
Failure of Immunotherapy—The Molecular and Immunological Origin of Immunotherapy Resistance in Lung Cancer


Abstract
1. Introduction
2. Resistance to Immunotherapy Associated with an Impaired Function of the Immune System
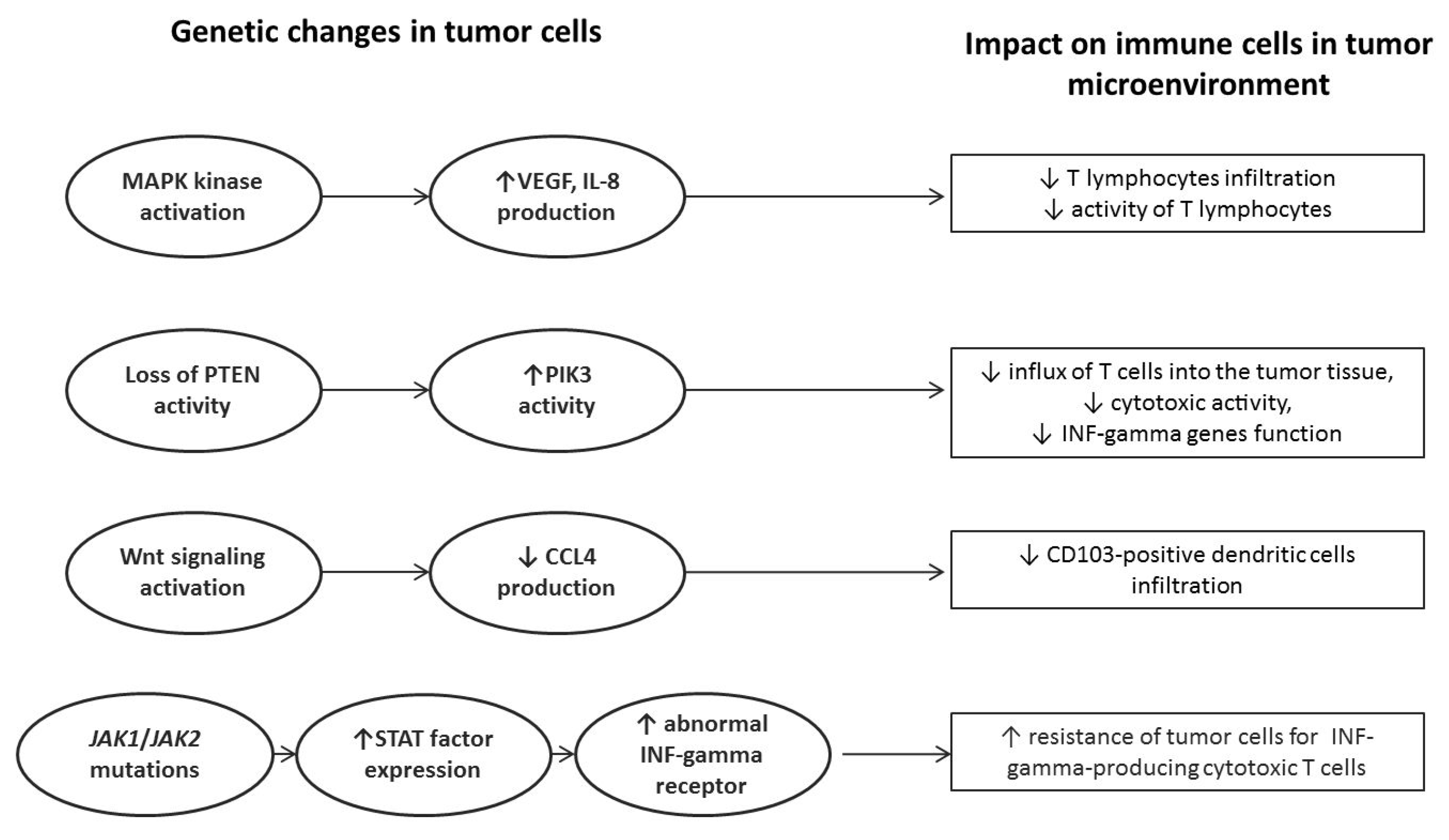
3. Genetic Background of Immunotherapy Failure
4. A Clinical Approach to Immunotherapy Resistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health 2019, 85, 8. [Google Scholar] [CrossRef]
- Michaelidou, K.; Agelaki, S.; Mavridis, K. Molecular markers related to immunosurveillance as predictive and monitoring tools in non-small cell lung cancer: Recent accomplishments and future promises. Expert Rev. Mol. Diagn. 2020, 20, 335–344. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef]
- Regzedmaa, O.; Zhang, H.; Liu, H.; Chen, J. Immune checkpoint inhibitors for small cell lung cancer: Opportunities and challenges. OncoTargets Ther. 2019, 12, 4605–4620. [Google Scholar] [CrossRef]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients With Advanced Non‒Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef]
- Borghaei, H.; Gettinger, S.; Vokes, E.E.; Chow, L.; Burgio, M.A.; de Castro Carpeno, J.; Pluzanski, A.; Arrieta, O.; Frontera, O.A.; Chiari, R.; et al. Five-Year Outcomes From the Randomized, Phase III Trials CheckMate 017 and 057: Nivolumab Versus Docetaxel in Previously Treated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 723–733. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non–Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol. 2019, 5, 1411–1420. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Jenkins, R.W.; A Barbie, D.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Hamm, C.A.; Pry, K.; Lu, J.; Bacus, S. Immune profiling reveals the diverse nature of the immune response in NSCLC and reveals signaling pathways that may influence the anti-tumor immune response. Exp. Mol. Pathol. 2019, 109, 1–15. [Google Scholar] [CrossRef]
- Lizotte, P.; Ivanova, E.V.; Awad, M.M.; Jones, R.E.; Keogh, L.; Liu, H.; Dries, R.; Almonte, C.; Herter-Sprie, G.S.; Santos, A.; et al. Multiparametric profiling of non–small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 2016, 1, e89014. [Google Scholar] [CrossRef] [PubMed]
- Raskovalova, T.; Lokshin, A.; Huang, X.; Su, Y.; Mandic, M.; Zarour, H.M.; Jackson, E.K.; Gorelik, E. Inhibition of Cytokine Production and Cytotoxic Activity of Human Antimelanoma Specific CD8+ and CD4+ T Lymphocytes by Adenosine-Protein Kinase A Type I Signaling. Cancer Res. 2007, 67, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Sun, I.-M.; Oh, M.-H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Sumitomo, M.; Takahara, K.; Zennami, K.; Nagakawa, T.; Maeda, Y.; Shiogama, K.; Yamamoto, Y.; Muto, Y.; Nukaya, T.; Takenaka, M.; et al. Tryptophan 2,3-dioxygenase in tumor cells is associated with resistance to immunotherapy in renal cell carcinoma. Cancer Sci. 2021, 112, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Attili, I.; Tarantino, P.; Passaro, A.; Stati, V.; Curigliano, G.; de Marinis, F. Strategies to overcome resistance to immune checkpoint blockade in lung cancer. Lung Cancer 2021, 154, 151–160. [Google Scholar] [CrossRef]
- Kelderman, S.; Schumacher, T.; Haanen, J.B. Acquired and intrinsic resistance in cancer immunotherapy. Mol. Oncol. 2014, 8, 1132–1139. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Eschweiler, S.; Clarke, J.; Ramírez-Suástegui, C.; Panwar, B.; Madrigal, A.; Chee, S.J.; Karydis, I.; Woo, E.; Alzetani, A.; Elsheikh, S.; et al. Intratumoral follicular regulatory T cells curtail anti-PD-1 treatment efficacy. Nat. Immunol. 2021, 22, 1052–1063. [Google Scholar] [CrossRef]
- Sprooten, J.; De Wijngaert, P.; Vanmeerbeerk, I.; Martin, S.; Vangheluwe, P.; Schlenner, S.; Krysko, D.V.; Parys, J.B.; Bultynck, G.; Vandenabeele, P.; et al. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells 2020, 9, 1823. [Google Scholar] [CrossRef]
- Wang, L.; Qin, X.; Liang, J.; Ge, P. Induction of Pyroptosis: A Promising Strategy for Cancer Treatment. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Wang, F.; Wang, S.; Zhou, Q. The resistance mechanisms of lung cancer immunotherapy. Front. Oncol. 2020, 10, 568059. [Google Scholar] [CrossRef]
- Candido, J.; Hagemann, T. Cancer-Related Inflammation. J. Clin. Immunol. 2012, 33, S79–S84. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cantor, H. The Path to Reactivation of Antitumor Immunity and Checkpoint Immunotherapy. Cancer Immunol. Res. 2014, 2, 926–936. [Google Scholar] [CrossRef][Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Nicoś, M.; Krawczyk, P.; Crosetto, N.; Milanowski, J. The Role of Intratumor Heterogeneity in the Response of Metastatic Non-Small Cell Lung Cancer to Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 569202. [Google Scholar] [CrossRef]
- Pourmir, I.; Gazeau, B.; de Saint Basile, H.; Fabre, E. Biomarkers of resistance to immune checkpoint inhibitors in non-small-cell lung cancer: Myth or reality? Cancer Drug Resist. 2020, 3, 276–286. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Boyero, L.; Sánchez-Gastaldo, A.; Alonso, M.; Noguera-Uclés, J.F.; Molina-Pinelo, S.; Bernabé-Caro, R. Primary and Acquired Resistance to Immunotherapy in Lung Cancer: Unveiling the Mechanisms Underlying of Immune Checkpoint Blockade Therapy. Cancers 2020, 12, 3729. [Google Scholar] [CrossRef]
- Tran, L.; Theodorescu, D. Determinants of Resistance to Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 1594. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Keenan, T.; Burke, K.P.; Van Allen, E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef]
- Offin, M.; Guo, R.; Wu, S.L.; Sabari, J.; Land, J.D.; Ni, A.; Montecalvo, J.; Halpenny, D.F.; Buie, L.W.; Pak, T.; et al. Immunophenotype and Response to Immunotherapy of RET-Rearranged Lung Cancers. JCO Precis. Oncol. 2019, 3, PO.18.00386. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.; Mezquita, L.; Thai, A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non–Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef]
- Sabari, J.; Leonardi, G.; Shu, C.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef]
- Dudnik, E.; Peled, N.; Nechushtan, H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Keren, S.; Popovits-Hadari, N.; Urban, D.; et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J. Thorac. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Cho, B.C.; Lopes, G.; Kowalski, D.M.; Kasahara, K.; Wu, Y.L.; Castro, G.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; Loboda, A.; et al. Relationship between STK11 and KAEP1 mutational status and efficacy in KEYNOTE-042: Pembrolizumab monotherapy versus platinum-based chemotherapy as first-line therapy for PD-L1-positive advanced NSCLC. Cancer Res. 2020, 80 (Suppl. 16), CT084. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Doshi, P.; Dobrin, R.; Szustakowski, J.; Walsh, A.M. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 2020, 5, e000706. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; TRACERx Consortium. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Frank, M.S.; Bodtger, U.; Høegholm, A.; Stamp, I.M.; Gehl, J. Re-biopsy after first line treatment in advanced NSCLC can reveal changes in PD-L1 expression. Lung Cancer 2020, 149, 23–32. [Google Scholar] [CrossRef]
- Liang, H.; Wang, M. Prospect of immunotherapy combined with anti-angiogenic agents in patients with advanced non-small cell lung cancer. Cancer Manag. Res. 2019, 11, 7707–7719. [Google Scholar] [CrossRef]
- Reck, M.; Mok, T.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Dafni, U.; Tsourti, Z.; Vervita, K.; Peters, S. Immune checkpoint inhibitors, alone or in combination with chemotherapy, as first-line treatment for advanced non-small cell lung cancer. A systematic review and network meta-analysis. Lung Cancer 2019, 134, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; A Rizvi, N.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; E Ready, N.; Gerber, D.; Chow, L.Q.; A Juergens, R.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Hellmann, M.; Kim, T.-W.; Lee, C.; Goh, B.-C.; Miller, W.; Oh, D.-Y.; Jamal, R.; Chee, C.-E.; Chow, L.; Gainor, J.; et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann. Oncol. 2019, 30, 1134–1142. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Faivre-Finn, C.; Vicente, D.; Kurata, T.; Planchard, D.; Paz-Ares, L.; Vansteenkiste, J.F.; Spigel, D.R.; Garassino, M.C.; Reck, M.; Senan, S.; et al. Four-Year Survival With Durvalumab After Chemoradiotherapy in Stage III NSCLC—An Update From the PACIFIC Trial. J. Thorac. Oncol. 2021, 16, 860–867. [Google Scholar] [CrossRef]
- Gerber, D.E.; Urbanic, J.J.; Langer, C.; Hu, C.; Chang, I.-F.; Lü, B.; Movsas, B.; Jeraj, R.; Curran, W.J.; Bradley, J.D. Treatment Design and Rationale for a Randomized Trial of Cisplatin and Etoposide Plus Thoracic Radiotherapy Followed by Nivolumab or Placebo for Locally Advanced Non–Small-Cell Lung Cancer (RTOG 3505). Clin. Lung Cancer 2017, 18, 333–339. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, J. T cell-redirecting bispecific antibodies in cancer immunotherapy: Recent advances. J. Cancer Res. Clin. Oncol. 2019, 145, 941–956. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Clinical Trial ID | Primary Outcome Measures | Intervention/Treatment |
|---|---|---|
| NCT04203095 | Correlation analysis between the ICIs resistance and CIN. | Diagnostic test: the level CIN in plasma cfDNA. Anti-PD-1 antibody treatment. |
| NCT04300062 | Molecular mechanism involving the progression of NSCLC and SCLC | Re-biopsy at the moment of cancer progression. Immunotherapy. |
| NCT03512847 | Concordance between specific gene profiles and treatment outcomes. Differences in molecular profiles pre- and post-treatment. Quantification of cfDNA during treatment linked to treatment outcome. | Comprehensive molecular profiling and whole exome sequencing (WES) of cfDNA. Immunotherapy/chemotherapy. |
| NCT04807114 | Identification and comparison the percentages of immune cell subtypes present in tumors from responding vs. non-responding the patients before start of ICIs therapy. | Tumor biopsies from patients before start of treatment. Anti-PD-1 monotherapy or combination of anti-PD-1 therapy and chemotherapy. |
| 5 | Identification of actionable mutations and intratumor heterogeneity in NSCLC patients with EGFR or HER2 genes mutations and relapse after afatinib treatment. Qualification to different treatment regimens and measurement of therapy effectiveness (ORR, PFS) | Biopsy sample at the time of NSCLC relapse. Arm 1: Patients without an actionable mutation—atezolizumab monotherapy or in combination with chemotherapy; Arm 2: Patients with BRAF V600 mutation—vemurafenib; Arm 3: Patients with ALK or RET genes rearrangement—alectinib; Arm 4: HER2 gene amplification—trastuzumab emtansine. |
| 5 | Correlations between specific genes mutations, immune microenvironment and ORR as well as PFS in patients received immunotherapy. | Sequencing of DNA from FFPE tissue and whole blood. IHC analysis of immune microenvironment. Anti-PD-1 or anti-PD-L1 antibody treatment. |
| Clinical Trial ID | Primary Endpoint | Treatment |
|---|---|---|
| NCT04782622 | ORR and PFS | apatinib + camrelizumab |
| NCT04691388 | PFS | Anlotinib + sinidilizumab |
| NCT04670107 | PFS and OS AE (adverse event caused by the combination) | anlotinib in combination with immune checkpoint inhibitors |
| NCT03833440 | The 12-week disease control rate | Arm A: durvalumab + monalizumab (IPH2201), Arm B: durvalumab + oleclumab (MEDI9447), Arm C: durvalumab + ceralasertib (AZD6738), Arm D: standard of third or fourth-line chemotherapy (docetaxel) |
| NCT03262779 | ORR | nivolumab + ipilimumab |
| NCT03600701 | ORR | atezolizumab + cobimetinib |
| NCT03841110 | Maximum tolerated dose | FT500 + cyclophosphamide + fludarabine FT500 + nivolumab + cyclophosphamide + fludarabine FT500 + pembrolizumab + cyclophosphamide + fludarabine FT500 + atezolizumab + cyclophosphamide + fludarabine FT500 + IL-2 (proleukin, aldesleukin) + nivolumab + cyclophosphamide + fludarabine FT500 + IL-2 (proleukin, aldesleukin) + pembrolizumab + cyclophosphamide + fludarabine FT500 + IL-2 (proleukin, aldesleukin) + atezolizumab + cyclophosphamide + fludarabine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Błach, J.; Wojas-Krawczyk, K.; Nicoś, M.; Krawczyk, P. Failure of Immunotherapy—The Molecular and Immunological Origin of Immunotherapy Resistance in Lung Cancer. Int. J. Mol. Sci. 2021, 22, 9030. https://doi.org/10.3390/ijms22169030
Błach J, Wojas-Krawczyk K, Nicoś M, Krawczyk P. Failure of Immunotherapy—The Molecular and Immunological Origin of Immunotherapy Resistance in Lung Cancer. International Journal of Molecular Sciences. 2021; 22(16):9030. https://doi.org/10.3390/ijms22169030
Chicago/Turabian StyleBłach, Justyna, Kamila Wojas-Krawczyk, Marcin Nicoś, and Paweł Krawczyk. 2021. "Failure of Immunotherapy—The Molecular and Immunological Origin of Immunotherapy Resistance in Lung Cancer" International Journal of Molecular Sciences 22, no. 16: 9030. https://doi.org/10.3390/ijms22169030
APA StyleBłach, J., Wojas-Krawczyk, K., Nicoś, M., & Krawczyk, P. (2021). Failure of Immunotherapy—The Molecular and Immunological Origin of Immunotherapy Resistance in Lung Cancer. International Journal of Molecular Sciences, 22(16), 9030. https://doi.org/10.3390/ijms22169030