
Adaptive Immunity and the Risk of Autoreactivity in COVID-19


 ,
, 
Abstract
:1. Introduction
2. Viral Infections and Autoimmunity
3. Autoantibodies Identified in COVID-19 Positive Patients
4. Autoimmunity Associated with SARS-CoV-2
4.1. Immune Thrombocytopenia and Vasculitis Post Infection or Vaccination
4.2. Autoimmune Haemolytic Anaemia and Cold Agglutinin Syndrome
4.3. Guillain−Barré Syndrome and Miller Fisher Syndrome
4.4. Systemic Lupus Erythematosus, Multiple Sclerosis and Systemic Rheumatoid Disease
4.5. Multisystem Inflammatory Syndrome in Children (MIS-C)
5. Viral Induced Autoimmune Mechanisms in COVID-19
6. Limitations and Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Who Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 17 August 2021).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- WHO. Coronavirus Disease (COVID-19) Advice for the Public. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/advice-for-public (accessed on 10 October 2020).
- Meng, X.; Deng, Y.; Dai, Z.; Meng, Z. COVID-19 and anosmia: A review based on up-to-date knowledge. Am. J. Otolaryngol. 2020, 41, 102581. [Google Scholar] [CrossRef]
- Han, H.; Xie, L.; Liu, R.; Yang, J.; Liu, F.; Wu, K.; Chen, L.; Hou, W.; Feng, Y.; Zhu, C. Analysis of heart injury laboratory parameters in 273 COVID-19 patients in one hospital in wuhan, China. J. Med. Virol. 2020, 92, 819–823. [Google Scholar] [CrossRef]
- Cheung, S.; Quiwa, J.C.; Pillai, A.; Onwu, C.; Tharayil, Z.J.; Gupta, R. Superior mesenteric artery thrombosis and acute intestinal ischemia as a consequence of COVID-19 infection. Am. J. Case Rep. 2020, 21, e925753. [Google Scholar] [CrossRef] [PubMed]
- Nalleballe, K.; Reddy Onteddu, S.; Sharma, R.; Dandu, V.; Brown, A.; Jasti, M.; Yadala, S.; Veerapaneni, K.; Siddamreddy, S.; Avula, A.; et al. Spectrum of neuropsychiatric manifestations in COVID-19. Brain Behav. Immun. 2020, 88, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Poyiadji, N.; Shahin, G.; Noujaim, D.; Stone, M.; Patel, S.; Griffith, B. COVID-19–associated acute hemorrhagic necrotizing encephalopathy: Imaging features. Radiology 2020, 296, E119–E120. [Google Scholar] [CrossRef] [Green Version]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-vessel stroke as a presenting feature of COVID-19 in the young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and methods for t- and b-cell epitope prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, N.; Eddy, D.; Huntley, C.; van Schalkwyk, M.C.I.; Shrotri, M.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. Antibody response to sars-cov-2 infection in humans: A systematic review. PLoS ONE 2021, 15, e0244126. [Google Scholar]
- Shrotri, M.; van Schalkwyk, M.C.I.; Post, N.; Eddy, D.; Huntley, C.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. T cell response to sars-cov-2 infection in humans: A systematic review. PLoS ONE 2021, 16, e0245532. [Google Scholar] [CrossRef]
- Hachim, A.; Kavian, N.; Cohen, C.A.; Chin, A.W.H.; Chu, D.K.W.; Mok, C.K.P.; Tsang, O.T.Y.; Yeung, Y.C.; Perera, R.A.P.M.; Poon, L.L.M.; et al. Orf8 and orf3b antibodies are accurate serological markers of early and late sars-cov-2 infection. Nat. Immunol. 2020, 21, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of t cell responses to sars-cov-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 2020, 181, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemazee, D. Mechanisms of central tolerance for b cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Maeda, Y.; Nishikawa, H.; Sugiyama, D.; Ha, D.; Hamaguchi, M.; Saito, T.; Nishioka, M.; Wing, J.B.; Adeegbe, D.; Katayama, I.; et al. Detection of self-reactive cd8(+) t cells with an anergic phenotype in healthy individuals. Science 2014, 346, 1536–1540. [Google Scholar] [CrossRef]
- Richards, D.M.; Ruggiero, E.; Hofer, A.-C.; Sefrin, J.P.; Schmidt, M.; von Kalle, C.; Feuerer, M. The contained self-reactive peripheral t cell repertoire: Size, diversity, and cellular composition. J. Immunol. 2015, 195, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Meffre, E.; Wardemann, H. B-cell tolerance checkpoints in health and autoimmunity. Curr. Opin. Immunol. 2008, 20, 632–638. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G.J. Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, P.S.; DeFranco, A.L. Making and breaking tolerance. Curr. Opin. Immunol. 2002, 14, 744–759. [Google Scholar] [CrossRef]
- Jackson, S.R.; Yuan, J.; Berrien-Elliott, M.M.; Chen, C.L.; Meyer, J.M.; Donlin, M.J.; Teague, R.M. Inflammation programs self-reactive cd8+ t cells to acquire t-box-mediated effector function but does not prevent deletional tolerance. J. Leukoc. Biol. 2014, 96, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Zharkova, O.; Celhar, T.; Cravens, P.D.; Satterthwaite, A.B.; Fairhurst, A.M.; Davis, L.S. Pathways leading to an immunological disease: Systemic lupus erythematosus. Rheumatology 2017, 56, i55–i66. [Google Scholar] [CrossRef] [Green Version]
- Høglund, R.A.; Maghazachi, A.A. Multiple sclerosis and the role of immune cells. World J. Exp. Med. 2014, 4, 27–37. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, J.F.; Pereira, R.M.; Shoenfeld, Y. The mosaic of autoimmunity: The role of environmental factors. Front. Biosci. (Elite Ed.) 2009, 1, 501–509. [Google Scholar]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and autoimmunity: A review on the potential interaction and molecular mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörner, T.; Radbruch, A. Antibodies and b cell memory in viral immunity. Immunity 2007, 27, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, S.; Halle, O.; Förster, R. Mechanisms and dynamics of t cell-mediated cytotoxicity in vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef]
- DiMaggio, D.; Anderson, A.; Bussel, J.B. Cytomegalovirus can make immune thrombocytopenic purpura refractory. Br. J. Haematol. 2009, 146, 104–112. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, H.; Chen, P.; Lin, Q.; Zhu, X.; Zhang, L.; Xue, X. Correlation between systemic lupus erythematosus and cytomegalovirus infection detected by different methods. Clin. Rheumatol. 2015, 34, 691–698. [Google Scholar] [CrossRef]
- Moon, U.Y.; Park, S.J.; Oh, S.T.; Kim, W.U.; Park, S.H.; Lee, S.H.; Cho, C.S.; Kim, H.Y.; Lee, W.K.; Lee, S.K. Patients with systemic lupus erythematosus have abnormally elevated epstein-barr virus load in blood. Arthritis Res. 2004, 6, R295–R302. [Google Scholar] [CrossRef] [Green Version]
- Yokochi, T.; Yanagawa, A.; Kimura, Y.; Mizushima, Y. High titer of antibody to the epstein-barr virus membrane antigen in sera from patients with rheumatoid arthritis and systemic lupus erythematosus. J. Rheumatol. 1989, 16, 1029–1032. [Google Scholar]
- Honkanen, H.; Oikarinen, S.; Nurminen, N.; Laitinen, O.H.; Huhtala, H.; Lehtonen, J.; Ruokoranta, T.; Hankaniemi, M.M.; Lecouturier, V.; Almond, J.W.; et al. Detection of enteroviruses in stools precedes islet autoimmunity by several months: Possible evidence for slowly operating mechanisms in virus-induced autoimmunity. Diabetologia 2017, 60, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Ramondetti, F.; Sacco, S.; Comelli, M.; Bruno, G.; Falorni, A.; Iannilli, A.; d’Annunzio, G.; Iafusco, D.; Songini, M.; Toni, S.; et al. Type 1 diabetes and measles, mumps and rubella childhood infections within the italian insulin-dependent diabetes registry. Diabet. Med. 2012, 29, 761–766. [Google Scholar] [CrossRef]
- Salmi, A.; Ziola, B.; Hovi, T.; Reunanen, M. Antibodies to coronaviruses oc43 and 229e in multiple sclerosis patients. Neurology 1982, 32, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.N.; Mounir, S.; Talbot, P.J. Human coronavirus gene expression in the brains of multiple sclerosis patients. Virology 1992, 191, 502–505. [Google Scholar] [CrossRef]
- Arbour, N.; Day, R.; Newcombe, J.; Talbot, P.J. Neuroinvasion by human respiratory coronaviruses. J. Virol. 2000, 74, 8913–8921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, P.J.; Paquette, J.S.; Ciurli, C.; Antel, J.P.; Ouellet, F. Myelin basic protein and human coronavirus 229e cross-reactive t cells in multiple sclerosis. Ann. Neurol. 1996, 39, 233–240. [Google Scholar] [CrossRef]
- Magdi, M.; Rahil, A. Severe immune thrombocytopenia complicated by intracerebral haemorrhage associated with coronavirus infection: A case report and literature review. Eur. J. Case Rep. Intern. Med. 2019, 6, 001155. [Google Scholar]
- Wong, R.S.M.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.K.; Wong, C.K.; Chan, P.K.S.; Ng, M.H.L.; Yu, L.M.; Hui, D.S.; et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 2003, 326, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Ng, M.H.; Li, C.K. Thrombocytopenia in patients with severe acute respiratory syndrome (review). Hematology 2005, 10, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, S.; Shen, H.; Jiang, L.; Zhang, M.; Xiao, D.; Liu, Y.; Ma, X.; Zhang, Y.; Guo, N.; et al. Cross-reaction of sars-cov antigen with autoantibodies in autoimmune diseases. Cell Mol. Immunol 2004, 1, 304–307. [Google Scholar]
- Zhou, Y.; Han, T.; Chen, J.; Hou, C.; Hua, L.; He, S.; Guo, Y.; Zhang, S.; Wang, Y.; Yuan, J.; et al. Clinical and autoimmune characteristics of severe and critical cases of COVID-19. Clin. Transl. Sci. 2020, 13, 1077–1086. [Google Scholar] [CrossRef]
- Vlachoyiannopoulos, P.G.; Magira, E.; Alexopoulos, H.; Jahaj, E.; Theophilopoulou, K.; Kotanidou, A.; Tzioufas, A.G. Autoantibodies related to systemic autoimmune rheumatic diseases in severely ill patients with COVID-19. Ann. Rheum. Dis. 2020, 79, 1661–1663. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type i ifns in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Franke, C.; Ferse, C.; Kreye, J.; Reincke, S.M.; Sanchez-Sendin, E.; Rocco, A.; Steinbrenner, M.; Angermair, S.; Treskatsch, S.; Zickler, D.; et al. High frequency of cerebrospinal fluid autoantibodies in COVID-19 patients with neurological symptoms. Brain Behav. Immun. 2021, 93, 415–419. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in critically ill COVID-19 patients. J. Thromb. Thrombolysis 2020, 50, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and antiphospholipid antibodies in patients with COVID-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Gatto, M.; Perricone, C.; Tonello, M.; Bistoni, O.; Cattelan, A.M.; Bursi, R.; Cafaro, G.; De Robertis, E.; Mencacci, A.; Bozza, S.; et al. Frequency and clinical correlates of antiphospholipid antibodies arising in patients with sars-cov-2 infection: Findings from a multicentre study on 122 cases. Clin. Exp. Rheumatol. 2020, 38, 754–759. [Google Scholar]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type i interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Banerjee, M. Immune thrombocytopenia secondary to COVID-19: A systematic review. SN Compr. Clin. Med. 2020, 1–11. [Google Scholar] [CrossRef]
- Bomhof, G.; Mutsaers, P.; Leebeek, F.W.G.; Te Boekhorst, P.A.W.; Hofland, J.; Croles, F.N.; Jansen, A.J.G. COVID-19-associated immune thrombocytopenia. Br. J. Haematol. 2020, 190, e61–e64. [Google Scholar] [CrossRef]
- Zulfiqar, A.-A.; Lorenzo-Villalba, N.; Hassler, P.; Andrès, E. Immune thrombocytopenic purpura in a patient with COVID-19. N. Engl. J. Med. 2020, 382, e43. [Google Scholar] [CrossRef] [PubMed]
- Camprodon Gómez, M.; González-Cruz, C.; Ferrer, B.; Barberá, M.J. Leucocytoclastic vasculitis in a patient with COVID-19 with positive sars-cov-2 pcr in skin biopsy. BMJ Case Rep. CP 2020, 13, e238039. [Google Scholar] [CrossRef] [PubMed]
- Oda, R.; Inagaki, T.; Ishikane, M.; Hotta, M.; Shimomura, A.; Sato, M.; Nakamoto, T.; Akiyama, Y.; Yamamoto, K.; Minamimoto, R.; et al. Case of adult large vessel vasculitis after sars-cov-2 infection. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Scully, M.; Singh, D.; Lown, R.; Poles, A.; Solomon, T.; Levi, M.; Goldblatt, D.; Kotoucek, P.; Thomas, W.; Lester, W. Pathologic antibodies to platelet factor 4 after chadox1 ncov-19 vaccination. N. Engl. J. Med. 2021, 384, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic thrombocytopenia after chadox1 ncov-19 vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Muir, K.-L.; Kallam, A.; Koepsell, S.A.; Gundabolu, K. Thrombotic Thrombocytopenia after Ad26.COV2.S Vaccination. N. Engl. J. Med. 2021, 384, 1964–1965. [Google Scholar] [CrossRef]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.-H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef]
- Greinacher, A.; Selleng, K.; Warkentin, T.E. Autoimmune heparin-induced thrombocytopenia. J. Thromb. Haemost. 2017, 15, 2099–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørvoll, I.H.; Horvei, K.D.; Ernstsen, S.L.; Laegreid, I.J.; Lund, S.; Grønli, R.H.; Olsen, M.K.; Jacobsen, H.K.; Eriksson, A.; Halstensen, A.M.; et al. An observational study to identify the prevalence of thrombocytopenia and anti-pf4/polyanion antibodies in norwegian health care workers after COVID-19 vaccination. J. Thromb. Haemost. 2021, 19, 1813–1818. [Google Scholar] [CrossRef]
- Thiele, T.; Ulm, L.; Holtfreter, S.; Schönborn, L.; Kuhn, S.O.; Scheer, C.; Warkentin, T.E.; Bröker, B.; Becker, K.; Aurich, K.; et al. Frequency of positive anti-PF4/polyanion antibody tests after COVID-19 vaccination with ChAdOx1 nCoV-19 and BNT162b2. Blood 2021, 138, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.E.; Wilson, S.; Thombare, A.; Weiss, S.; Ma, A. Cold agglutinin syndrome as a complication of COVID-19 in two cases. Clin. Infect. Pract. 2020, 7, 100041. [Google Scholar] [CrossRef] [PubMed]
- Maslov, D.V.; Simenson, V.; Jain, S.; Badari, A. COVID-19 and cold agglutinin hemolytic anemia. TH Open 2020, 4, e175–e177. [Google Scholar] [CrossRef]
- Lazarian, G.; Quinquenel, A.; Bellal, M.; Siavellis, J.; Jacquy, C.; Re, D.; Merabet, F.; Mekinian, A.; Braun, T.; Damaj, G.; et al. Autoimmune haemolytic anaemia associated with COVID-19 infection. Br. J. Haematol. 2020, 190, 29–31. [Google Scholar] [CrossRef]
- Patil, N.R.; Herc, E.S.; Girgis, M. Cold agglutinin disease and autoimmune hemolytic anemia with pulmonary embolism as a presentation of COVID-19 infection. Hematol. Oncol. Stem Cell Ther. 2020. [Google Scholar] [CrossRef]
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain–barré syndrome associated with sars-cov-2. N. Engl. J. Med. 2020, 382, 2574–2576. [Google Scholar] [CrossRef]
- Zito, A.; Alfonsi, E.; Franciotta, D.; Todisco, M.; Gastaldi, M.; Cotta Ramusino, M.; Ceroni, M.; Costa, A. COVID-19 and guillain-barré syndrome: A case report and review of literature. Front. Neurol. 2020, 11, 909. [Google Scholar] [CrossRef]
- Senel, M.; Abu-Rumeileh, S.; Michel, D.; Garibashvili, T.; Althaus, K.; Kassubek, J.; Otto, M. Miller-fisher syndrome after COVID-19: Neurochemical markers as an early sign of nervous system involvement. Eur. J. Neurol. 2020, 27, 2378–2380. [Google Scholar] [CrossRef]
- Gutiérrez-Ortiz, C.; Méndez-Guerrero, A.; Rodrigo-Rey, S.; San Pedro-Murillo, E.; Bermejo-Guerrero, L.; Gordo-Mañas, R.; de Aragón-Gómez, F.; Benito-León, J. Miller fisher syndrome and polyneuritis cranialis in COVID-19. Neurology 2020, 95, e601–e605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamani, B.; Moeini Taba, S.-M.; Shayestehpour, M. Systemic lupus erythematosus manifestation following COVID-19: A case report. J. Med. Case Rep. 2021, 15, 1–4. [Google Scholar] [CrossRef]
- Mantovani Cardoso, E.; Hundal, J.; Feterman, D.; Magaldi, J. Concomitant new diagnosis of systemic lupus erythematosus and COVID-19 with possible antiphospholipid syndrome. Just a coincidence? A case report and review of intertwining pathophysiology. Clin. Rheumatol. 2020, 39, 2811–2815. [Google Scholar] [CrossRef]
- Palao, M.; Fernández-Díaz, E.; Gracia-Gil, J.; Romero-Sánchez, C.M.; Díaz-Maroto, I.; Segura, T. Multiple sclerosis following sars-cov-2 infection. Mult. Scler. Relat. Disord. 2020, 45, 102377. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Ghannam, M.; Manousakis, G. A first presentation of multiple sclerosis with concurrent COVID-19 infection. Eneurologicalsci 2021, 22, 100299. [Google Scholar] [CrossRef] [PubMed]
- Yavari, F.; Raji, S.; Moradi, F.; Saeidi, M. Demyelinating changes alike to multiple sclerosis: A case report of rare manifestations of COVID-19. Case Rep. Neurol. Med. 2020, 2020, 6682251. [Google Scholar]
- Hsu, T.Y.T.; D’Silva, K.M.; Patel, N.J.; Fu, X.; Wallace, Z.S.; Sparks, J.A. Incident systemic rheumatic disease following COVID-19. Lancet Rheumatol. 2021, 3, e402–e404. [Google Scholar] [CrossRef]
- Jones, V.G.; Mills, M.; Suarez, D.; Hogan, C.A.; Yeh, D.; Segal, J.B.; Nguyen, E.L.; Barsh, G.R.; Maskatia, S.; Mathew, R. COVID-19 and kawasaki disease: Novel virus and novel case. Hosp. Pediatr. 2020, 10, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Hicar, M.D. Antibodies and immunity during kawasaki disease. Front. Cardiovasc. Med. 2020, 7, 94. [Google Scholar] [CrossRef]
- Toubiana, J.; Poirault, C.; Corsia, A.; Bajolle, F.; Fourgeaud, J.; Angoulvant, F.; Debray, A.; Basmaci, R.; Salvador, E.; Biscardi, S.; et al. Kawasaki-like multisystem inflammatory syndrome in children during the COVID-19 pandemic in paris, france: Prospective observational study. BMJ 2020, 369, m2094. [Google Scholar] [CrossRef]
- Organisation, W.H. Multisystem Inflammatory Syndrome in Children and Adolescents Temporally Related to COVID-19. Available online: https://www.who.int/news-room/commentaries/detail/multisystem-inflammatory-syndrome-in-children-and-adolescents-with-covid-19 (accessed on 19 June 2021).
- Consiglio, C.R.; Cotugno, N.; Sardh, F.; Pou, C.; Amodio, D.; Rodriguez, L.; Tan, Z.; Zicari, S.; Ruggiero, A.; Pascucci, G.R.; et al. The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell 2020, 183, 968–981. [Google Scholar] [CrossRef]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (mis-c). Cell 2020, 183, 982–995. [Google Scholar] [CrossRef]
- Gregorova, M.; Morse, D.; Brignoli, T.; Steventon, J.; Hamilton, F.; Albur, M.; Arnold, D.; Thomas, M.; Halliday, A.; Baum, H.; et al. Post-acute COVID-19 associated with evidence of bystander t-cell activation and a recurring antibiotic-resistant bacterial pneumonia. Elife 2020, 9, e63430. [Google Scholar] [CrossRef]
- Bergamaschi, L.; Mescia, F.; Turner, L.; Hanson, A.L.; Kotagiri, P.; Dunmore, B.J.; Ruffieux, H.; De Sa, A.; Huhn, O.; Morgan, M.D.; et al. Longitudinal analysis reveals that delayed bystander cd8+ t cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity 2021, 54, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, D. From anti-sars-cov-2 immune responses to COVID-19 via molecular mimicry. Antibodies 2020, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Angileri, F.; Légaré, S.; Marino Gammazza, A.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. Is molecular mimicry the culprit in the autoimmune haemolytic anaemia affecting patients with COVID-19? Br. J. Haematol. 2020, 190, e92–e93. [Google Scholar] [CrossRef]
- Marino Gammazza, A.; Légaré, S.; Lo Bosco, G.; Fucarino, A.; Angileri, F.; Conway de Macario, E.; Macario, A.J.; Cappello, F. Human molecular chaperones share with sars-cov-2 antigenic epitopes potentially capable of eliciting autoimmunity against endothelial cells: Possible role of molecular mimicry in COVID-19. Cell Stress Chaperones 2020, 25, 737–741. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. On the molecular determinants of the sars-cov-2 attack. Clin. Immunol. 2020, 215, 108426. [Google Scholar] [CrossRef]
- Angileri, F.; Legare, S.; Marino Gammazza, A.; Conway de Macario, E.; Jl Macario, A.; Cappello, F. Molecular mimicry may explain multi-organ damage in COVID-19. Autoimmun. Rev. 2020, 19, 102591. [Google Scholar] [CrossRef]
- Lucchese, G.; Flöel, A. Molecular mimicry between sars-cov-2 and respiratory pacemaker neurons. Autoimmun. Rev. 2020, 19, 102556. [Google Scholar] [CrossRef]
- Lyons-Weiler, J. Pathogenic priming likely contributes to serious and critical illness and mortality in COVID-19 via autoimmunity. J. Transl. Autoimmun. 2020, 3, 100051. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. COVID-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Kharrazian, D. Potential antigenic cross-reactivity between sars-cov-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immunol. 2020, 217, 108480. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of human monoclonal antibodies to sars-cov-2 proteins with tissue antigens: Implications for autoimmune diseases. Front. Immunol. 2021, 11, 3679. [Google Scholar] [CrossRef] [PubMed]
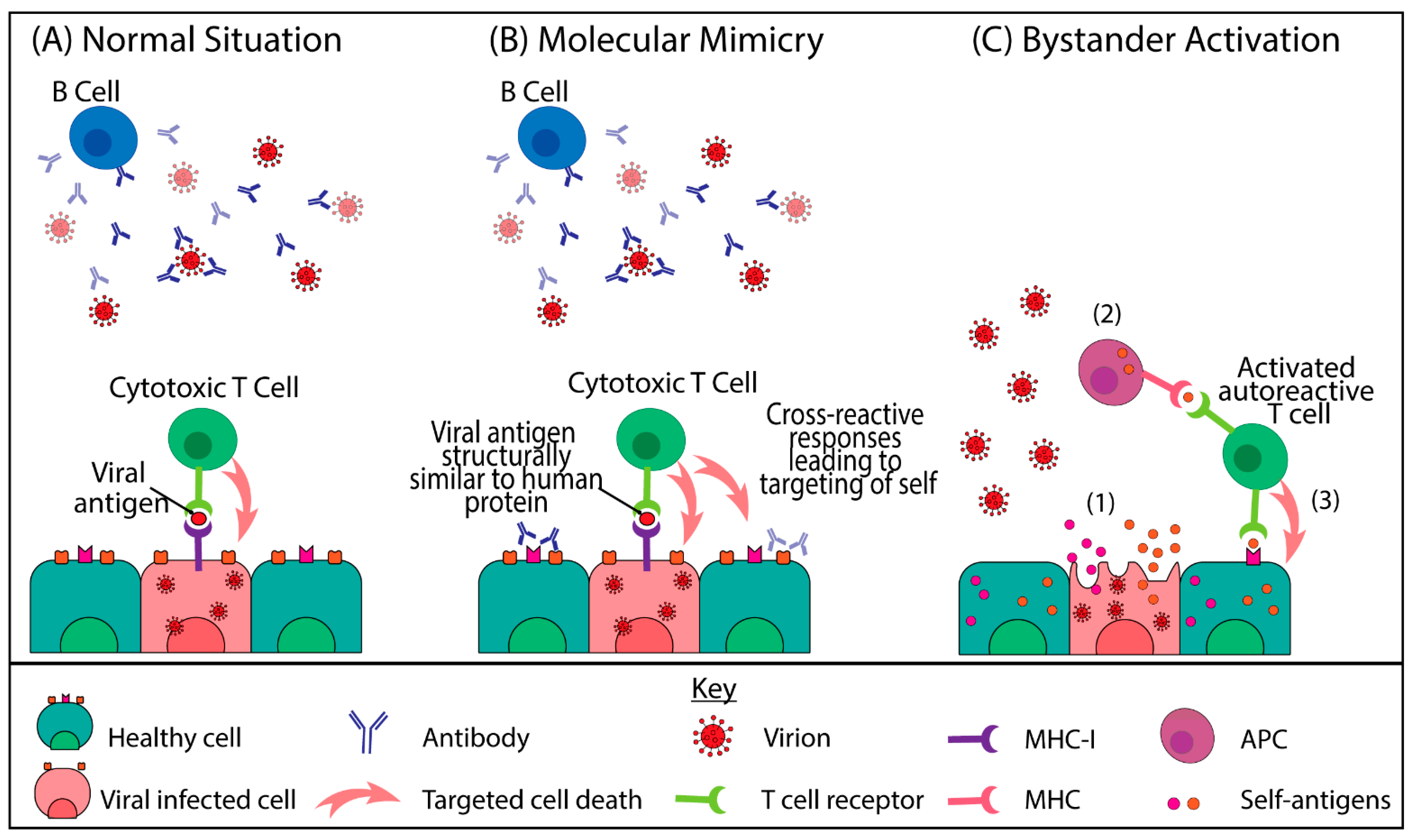

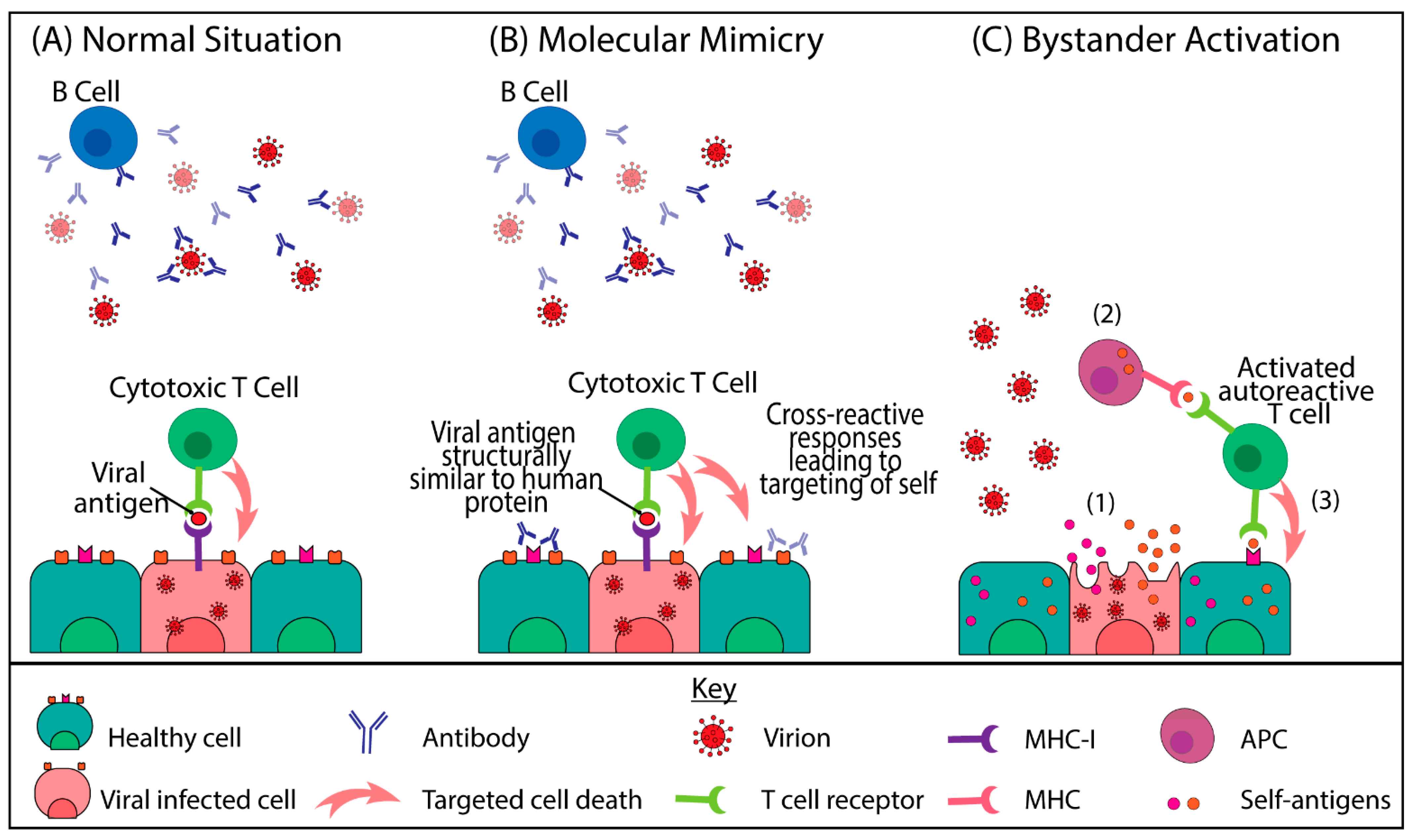{kind=link}
| Protein Family/Disease Association | Protein | Reference |
|---|---|---|
| Autoimmune haemolytic anaemia | Ankyrin 1 (ANK1) | [87] |
| Human molecular chaperones | 17 proteins listed, including: heat shock proteins, DNAJ homologs, etc. | [88] |
| Pulmonary surfactant related proteins | 23 proteins listed, e.g.,
| [89] |
| Anosmia | Odorant Receptor 7D4 (OR7D4) | [90] |
| Leukopenia | Poly (ADP-Ribose) Polymerase Family Member 9 (PARP9) | [90] |
| Vascular damage | Solute Carrier Family 12 Member 6 (SLC12A6) | [90] |
| Brainstem (pre-Bözinger complex) proteins |
| [91] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moody, R.; Wilson, K.; Flanagan, K.L.; Jaworowski, A.; Plebanski, M. Adaptive Immunity and the Risk of Autoreactivity in COVID-19. Int. J. Mol. Sci. 2021, 22, 8965. https://doi.org/10.3390/ijms22168965
Moody R, Wilson K, Flanagan KL, Jaworowski A, Plebanski M. Adaptive Immunity and the Risk of Autoreactivity in COVID-19. International Journal of Molecular Sciences. 2021; 22(16):8965. https://doi.org/10.3390/ijms22168965
Chicago/Turabian StyleMoody, Rhiane, Kirsty Wilson, Katie L. Flanagan, Anthony Jaworowski, and Magdalena Plebanski. 2021. "Adaptive Immunity and the Risk of Autoreactivity in COVID-19" International Journal of Molecular Sciences 22, no. 16: 8965. https://doi.org/10.3390/ijms22168965
APA StyleMoody, R., Wilson, K., Flanagan, K. L., Jaworowski, A., & Plebanski, M. (2021). Adaptive Immunity and the Risk of Autoreactivity in COVID-19. International Journal of Molecular Sciences, 22(16), 8965. https://doi.org/10.3390/ijms22168965