach in MM |
| Caterina Musolino, Alessandro Allegra, Mirella Profita et al. | 2013 | Research paper | To demonstrate decreased concentrations of IL-33 in patients with MM | Reduced IL-33 levels might prevent an adequate Th2 response towards MM idiotype proteins, and compromise the anti-tumour surveillance |
| Xiaoqing Zhu, Yinghua Zhao, Yuxue Jiang et al | 2017 | Research paper | Curdlan potently inhibited RANKL-induced osteoclast differentiation and the resultant bone resorption | IL-33 receptor, partially abrogated curdlan-induced inhibition of NFATc1 expression and osteoclast differentiation |
| Adrien Bosseboeuf, Sophie Allain-Maillet, Nicolas Mennesson et al | 2017 | Research paper | Characterised the sialylation of purified mc and pc IgGs from 148 MGUS and MM patients, in comparison to pc IgGs from healthy volunteers | Hyposialylation of mc IgGs contribute to the pathogenesis of MGUS and MM |
| Kim De Veirman, NathanDe Beule, Ken Maes et al | 2017 | Research paper | To evaluated the role of extracellular S100A9 and the therapeutic relevance of S100A9 inhibition in multiple myeloma (MM), using the immunocompetent murine 5T33MM model | Extracellular S100A9 promotes MM and that inhibition of S100A9 may have therapeutic benefit |
| H Y Bao, Y Wang, J N Wang, M Song, Q Q Meng, X Han | 2017 | Research paper | To investigate the expression levels of S100A6, Notch1 in multiple myeloma (MM) patients and its clinical significance | S100A6 and Notch1 were closely associated with MM progress and intramedullary metastasis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murdaca, G.; Allegra, A.; Paladin, F.; Calapai, F.; Musolino, C.; Gangemi, S. Involvement of Alarmins in the Pathogenesis and Progression of Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 9039. https://doi.org/10.3390/ijms22169039
Murdaca G, Allegra A, Paladin F, Calapai F, Musolino C, Gangemi S. Involvement of Alarmins in the Pathogenesis and Progression of Multiple Myeloma. International Journal of Molecular Sciences. 2021; 22(16):9039. https://doi.org/10.3390/ijms22169039
Chicago/Turabian StyleMurdaca, Giuseppe, Alessandro Allegra, Francesca Paladin, Fabrizio Calapai, Caterina Musolino, and Sebastiano Gangemi. 2021. "Involvement of Alarmins in the Pathogenesis and Progression of Multiple Myeloma" International Journal of Molecular Sciences 22, no. 16: 9039. https://doi.org/10.3390/ijms22169039
APA StyleMurdaca, G., Allegra, A., Paladin, F., Calapai, F., Musolino, C., & Gangemi, S. (2021). Involvement of Alarmins in the Pathogenesis and Progression of Multiple Myeloma. International Journal of Molecular Sciences, 22(16), 9039. https://doi.org/10.3390/ijms22169039