Developing a new drug that gains marketing approval is estimated to cost USD 2.6 billion, and the approval rate for drugs entering clinical development is less than 12% [
1,
2]. Such massive investments and high risks drive scientists to explore novel and more efficient approaches in drug discovery. Under such circumstances, computer-aided drug design methods, especially the recent deep learning-based approaches, have been rapidly developing and have made key contributions to the development of drugs that are in either clinical use or clinical trials. Among the broad range of drug design phases that computational approaches involve, the prediction of drug–target affinity (DTA) is one of the most important steps, as an accurate and efficient DTA prediction algorithm could effectively speed up the process of virtual screening of potential drug molecules, minimizing unnecessary biological and chemical experiments by refining the search space for potential drugs.
Computational approaches for DTA prediction generally comprise two major steps. First, features of drugs or proteins, or representations/descriptors as alternative expressions, are obtained from raw input data by feature extraction methods. Compared to the original input data, the embedded representations are normally more applicable to the subsequent phase and can achieve better performance. The next step, as previously mentioned, is the classification/regression procedure, where the representations act as inputs and the network outputs as either data labels (i.e., active or inactive) or specific values (i.e., the affinity for each drug–target pair). For the feature extraction methods, earlier research represented drugs and proteins based on human experience or skillfully designed mathematical descriptors, i.e., hand-crafted features [
3,
4]. In this regard, KronRLS uses pairwise kernels that are computed as the Kronecker product of the compound kernel and the protein kernel for the representations [
5]. In the SimBoost model, He et al. defined three types of features separately for the drug, target, and the drug–target pair, each of which contained multiple hand-crafted features [
6]. These approaches, despite achieving good performance in the DTA prediction task, depend on chemical insights or expert experiences, which, in turn, restrict further optimizations of these models.
With the rapid advancements in deep learning in the last decade, various data-driven methods were proposed for the description of drugs and target proteins [
7,
8,
9,
10,
11]. These deep learning approaches differ from hand-crafted features, and features can be extracted automatically through deep learning methods and are proved to be more effective. For deep learning approaches specifically in the DTA area, they can be categorized into non-structure-based and structure-based methods. The former learns the representations from sequential data, which are fingerprints of molecule and acid sequences of protein. For example, DeepDTA [
12] used only 1D representations of targets and proteins, and convolutional neural networks containing three layers were applied to both acid sequence and drug SMILES to obtain the representations. Similarly, WideDTA [
13] also relied only on the 1D representation, but it differed from DeepDTA in which the drug SMILES and protein sequence were represented as words (instead of characters) that correspond to an eight-character sequence and a three-residual sequence, respectively. In addition, the ligand maximum common substructure (LMCS) of drugs and motifs and domains of proteins (PDM) were utilized and formed a four-branch architecture together with the ligand SMILES and protein sequence branches. DeepCPI [
14] leveraged techniques from natural language processing to learn low-dimensional feature representations, including latent semantic analysis for drug embedding and Work2vec for protein embedding. On the other hand, the structure-based methods utilized two-dimensional topology (i.e., graph) [
15] or three-dimensional structures [
16] for representation extraction. As a type of non-Euclidean data, the molecular graph is irregular with variable size, which makes it difficult to apply traditional deep learning methods such as convolutional neural network (CNN) to it. This type of data graph differs from Euclidean structural data, which are not applicable to many basic operations of traditional deep learning methods. In this regard, the graph neural network (GNN) was proposed to handle graph data, and it put no limit on the size of the input graph, thus providing a flexible format to extract in-depth information within the graph [
17]. Following this work, a number of variants of the GNN have emerged in recent years, such as the graph convolutional network (GCN) [
18], the graph attention network (GAT) [
19], and the gated graph neural network (GGNN) [
20], and systems based on these GNN variants have demonstrated ground-breaking performance in many relevant application tasks [
21]. Focusing on the drug–target prediction tasks, Tsubaki et al. proposed the application of the GNN to DTA (or compound–protein interaction, CPI) prediction, where the compounds were represented as graphs, and, consequently, the 2D structural information could be kept and extracted using the GNN. The
r-radius subgraphs and
n-length subsequence were introduced and were proved to be crucial in improving model performance [
22]. Similarly, Gao et al. utilized the GNN for drug representation, whereas the protein descriptors were obtained using long short-term memory (LSTM) [
23]. GraphDTA also introduced graph representation to take advantage of the 2D structural information of the drug molecular graph [
24]. GraphDTA also discarded the CNN in the drug branch, and it used a three-layer GCN as an alternative for drug representation, while keeping the CNN in the protein branch as in DeepDTA. GraphDTA provided better results than those of the baseline 1D approaches, suggesting a prominent role of structural information. Chemical context can also be considered in order to provide additional features other than the molecular graph itself; for example, DeepGS used embedding techniques of Smi2Vec and Prot2Vec to exploit the chemical context within the drug SMILES and amino sequences. This chemical context was then combined with graph-derived features for DTA prediction [
25].
The performance of either the 1D or structure-based representation can be enhanced by introducing attention mechanisms. The attention mechanisms allow the network to focus on the most relevant parts of the input and have been proven to be useful for various tasks [
19,
26]. For instances, AttentionDTA added an additional attention block following the two branches of the drug and protein, and, therefore, the learned features could be further weighted according to the attention score before they were fed into the fully connected classifying layers [
27]. Lim et al. proposed a distance-aware attention algorithm that could capture the most relevant intermolecular interactions within the 3D protein–ligand complex. Such attention mechanisms were proved to be effective when applied to DTA prediction tasks with structural information of a complex [
16]. Recently, Lee et al. proposed a novel attention structure that introduced self-attention mechanisms for node pooling named self-attention graph pooling (SAGPool), and it achieved state-of-the-art performances in many graph learning tasks [
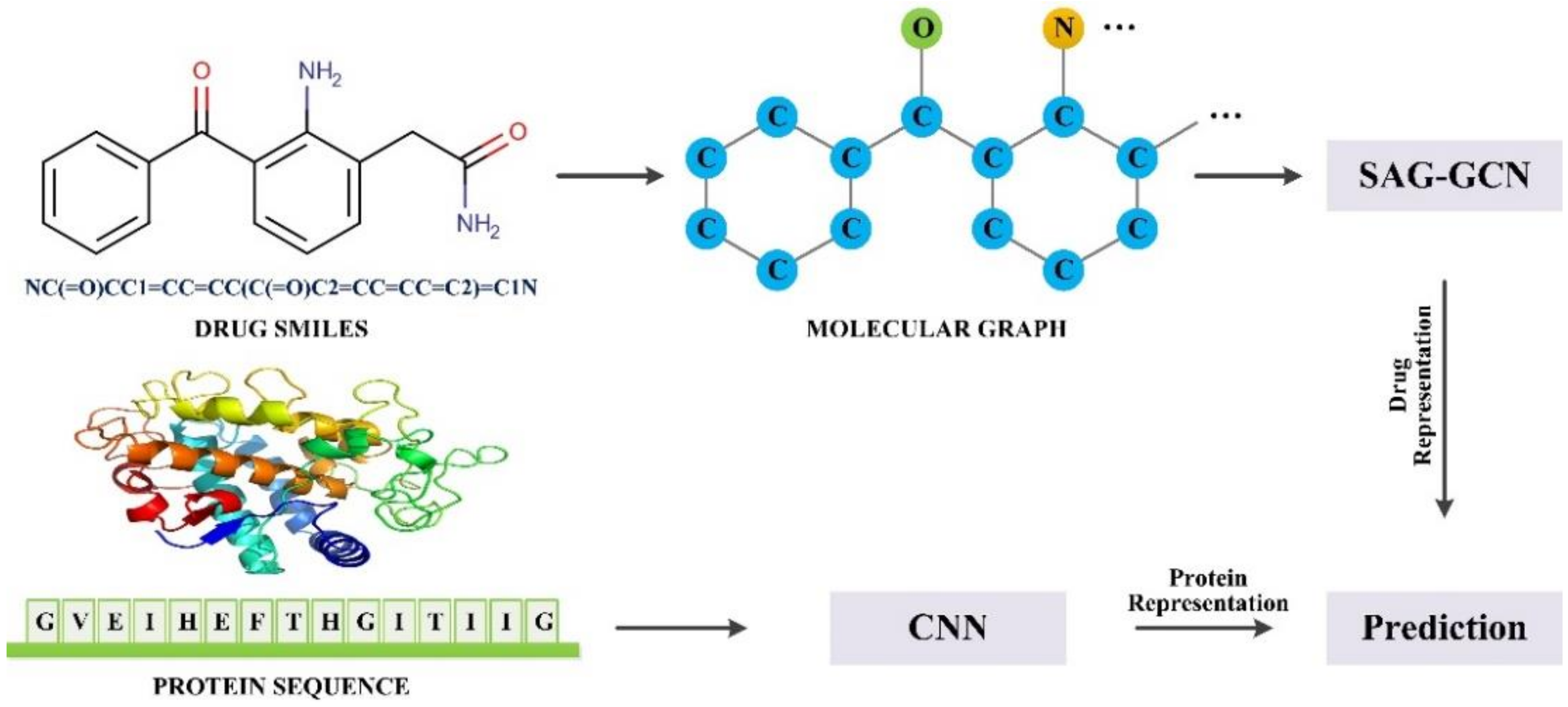
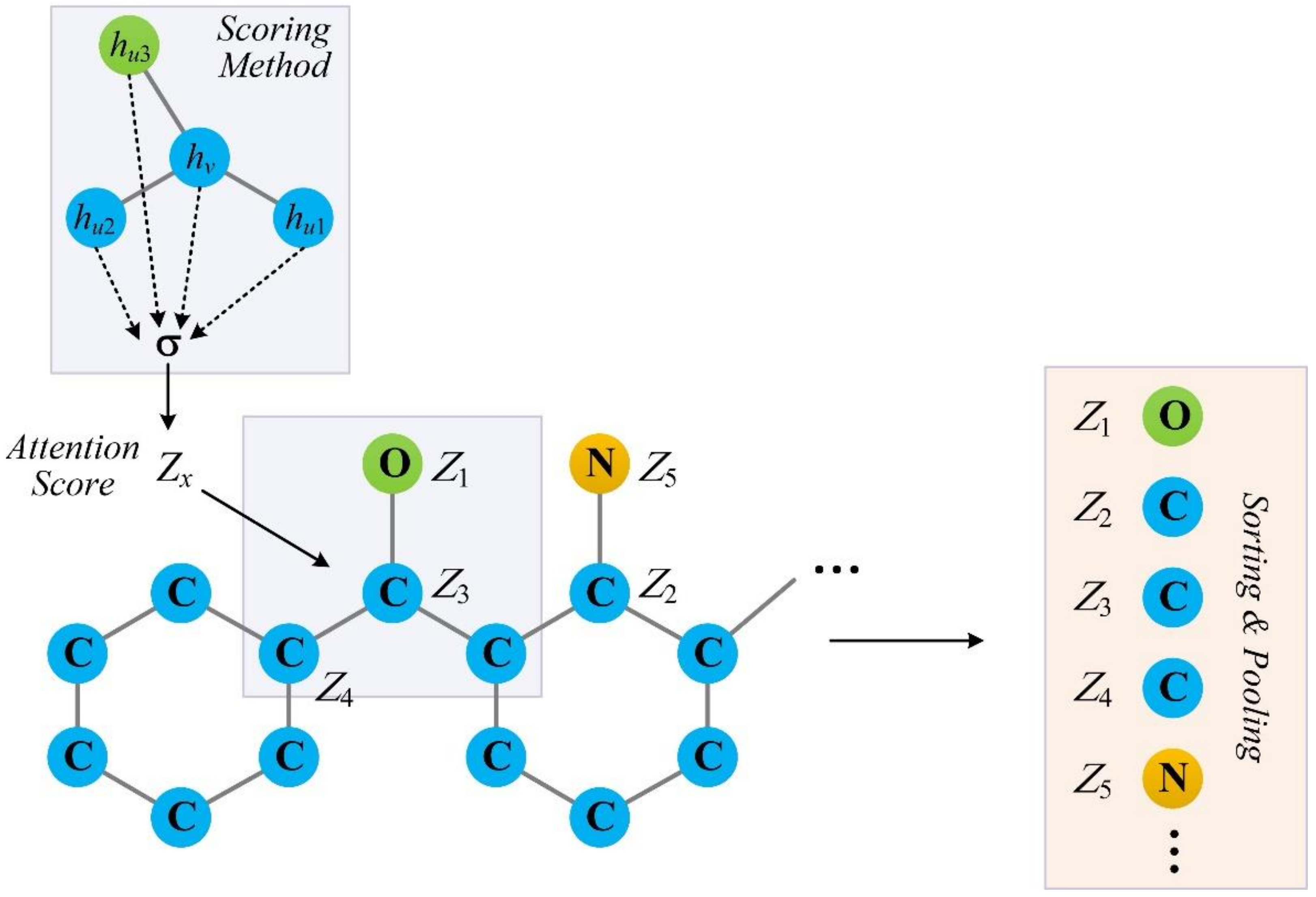
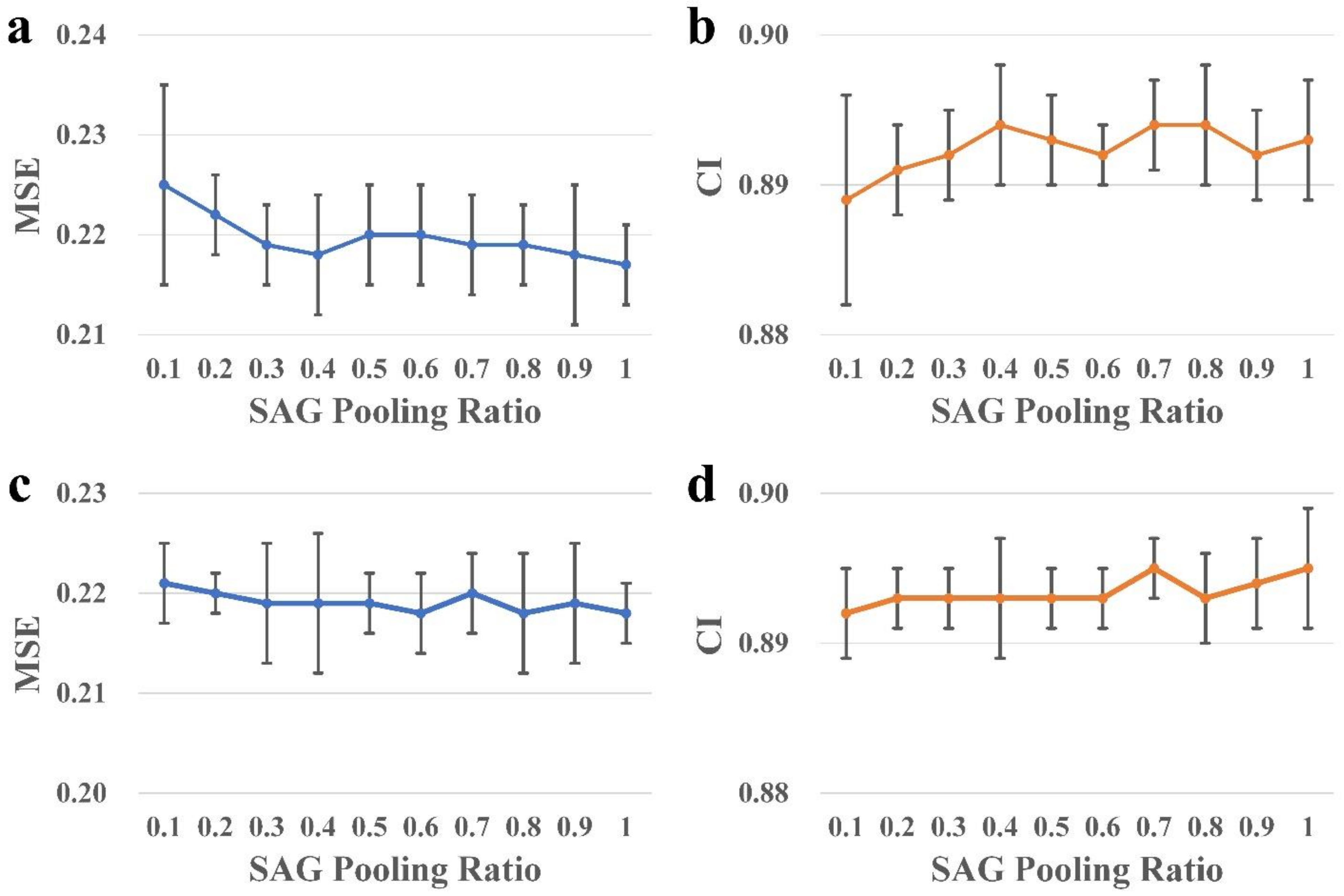
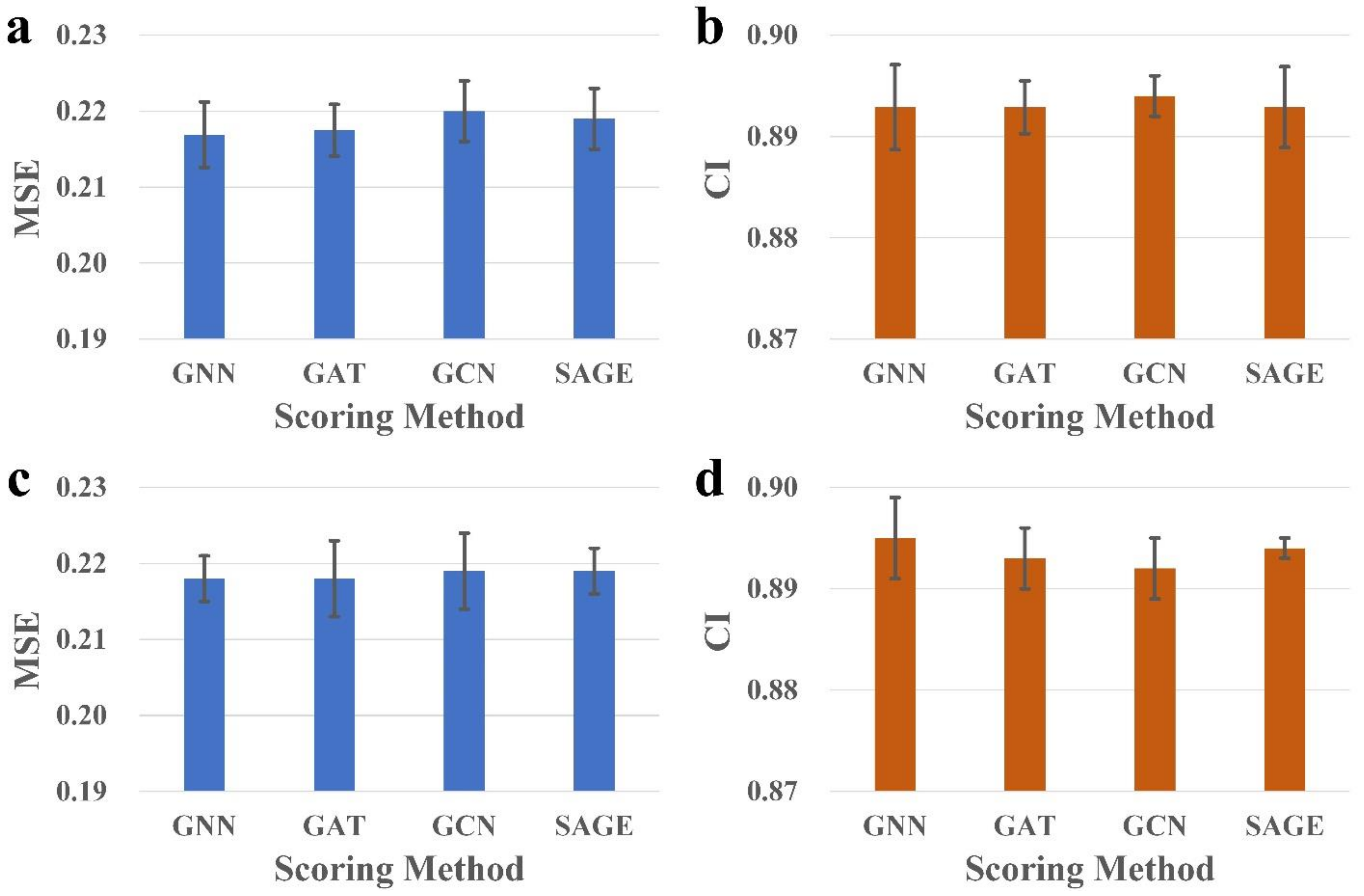
28]. Inspired by this work, we implemented an SAG-DTA network in this study, which adopted a self-attention graph pooling approach to molecular graph representation. Two architectures, namely, global pooling and hierarchical pooling, were implemented and evaluated, with a detailed comparison of the pooling ratio and scoring method for each architecture.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}