
Contemporary Approach to the Porosity of Dental Materials and Methods of Its Measurement

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Porosity of Dental Composite
3. Porosity of Dental Ceramics
4. Porosity of Titanium and Its Alloys
5. Methods Used to Assess Porosity in Materials
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Zdravkov, B.D.; Čermák, J.J.; Šefara, M.; Janků, J. Pore classification in the characterization of porous materials: A perspective. Cent. Eur. J. Chem. 2007, 5, 385–395. [Google Scholar] [CrossRef]
- Yingchao, Z.; Haihuan, G.; Dan, F.; Tengjiaozi, F.; Danfeng, C.; Zuosen, S.; Song, Z.; Zhanchen, C. New strategy for overcoming microleakage: An elastic layer for dental caries restoration. J. Mater. Chem. B 2015, 3, 4401–4405. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Sul, J.-H.; Stenzel, M.H.; Farrar, P.; Prusty, B.G. Experimental cum computational investigation on interfacial and mechanical behavior of short glass fiber reinforced dental composites. Compos. B Eng. 2020, 200, 08294. [Google Scholar] [CrossRef]
- Cho, K.; Yasir, M.; Jung, M.; Willcox, M.D.P.; Stenzel, M.H.; Rajan, G.; Farrar, P.; Prusty, B.G. Hybrid engineered dental composites by multiscale reinforcements with chitosan- integrated halloysite nanotubes and S-glass fibers. Compos. B Eng. 2020, 202, 108448. [Google Scholar] [CrossRef]
- Nilsen, B.; Jensen, E.; Ortengren, U.; Michelsen, V.B. Analysis of organic components in resin-modified pulp capping materials: Critical considerations. Eur. J. Oral Sci. 2017, 125, 112–183. [Google Scholar] [CrossRef]
- Hirata, R.; Pacheco, R.; Caceres, E.; Janal, M.N.; Romero, M.F.; Giannini, M.; Coelho, P.G.; Rueggeberg, F.A. Effect of sonic resin composite delivery on void formation assessed by micro-computed tomography. Oper. Dent. 2018, 43, 144–150. [Google Scholar] [CrossRef]
- Demirel, G.; Baltacıoglu, I.H.; Kolsuz, M.E.; Ocak, M.; Orhan, K. Microcomputed tomography evaluation of internal void formation of bulk-fill resin composites in Class II restorations. Polym. Compos. 2019, 40, 2984–2992. [Google Scholar] [CrossRef]
- Baudin, C.; Osorio, R.; Toledano, M.; de Aza, S. Work of fracture of a composite resin: Fracture-toughening mechanisms. J. Biomed. Mater. Res. A 2009, 89, 751–758. [Google Scholar] [CrossRef]
- Murray, P.E.; Garcia Godoy, C.; Garcia Godoy, F. How is the biocompatibility of dental biomaterials evaluated? Med. Oral Patol. Oral Cir. Bucal. 2007, 12, E258–E266. [Google Scholar]
- Maske, T.T.; Hollanders, A.C.C.; Kuper, N.K.; Bronkhorst, E.M.; Cenci, M.S.; Huysmans, M.C.D.N.J.M. A threshold gap size for in situ secondary caries lesion development. J. Dent. 2019, 80, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Oysaed, H.; Ruyter, I.E. Water sorption and filler characteristics of composites for use in posterior teeth. J. Dent. Res. 1987, 65, 1315–1318. [Google Scholar] [CrossRef]
- Santerre, J.P.; Shajii, L.; Leung, B.W. Relation of dental composite formulations to their degradation and the release of hydrolyzed polymeric-resin-derived products. Crit. Rev. Oral Biol. Med. 2001, 12, 136–151. [Google Scholar] [CrossRef]
- Curtis, A.R.; Shortall, A.C.; Marquis, P.M.; Palin, W.M. Water uptake and strength characteristics of a nanofilled resin-based composite. J. Dent. 2008, 36, 186–193. [Google Scholar] [CrossRef]
- Fano, V.; Ortalli, I.; Pozela, K. Porosity in composite resins. Biomaterials 1995, 16, 1291–1295. [Google Scholar] [CrossRef]
- Gotfredsen, P.; Hörsted, P.; Kragstrup, J. Porosity of restorative resins. Scand. J. Dent. Res. 1983, 91, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Van Dijken, J.W.; Ruyter, I.E.; Holland, R.I. Porosity in posteriori composite resins. Scand. J. Dent. Res. 1986, 94, 471–478. [Google Scholar] [CrossRef]
- Balthazard, R.; Jager, S.; Dahoun, A.; Gerdolle, D.; Engels-Deutsch, M.; Mortier, E. High-resolution tomography study of the porosity of three restorative resin composites. Clin. Oral. Investig. 2014, 18, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, B.W.; Mouhat, M.; Jokstad, A. Quantification of porosity in composite resins delivered by injectable syringes using X-ray microtomography. Biomater. Investig. Dent. 2020, 7, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.J.; Rosatto, C.M.P.; Carvalho, V.F.; Bicalho, A.A.; Henriques, J.C.G.; Faria-e-Silva, A.L. Radiopacity and Porosity of Bulk-fill and Conventional Composite Posterior Restorations—Digital X-ray Analysis. Oper. Dent. 2017, 42, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Gonzagaa, C.C.; Cesara, P.F.; Okadaa, C.Y.; Fredericcib, C.; Netob, F.B.; Yoshimurab, H.N. Mechanical Properties and Porosity of Dental Glass-Ceramics Hot-Pressed at Different Temperatures. Mat. Res. 2008, 11, 301–306. [Google Scholar] [CrossRef]
- Bajraktarova-Valjakova, E.; Korunoska-Stevkovska, V.; Kapusevska, B.; Gigovski, N.; Bajraktarova-Misevska, C.; Grozdanov, A. Contemporary Dental Ceramic Materials, A Review: Chemical Composition, Physical and Mechanical Properties, Indications for Use. Maced. J. Med. Sci. 2018, 6, 1742–1755. [Google Scholar] [CrossRef]
- Ho, G.W.; Matinlinna, J.P. Insights on Ceramics as Dental Materials. Part I: Ceramic Material Types in Dentistry. Silicon 2011, 3, 109–115. [Google Scholar] [CrossRef]
- El-Mowafy, O.; Brochu, J.F. Longevity and clinical performance of IPS-Empress ceramic restorations—A literature review. J. Can. Dent. Assoc. 2002, 68, 233–237. [Google Scholar] [PubMed]
- Shenoy, A.; Shenoy, N. Dental ceramics: An update. J. Conserv. Dent. 2010, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Okoński, P.; Lasek, K.; Mierzwińska-Nastalska, E. Kliniczne zastosowanie wybranych materiałów ceramicznych. Protetyka Stomatologiczna 2012, 62, 181–189. [Google Scholar] [CrossRef]
- Wydra, M.; Grelowska, I. Materiały ceramiczne w stomatologii. Szkło i Ceramika 2017, 6, 11–15. [Google Scholar]
- Cattell, M.J.; Clarke, R.L.; Lynch, E.J. The biaxial flexural strength and reliability of four dental ceramics—Part II. J. Dent. 1997, 25, 409–414. [Google Scholar] [CrossRef]
- Gorman, C.M.; McDevitt, W.E.; Hill, R.G. Comparison of two heat-pressed all-ceramic dental materials. Dent. Mater. 2000, 16, 389–395. [Google Scholar] [CrossRef]
- Holand, W.; Beall, G. Glass-Ceramic Technology; The American Ceramic Society: Westerville, OH, USA, 2002; p. 385. [Google Scholar]
- Yoshimura, H.N.; Cruz, A.C.D.; Zhou, Y.; Tanaka, H. Sintering of 6H(α)-SiC and 3C(β)-SiC powders with B4C and C additives. J. Mater. Sci. 2002, 37, 1541–1546. [Google Scholar] [CrossRef]
- Kassis, N.; Frischat, G.H. Vapor pressure of simple silicate glass melts. J. Am. Ceram. Soc. 1981, 64, C28–C29. [Google Scholar] [CrossRef]
- De Carvalho, R.F.; Martins, M.E.; de Queiroz, J.R.; Leite, F.P.; Ozcan, M. Influence of silane heat treatment on Bond strength of resin cement to a feldspathic ceramic. Dent. Mater. J. 2011, 30, 392–397. [Google Scholar] [CrossRef]
- Vargas, M.A.; Bergeron, C.; Diaz-Arnold, A. Cementing all-ceramic restorations: Recommendations for success. J. Am. Dent. Assoc. 2011, 142, 20S–24S. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.C.; Darvell, B.W. Sintering of dental porcelain: Effect of time and temperature on appearance and porosity. Dent. Mater. 2002, 18, 163–173. [Google Scholar] [CrossRef]
- Falkensammer, F.; Jonke, E.; Bertl, M.; Freudenthaler, J.; Bantleon, H.P. Rebonding performance of different ceramic brackets conditioned with a new silane coupling agent. Eur. J. Orthod. 2013, 35, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Osorio, E.; Toledano, M.; da Silveira, B.L.; Osorio, R. Effect of different surface treatments on In-Ceram Alumina roughness. An AFM study. J. Dent. 2010, 38, 118–122. [Google Scholar] [CrossRef]
- Valian, A.; Moravej-Salehi, E. Surface treatment of feldspathic porcelain: Scanning electron microscopy analysis. J. Adv. Prosthodont. 2014, 6, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Borges, G.A.; Sophr, A.M.; de Goes, M.F.; Sobrinho, L.C.; Chan, D.C. Effect of etching and airborne particle abrasion on the microstructure of different dental ceramics. J. Prosthet. Dent. 2003, 89, 479–488. [Google Scholar] [CrossRef]
- Bottino, M.C.; Ozcan, M.; Coelho, P.G.; Valandro, L.F.; Bressiani, J.C.; Bressiani, A.H. Micro-morphological changes prior to adhesive bonding: High-alumina and glassy-matrix ceramics. Braz. Oral. Res. 2008, 22, 158–163. [Google Scholar] [CrossRef]
- Kukiattrakoon, B.; Thammasitboon, K. Optimal acidulated phosphate fluoride gel etching time for surface treatment of feldspathic porcelain: On shear bond strength to resin composite. Eur. J. Dent. 2012, 6, 63–69. [Google Scholar] [CrossRef]
- Amin Salehi, E.; Heshmat, H.; Moravej-Salehi, E.; Kharazifard, M. In vitro evaluation of the effect of different sandblasting times on the bond strength of feldspathic porcelain to composite resin. J. Islam. Dent. Assoc. Iran 2013, 25, 22–30. [Google Scholar]
- Kern, M.; Barloi, A.; Yang, B. Surface conditioning influences zirconia ceramic bonding. J. Dent. Res. 2009, 88, 817–822. [Google Scholar] [CrossRef]
- Saraç, Y.S.; Elekdag-Turk, S.; Saraç, D.; Turk, T. Surface conditioning methods and polishing techniques effect on surface roughness of a feldspar ceramic. Angle Orthod. 2007, 77, 723–728. [Google Scholar] [CrossRef]
- Baratto, S.S.; Spina, D.R.; Gonzaga, C.C.; Cunha, L.F.; Furuse, A.Y.; Baratto Filho, F.; Correr, G.M. Silanated Surface Treatment: Effects on the Bond Strength to Lithium Disilicate Glass-Ceramic. Braz. Dent. J. 2015, 26, 474–477. [Google Scholar] [CrossRef]
- Sannino, G.; Germano, F.; Arcuri, L.; Bigelli, E.; Arcuri, C.; Barlattani, A. CEREC CAD/CAM chairside system. Oral Implantol. 2015, 7, 57–70. [Google Scholar]
- Sidambe, A.T. Biocompatibility of Advanced Manufactured Titanium Implants-A Review. Materials 2014, 7, 8168–8188. [Google Scholar] [CrossRef] [PubMed]
- Sidambe, A.T.; Figueroa, I.A.; Hamilton, H.G.C.; Todd, I. Metal injection moulding of CP-Ti components for biomedical applications. J. Mater. Process. Technol. 2012, 212, 1591–1597. [Google Scholar] [CrossRef]
- Wally, Z.J.; van Grunsven, W.; Claeyssens, F.; Goodall, R.; Reilly, G.C. Porous Titanium for Dental Implant Applications. Metals 2015, 5, 1902–1920. [Google Scholar] [CrossRef]
- Mangano, F.; Chambrone, L.; van Noort, R.; Miller, C.; Hatton, P.; Mangano, C. Direct metal laser sintering titanium dental implants: A review of the current literature. Int. J. Biomater. 2014, 2014, 461534. [Google Scholar] [CrossRef] [PubMed]
- Bidan, C.M.; Kommareddy, K.P.; Rumpler, M.; Kollmannsberger, P.; Fratzl, P.; Dunlop, J.W. Geometry as a factor for tissue growth: Towards shape optimization of tissue engineering scaffolds. Adv. Healthc. Mater. 2013, 2, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Tison, C.K.; Chatterjee, K.; Pine, P.S.; McDaniel, J.H.; Salit, M.L.; Young, M.F.; Simon, C.G., Jr. The determination of stem cell fate by 3D scaffold structures through the control of cell shape. Biomaterials 2011, 32, 9188–9196. [Google Scholar] [CrossRef]
- Cheng, A.; Humayun, A.; Cohen, D.J.; Boyan, B.D.; Schwartz, Z. Additively manufactured 3D porous Ti–6Al–4V constructs mimic trabecular bone structure and regulate osteoblast proliferation, differentiation and local factor production in a porosity and surface roughness dependent manner. Biofabrication 2014, 6, 045007. [Google Scholar] [CrossRef]
- Mour, M.; Das, D.; Winkler, T.; Hoenig, E.; Mielke, G.; Morlock, M.M.; Schilling, A.F. Advances in Porous Biomaterials for Dental and Orthopaedic Applications. Materials 2010, 3, 2947–2974. [Google Scholar] [CrossRef]
- De Wild, M.; Schumacher, R.; Mayer, K.; Schkommodau, E.; Thoma, D.; Bredell, M.; Gujer, A.K.; Grätz, K.W.; Weber, F.E. Bone regeneration by the osteoconductivity of porous titanium implants manufactured by selective laser melting: A histological and micro computed tomography study in the rabbit. Tissue Eng. Part A 2013, 19, 2645–2654. [Google Scholar] [CrossRef]
- Rodriguez-Contrera, A.; Punset, M.; Calero, J.A.; Gil, F.J.; Ruperez, E.; Manero, J.M. Powder metallurgy with space holder for porous titanium implants: A review. J. Mater. Sci. Technol. 2021, 76, 129–149. [Google Scholar] [CrossRef]
- Bai, F.; Wang, Z.; Lu, J.; Liu, J.; Chen, G.; Lv, R.; Wang, J.; Lin, K.; Zhang, J.; Huang, X. The correlation between the internal structure and vascularization of controllable porous bioceramic materials in vivo: A quantitative study. Tissue Eng. Part A 2010, 16, 3791–3803. [Google Scholar] [CrossRef]
- Shibli, J.A.; Mangano, C.; D’avila, S.; Piattelli, A.; Pecora, G.E.; Mangano, F.; Onuma, T.; Cardoso, L.A.; Ferrari, D.S.; Aguiar, K.C.; et al. Influence of direct laser fabrication implant topography on type IV bone: A histomorphometric study in humans. J. Biomed. Mater. Res. A 2010, 93, 607–614. [Google Scholar] [CrossRef]
- Wen, C.E.; Yamada, Y.; Nouri, A.; Hodgson, P.D. Porous Titanium with Porosity Gradients for Biomedical Applications. In Materials Science Forum; Trans Tech Publications Ltd.: Zurich, Switzerland, 2007; Volume 539, pp. 720–725. [Google Scholar]
- Ghouse, S.; Reznikov, N.; Boughton, O.R.; Babu, S.; Ng, K.C.G.; Blunn, G.; Cobb, J.P.; Stevens, M.M.; Jeffers, J.R.T. The design and in vivo testing of a locally stiffness-matched porous scaffold. Appl. Mater. Today 2019, 15, 377–388. [Google Scholar] [CrossRef]
- Li, L.; Shi, J.; Zhang, K.; Yang, L.; Yu, F.; Zhu, L.; Liang, H.; Wang, X.; Jiang, Q. Early osteointegration evaluation of porous Ti6Al4V scaffolds designed based on triply periodic minimal surface models. J. Orthop. Transl. 2019, 19, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Balla, V.K.; Bodhak, S.; Bose, S.; Bandyopadhyay, A. Porous tantalum structures for bone implants: Fabrication, mechanical and in vitro biological properties. Acta Biomater. 2010, 6, 3349–3359. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Espana, F.; Balla, V.K.; Bose, S.; Ohgami, Y.; Davies, N.M. Influence of porosity on mechanical properties and in vivo response of Ti6Al4V implants. Acta Biomater. 2010, 6, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Heary, R.F.; Parvathreddy, N.; Sampath, S.; Agarwal, N. Elastic modulus in the selection of interbody implants. J. Spine Surg. 2017, 3, 163–167. [Google Scholar] [CrossRef]
- Laptev, A.; Vyal, O.; Bram, M.; Buchkremer, H.P.; Stöver, D. Green strength of powder compacts provided for production of highly porous titanium parts. Powder Metall. 2005, 48, 358–364. [Google Scholar] [CrossRef]
- Zhang, K.; Fan, Y.; Dunne, N.; Li, X. Effect of microporosity on scaffolds for bone tissue engineering. Regen. Biomater. 2018, 5, 115–124. [Google Scholar] [CrossRef]
- Junker, R.; Dimakis, A.; Thoneick, M.; Jansen, J.A. Effects of implant surface coatings and composition on bone integration: A systematic review. Clin. Oral Implant. Res. 2009, 20, 185–206. [Google Scholar] [CrossRef]
- Gaviria, L.; Salcido, J.P.; Guda, T.; Ong, J.L. Current trends in dental implants. J. Korean Assoc. Oral Maxillofac. Surg. 2014, 40, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Le Guéhennec, L.; Soueidan, A.; Layrolle, P.; Amouriq, Y. Surface treatments of titanium dental implants for rapid osseointegration. Dent. Mater. 2007, 23, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Çelen, S.; Özden, H. Laser-induced novel patterns: As smart strain actuators for new-age dental implant surfaces. Appl. Surf. Sci. 2012, 263, 579–585. [Google Scholar] [CrossRef]
- Hajnos, M.; Lipiec, J.; Świeboda, R.; Sokołowska, Z.; Witkowska-Walczak, B. Complete characterization of pore size distribution of tilled and orchard soil using water retention curve, mercury porosimetry, nitrogen adsorption, and water desorption methods. Geoderma 2006, 135, 307–314. [Google Scholar] [CrossRef]
- Volzone, C.; Zagorodny, N. Mercury intrusion porosimetry (MIP) study of archaeological pottery from Hualfin Valley, Catamarca, Argentina. Appl. Clay Sci. 2014, 91–92, 12–15. [Google Scholar] [CrossRef]
- Li, B.; Mao, J.; Nawa, T.; Han, T. Mesoscopic damage model of concrete subjected to freeze-thaw cycles using mercury intrusion porosimetry and differential scanning calorimetry (MIP-DSC). Constr. Build. Mater. 2017, 147, 79–90. [Google Scholar] [CrossRef]
- Moro, F.; Böhni, H. Ink-bottle effect in mercury intrusion porosimetry of cement-based materials. J. Colloid Interface Sci. 2002, 246, 135–149. [Google Scholar] [CrossRef]
- Anovitz, L.M.; Cole, D.R. Characterization and analysis of porosity and pore structures. Rev. Miner. Geochem. 2015, 80, 61–164. [Google Scholar] [CrossRef]
- Vázquez, E.V.; Ferreiro, J.P.; Miranda, J.G.V.; González, A.P. Multifractal Analysis of Pore Size Distributions as Affected by Simulated Rainfall. Vadose Zone J. 2008, 7, 500–511. [Google Scholar] [CrossRef]
- Dal Ferro, N.; Delmas, P.; Duwig, C.; Simonetti, G.; Morari, F. Coupling X-ray microtomography and mercury intrusion porosimetry to quantify aggregate structures of a cambisol under different fertilisation treatments. Soil Tillage Res. 2012, 119, 13–21. [Google Scholar] [CrossRef]
- Boulin, P.F.; Angulo-Jaramillo, R.; Daian, J.F.; Talandier, J.; Berne, P. Pore gas connectivity analysis in Callovo-Oxfordian argillite. Appl. Clay Sci. 2008, 42, 276–283. [Google Scholar] [CrossRef]
- Tibbetts, C.M.; Tao, C.; Paris, J.M.; Ferraro, C.C. Mercury intrusion porosimetry parameters for use in concrete penetrability qualification using the Katz-Thompson relationship. Constr. Build. Mater. 2020, 263, 119834. [Google Scholar] [CrossRef]
- Kumar, R.; Bhattacharjee, B. Study on some factors affecting the results in the use of MIP method in concrete research. Cem. Concr. Res. 2003, 33, 417–424. [Google Scholar] [CrossRef]
- Jozefaciuk, G.; Czachor, H.; Lamorski, K.; Hajnos, M.; Swieboda, R.; Franus, W. Effect of humic acids, sesquioxides and silica on the pore system of silt aggregates measured by water vapour desorption, mercury intrusion and microtomography. Eur. J. Soil Sci. 2015, 66, 992–1001. [Google Scholar] [CrossRef]
- Otalvaro, I.F.; Neto, M.P.C.; Delage, P.; Caicedo, B. Relationship between soil structure and water retention properties in a residual compacted soil. Eng. Geol. 2016, 205, 73–80. [Google Scholar] [CrossRef]
- Rabot, E.; Wiesmeier, M.; Schlüter, S.; Vogel, H.J. Soil structure as an indicator of soil functions: A review. Geoderma 2018, 314, 122–137. [Google Scholar] [CrossRef]
- Korat, L.; Ducman, V.; Legat, A.; Mirtič, B. Characterisation of the pore-forming process in lightweight aggregate based on silica sludge by means of X-ray micro-tomography (micro-CT) and mercury intrusion porosimetry (MIP). Ceram. Int. 2013, 39, 6997–7005. [Google Scholar] [CrossRef]
- Washburn, E.W. The dynamics of capillary low. Phys. Rev. 1921, 17, 273–283. [Google Scholar] [CrossRef]
- Zhou, J.; Ye, G.; van Breugel, K. Characterization of pore structure in cement-based materials using pressurization-depressurization cycling mercury intrusion porosimetry (PDC-MIP). Cem. Concr. Res. 2010, 40, 1120–1128. [Google Scholar] [CrossRef]
- Diamond, S. Mercury porosimetry. An inappropriate method for the measurement of pore size distributions in cement-based materials. Cem. Concr. Res. 2000, 30, 1517–1525. [Google Scholar] [CrossRef]
- Salejova, G.; Grof, Z.; Solcova, O.; Schneider, P.; Kosek, J. Strategy for predicting effective transport properties of complex porous structures. Comput. Chem. Eng. 2011, 35, 200–211. [Google Scholar] [CrossRef]
- Giesche, H. Mercury porosimetry: A general (practical) overview. Part. Part. Syst. Charact. 2006, 23, 9–19. [Google Scholar] [CrossRef]
- Thompson, M.L.; McBride, J.F.; Horton, R. Effects of Drying Treatments on Porosity of Soil Materials. Soil Sci. Soc. Am. J. 1985, 49, 1360–1364. [Google Scholar] [CrossRef]
- Galle, C. Effect of drying on cement-based materials pore structure as identified by mercury intrusion porosimetry a comparative study between oven-, vacuum-, and freeze-drying. Cem. Concr. Res. 2001, 31, 1467–1477. [Google Scholar] [CrossRef]
- Yu, S.; Bo, J.; Pei, S.; Jiahao, W. Matrix compression and multifractal characterization for tectonically deformed coals by Hg porosimetry. Fuel 2018, 211, 661–675. [Google Scholar] [CrossRef]
- Cai, Y.; Li, Q.; Liu, D.; Zhou, Y.; Lv, D. Insights into matrix compressibility of coals by mercury intrusion porosimetry and N2 adsorption. Int. J. Coal. Geol. 2019, 200, 199–212. [Google Scholar] [CrossRef]
- Yang, Q.; Xue, J.; Li, W.; Du, X.; Ma, Q.; Zhan, K.; Chen, Z. Comprehensive evaluation and interpretation of mercury intrusion porosimetry data of coals based on fractal theory, Tait equation and matrix compressibility. Fuel 2021, 298, 120823. [Google Scholar] [CrossRef]
- Bogas, J.A.; Mauricio, A.; Pereira, M.F.C. Microstructural analysis of iberian expanded clay aggregates. Microsc. Microanal. 2012, 18, 1190–1208. [Google Scholar] [CrossRef]
- Li, X.; Kang, Y.; Haghighi, M. Investigation of pore size distributions of coals with different structures by nuclear magnetic resonance (NMR) and mercury intrusion porosimetry (MIP). Meas. J. Int. Meas. Confed. 2018, 116, 122–128. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004. [Google Scholar] [CrossRef]
- Condon, B.J. Surface Area and Porosity Determination by Physisorption: Measurements and Theory; Elsevier Science: Amsterdam, The Netherlands, 2006. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.T.S. Raporting Physisorption data for Gas/Solid Systems. In Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]
- Macht, F.; Eusterhues, K.; Pronk, G.J.; Totsche, K.U. Specific surface area of clay minerals: Comparison between atomic force microscopy measurements and bulk-gas (N2) and -liquid (EGME) adsorption methods. Appl. Clay Sci. 2011, 53, 20–26. [Google Scholar] [CrossRef]
- Séquaris, J.M.; Guisado, G.; Magarinos, M.; Moreno, C.; Burauel, P.; Narres, H.D.; Vereecken, H. Organic-carbon fractions in an agricultural topsoil assessed by the determination of the soil mineral surface area. J. Plant Nutr. Soil Sci. 2010, 173, 699–705. [Google Scholar] [CrossRef]
- Sdanghi, G.; Canevesi, R.L.S.; Celzard, A.; Thommes, M.; Fierro, V. Characterization of Carbon Materials for Hydrogen Storage and Compression. J. Carbon Res. 2020, 6, 46. [Google Scholar] [CrossRef]
- Wang, X. Lacustrine Shale Gas: Case Study from the Ordos Basin; Gulf Professional Publishing: San Diego, CA, USA, 2017. [Google Scholar] [CrossRef]
- Lu, S.; Yu, X.; Zong, Y. Nano-microscale porosity and pore size distribution in aggregates of paddy soil as affected by long-term mineral and organic fertilization under rice-wheat cropping system. Soil Tillage Res. 2019, 186, 191–199. [Google Scholar] [CrossRef]
- Jagiello, J.; Kenvin, J.; Celzard, A.; Fierro, V. Enhanced resolution of ultra micropore size determination of biochars and activated carbons by dual gas analysis using N2 and CO2 with 2D-NLDFT adsorption models. Carbon 2019, 144, 206–215. [Google Scholar] [CrossRef]
- Yi, M.; Cheng, Y.; Wang, Z.; Wang, C.; Hu, B.; He, X. Effect of particle size and adsorption equilibrium time on pore structure characterization in low pressure N2 adsorption of coal: An experimental study. Adv. Powder Technol. 2020, 31, 4275–4281. [Google Scholar] [CrossRef]
- Landers, J.; Gor, G.Y.; Neimark, A.V. Density functional theory methods for characterization of porous materials. Colloids Surfaces A Physicochem. Eng. Asp. 2013, 437, 3–32. [Google Scholar] [CrossRef]
- Benavente, D.; Such-Basañez, I.; Fernandez-Cortes, A.; Pla, C.; Cazorla-Amoros, D.; Cañaveras, J.C.; Sanchez-Moral, S. Comparative analysis of water condensate porosity using mercury intrusion porosimetry and nitrogen and water adsorption techniques in porous building stones. Constr. Build. Mater. 2021, 288, 123131. [Google Scholar] [CrossRef]
- Echeverría, J.C.; Morera, M.T.; Mazkiarán, C.; Garrido, J.J. Characterization of the porous structure of soils: Adsorption of nitrogen (77 K) and carbon dioxide (273 L), and mercury porosimetry. Eur. J. Soil Sci. 1999, 50, 497–503. [Google Scholar] [CrossRef]
- Heister, K. The measurement of the specific surface area of soils by gas and polar liquid adsorption methods-limitations and potentials. Geoderma 2014, 216, 75–87. [Google Scholar] [CrossRef]
- Krause, K.M.; Thommes, M.; Brett, M.J. Pore analysis of obliquely deposited nanostructures by krypton gas adsorption at 87 K. Microporous Mesoporous Mater. 2011, 143, 166–173. [Google Scholar] [CrossRef]
- Kuwabara, H.; Suzuki, T.; Kaneko, K. Ultramicropores in Microporous Carbon Fibres evidenced by Helium Adsorption at 4.2 K. J. Chem. Soc. Faraday Trans. 1991, 87, 1915–1916. [Google Scholar] [CrossRef]
- Barna, G.; Makó, A.; Takács, T.; Skic, K.; Füzy, A.; Horel, Á. Biochar alters soil physical characteristics, arbuscular mycorrhizal fungi colonization, and glomalin production. Agronomy 2020, 10, 1933. [Google Scholar] [CrossRef]
- Yukselen-Aksoy, Y.; Kaya, A. Method dependency of relationships between specific surface area and soil physicochemical properties. Appl. Clay Sci. 2010, 50, 182–190. [Google Scholar] [CrossRef]
- Skic, K.; Boguta, P.; Sokołowska, Z. Analysis of the sorption properties of different soils using water vapour adsorption and potentiometric titration methods. Int. Agrophys. 2016, 30, 369–374. [Google Scholar] [CrossRef]
- Jozefaciuk, G.; Toth, T.; Szendrei, G. Surface and micropore properties of saline soil profiles. Geoderma 2006, 135, 1–15. [Google Scholar] [CrossRef]
- Zaffar, M.; Lu, S.G. Pore size distribution of clayey soils and its correlation with soil organic matter. Pedosphere 2015, 25, 240–249. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquérol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Sigmund, G.; Hüffer, T.; Hofmann, T.; Kah, M. Biochar total surface area and total pore volume determined by N2 and CO2 physisorption are strongly influenced by degassing temperature. Sci. Total Environ. 2017, 580, 770–775. [Google Scholar] [CrossRef]
- Mergia, K.; Stefanopoulos, K.L.; Ordás, N.; García-Rosales, C. A comparative study of the porosity of doped graphites by small angle neutron scattering, nitrogen adsorption and helium pycnometry. Microporous Mesoporous Mater. 2010, 134, 141–149. [Google Scholar] [CrossRef]
- Huxham, I.M.; Rowatt, B.; Sherrington, D.C.; Tetley, L. Molecular architectural changes in hydrated macroporous styrene-divinylbenzene resin sorbents revealed by transmission electron microscopy using image analysis. Polymer 1992, 33, 2768–2777. [Google Scholar] [CrossRef]
- Takase, A.; McNulty, T.; Fitzgibbons, T. Foam Porosity Calculation by X-Ray Computed Tomography and Errors Caused by Insufficient Resolution. Microsc. Microanal. 2018, 24, 546–547. [Google Scholar] [CrossRef][Green Version]
- Chen, F.; Lu, S.; Ding, X.; Zhao, H.; Ju, Y. Total porosity measured for shale gas reservoir samples: A case from the lower Silurian Longmaxi formation in southeast Chongqing, China. Minerals 2019, 9, 5. [Google Scholar] [CrossRef]
- Bahadur, J.; Medina, C.R.; He, L.; Melnichenko, Y.B.; Rupp, J.A.; Blach, T.P.; Mildner, D.F.R. Determination of closed porosity in rocks by small-angle neutron scattering. J. Appl. Crystallogr. 2016, 49, 2021–2030. [Google Scholar] [CrossRef]
- Radlinski, A.P.; Mastalerz, M.; Hinde, A.L.; Hainbuchner, M.; Rauch, H.; Baron, M.; Lin, J.S.; Fan, L.; Thiyagarajan, P. Application of SAXS and SANS in evaluation of porosity, pore size distribution and surface area of coal. Int. J. Coal Geol. 2004, 59, 245–271. [Google Scholar] [CrossRef]
- He, L.; Melnichenko, Y.B.; Mastalerz, M.; Sakurovs, R.; Radlinski, A.P.; Blach, T. Pore accessibility by methane and carbon dioxide in coal as determined by neutron scattering. Energy Fuels 2012, 26, 1975–1983. [Google Scholar] [CrossRef]
- Calo, J.M.; Hall, P.J.; Houtmann, S.; Lozano Castello, D.; Winans, R.E.; Seifert, S. “Real time” determination of porosity development in carbons: A combined SAXS/TGA approach. Stud. Surf. Sci. Catal. 2002, 144, 59–66. [Google Scholar] [CrossRef]
- Conceição, A.L.C.; Perlich, J.; Haas, S.; Funari, S.S. SAXS-CT: A nanostructure resolving microscopy for macroscopic biologic specimens. Biomed. Phys. Eng. Express 2020, 6, 035012. [Google Scholar] [CrossRef]
- Leppänen, K.; Bjurhager, I.; Peura, M.; Kallonen, A.; Suuronen, J.P.; Penttilä, P.; Love, J.; Fagerstedt, K.; Serimaa, R. X-ray scattering and microtomography study on the structural changes of never-dried silver birch, European aspen and hybrid aspen during drying. Holzforschung 2011, 65, 865–873. [Google Scholar] [CrossRef]
- Kruth, J.P.; Bartscher, M.; Carmignato, S.; Schmitt, R.; De Chiffre, L.; Weckenmann, A. Computed tomography for dimensional metrology. CIRP Ann. Manuf. Technol. 2011, 60, 821–842. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, L.; Fuh, J.Y.H.; Lee, H.P. Effect of porosity on mechanical properties of 3D printed polymers: Experiments and micromechanical modeling based on X-ray computed tomography analysis. Polymers 2019, 11, 1154. [Google Scholar] [CrossRef]
- Ziółkowski, G.; Szymczyk, P.; Pawlak, A.; Kurzynowski, T.; Dybała, B.; Chlebus, E. Porosity Detection by Computed Tomography. Pomiary Autom. Robot. 2017, 21, 27–34. [Google Scholar] [CrossRef]
- Sinico, M.; Jadhav, S.D.; Witvrouw, A.; Vanmeensel, K.; Dewulf, W. A Micro-Computed Tomography Comparison of the Porosity in Additively Fabricated CuCr1 Alloy Parts Using Virgin and Surface-Modified Powders. Materials 2021, 14, 1995. [Google Scholar] [CrossRef]
- Akin, S.; Demiral, M.; Okandan, E. A novel method of porosity measurement utilizing computerized tomography. In Situ 1996, 20, 347–365. [Google Scholar]
- Lu, S.; Landis, E.N.; Keane, D.T. X-ray microtomographic studies of pore structure and permeability in Portland cement concrete. Mater. Struct. Constr. 2006, 39, 611–620. [Google Scholar] [CrossRef]
- Nehler, M.; Stoeckhert, F.; Oelker, A.; Renner, J.; Saenger, E. Evaluating porosity estimates for sandstones based on X-ray micro-tomographic images. Solid Earth Discuss. 2019, 1–45. [Google Scholar] [CrossRef]
- Hermanek, P.; Carmignato, S. Porosity measurements by X-ray computed tomography: Accuracy evaluation using a calibrated object. Precis. Eng. 2017, 49, 377–387. [Google Scholar] [CrossRef]
- Widiarini, P.; Suparta, G.B. Testing on porosity of composite material composed by ultrafine amorphous silica (UFAS) from rice husk using X-ray micro-computed tomography. J. Phys. Conf. Ser. 2018, 1040, 012049. [Google Scholar] [CrossRef]
- Prokop, J.; Švéda, L.; Jančárek, A.; Pína, L. Porosity measurement method by X-ray computed tomography. Key Eng. Mater. 2009, 409, 402–405. [Google Scholar] [CrossRef]
- Wilczek, A.; Długosz, P.; Hebda, M. Porosity Characterization of Aluminium Castings by Using Particular Non-destructive Techniques. J. Nondestruct. Eval. 2015, 34, 26. [Google Scholar] [CrossRef]
- Boone, M.N.; Vlassenbroeck, J.; Peetermans, S.; Van Loo, D.; Dierick, M.; Van Hoorebeke, L. Secondary radiation in transmission-type X-ray tubes: Simulation, practical issues and solution in the context of X-ray microtomography. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2012, 661, 7–12. [Google Scholar] [CrossRef]
- Van de Casteele, E.; Van Dyck, D.; Sijbers, J.; Raman, E. An energy-based beam hardening model in tomography. Phys. Med. Biol. 2002, 47, 4181–4190. [Google Scholar] [CrossRef]
- Soulaine, C.; Gjetvaj, F.; Garing, C.; Roman, S.; Russian, A.; Gouze, P.; Tchelepi, H.A. The Impact of Sub-Resolution Porosity of X-ray Microtomography Images on the Permeability. Transp. Porous Media 2016, 113, 227–243. [Google Scholar] [CrossRef]
- Shoukroun, D.; Massimi, L.; Endrizzi, M.; Bate, D.; Fromme, P.; Olivo, A. Composite porosity characterization using X-ray edge illumination phase contrast and ultrasonic techniques. In Proceedings of the SPIE 11593 Health Monitoring of Structural and Biological Systems XV, Online Conference, 22 March 2021. [Google Scholar]
- Espinal, L. Porosity and Its Measurement. In Characterization of Materials; Kaufmann, E.N., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2012; pp. 1–9. [Google Scholar]
- Stefanidou, M. Methods for porosity measurement in lime-based mortars. Constr. Build. Mater. 2010, 24, 2572–2578. [Google Scholar] [CrossRef]
- Ghasemi-Mobarakeh, L.; Semnani, D.; Morsked, M. Crystallization behavior of poly(ε-caprolactone)/layered double hydroxide nanocomposites. J. Appl. Polym. Sci. 2010, 106, 2536–2542. [Google Scholar] [CrossRef]
- Erdman, N.; Drenzek, N. Integrated preparation and imaging techniques for the microstructural and geochemical characterization of shale from scanning electron microscopy. Am. Assoc. Petrol. Geol. Memoir. 2013, 102, 7–14. [Google Scholar]
- Reese, J.P. New Nanotechnology Research; Nova Science Publishers Inc.: New York, NY, USA, 2006; pp. 1–246. [Google Scholar]
- Al-Abboodi, A.; Zhang, S.; Al-Saady, M.; Ong, J.W.; Chan, P.P.Y.; Fu, J. Printing in situ tissue sealant with visible-light-crosslinked porous hydrogel. Biomed. Mater. 2019, 14, 045010. [Google Scholar] [CrossRef]
- Alvarez, J.; Saudino, G.; Musteata, V.; Madhavan, P.; Genovese, A.; Behzad, A.R.; Sougrat, R.; Boi, C.; Peinemann, K.V.; Nunes, S.P. 3D Analysis of Ordered Porous Polymeric Particles using Complementary Electron Microscopy Methods. Sci. Rep. 2019, 9, 13987. [Google Scholar] [CrossRef]
- Arzate-Vázquez, I.; Méndez-Méndez, J.V.; Flores-Johnson, E.A.; Nicolás-Bermúdez, J.; Chanona-Pérez, J.J.; Santiago-Cortés, E. Study of the porosity of calcified chicken eggshell using atomic force microscopy and image processing. Micron 2019, 118, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, B.; Wang, W.; Wang, Z.; Zhang, L. The use of AFM in quantitative analysis of pore characteristics in coal and coal-bearing shale. Mar. Pet. Geol. 2019, 105, 331–337. [Google Scholar] [CrossRef]
- Yan, W.; Sun, J.; Sun, Y.; Golsanami, N. A robust NMR method to measure porosity of low porosity rocks. Microporous Mesoporous Mater. 2018, 269, 113–117. [Google Scholar] [CrossRef]
- Aghda, S.M.F.; Taslimi, M.; Fahimifar, A. Adjusting porosity and permeability estimation by nuclear magnetic resonance: A case study from a carbonate reservoir of south of Iran. J. Pet. Explor. Prod. Technol. 2018, 8, 1113–1127. [Google Scholar] [CrossRef]
- De Terris, T.; Andreau, O.; Peyre, P.; Adamski, F.; Koutiri, I.; Gorny, C.; Dupuy, C. Optimization and comparison of porosity rate measurement methods of Selective Laser Melted metallic parts. Addit. Manuf. 2019, 28, 802–813. [Google Scholar] [CrossRef]
- Wits, W.W.; Carmignato, S.; Zanini, F.; Vaneker, T.H.J. Porosity testing methods for the quality assessment of selective laser melted parts. CIRP Ann. Manuf. Technol. 2016, 65, 201–204. [Google Scholar] [CrossRef]
- Wang, P.; Tan, X.; He, C.; Nai, M.L.S.; Huang, R.; Tor, S.B.; Wei, J. Scanning optical microscopy for porosity quantification of additively manufactured components. Addit. Manuf. 2018, 21, 350–358. [Google Scholar] [CrossRef]
- Hossen, M.R.; Talbot, M.W.; Kennard, R.; Bousfield, D.W.; Mason, M.D. A comparative study of methods for porosity determination of cellulose based porous materials. Cellulose 2020, 27, 6849–6860. [Google Scholar] [CrossRef]



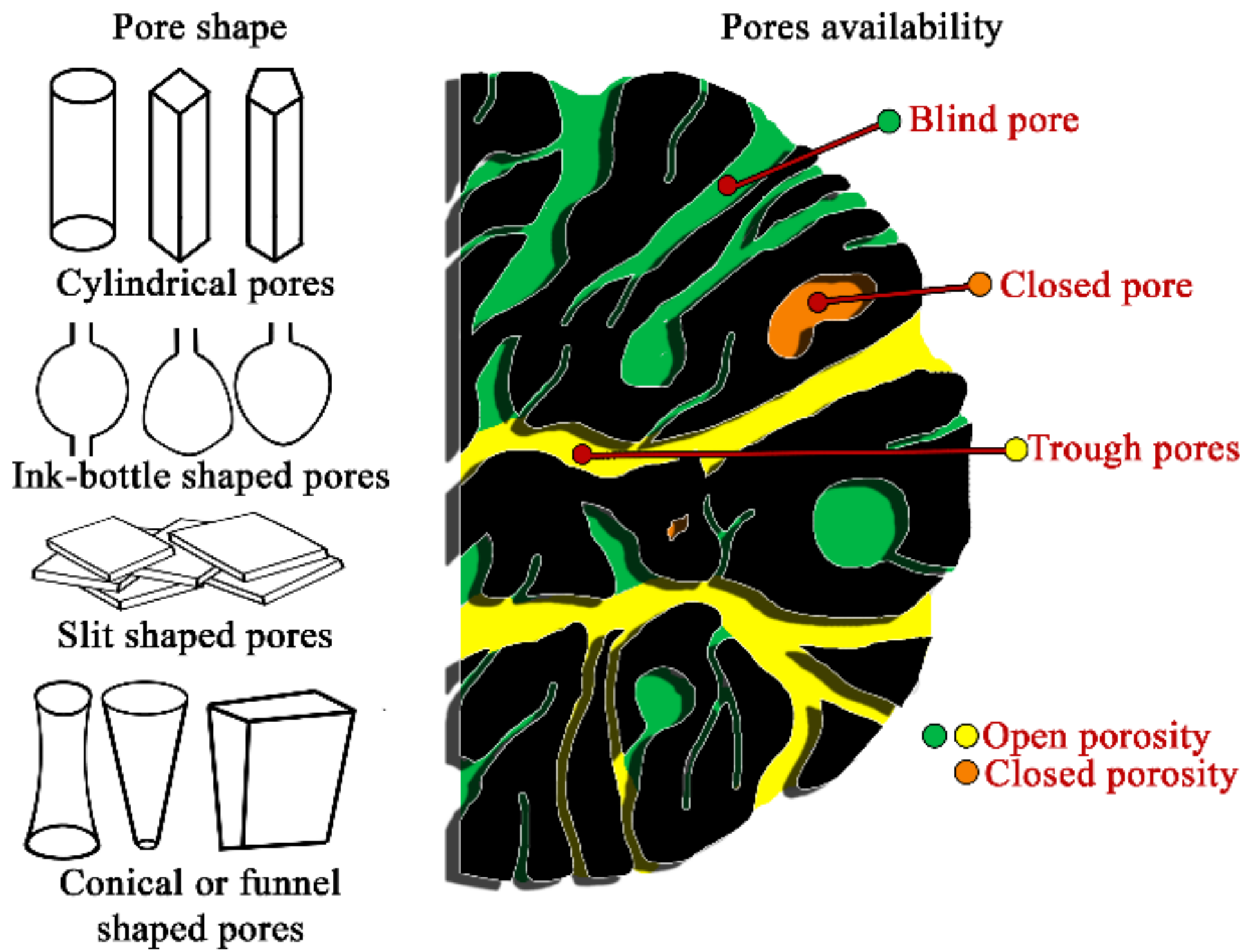{kind=link}
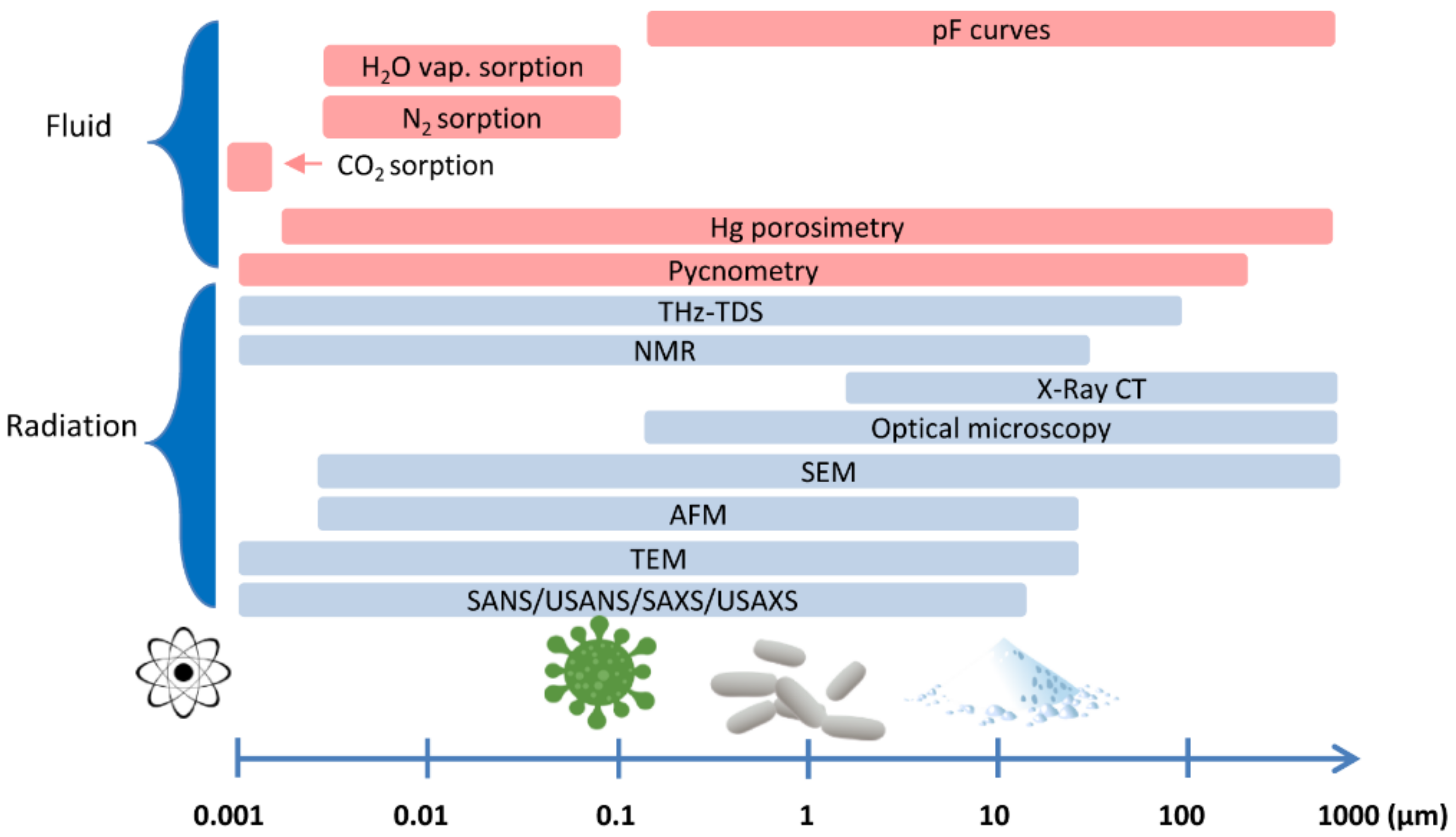{kind=link}
| Type of Pores, [nm] | ||||||
|---|---|---|---|---|---|---|
| Classification by Pore Size | Macro- | Meso- | Micro- | Supermicro- | Ultramicro- | Submicro- |
| Dubinin, 1979 | d > 200–400 | 200–400 > d > 3- 3.2 | d < 0.6–0.7 | 3–3.2 < d < 1.2–1.4 | - | - |
| IUPAC, 1972 | d > 50 | 2–50 | d < 2 | 0.7–2 | d < 0.7 | |
| Cheremskoj, 1985 | > 2000 | - | 2000 > d > 200 | - | <2–4 | <200 |
| Classification by Pore Functions | Transmission | Storage | Residual | Bonding | ||
| Greenland and Pereira, 1977 | 50,000–500,000 | 500–50,000 | d < 500 | d < 5 | ||
| Properties | Dental Ceramics | |||||
|---|---|---|---|---|---|---|
| Feldspar Ceramic | Leucite- Reinforced Glass Ceramic | Lithium Disilicate Ceramic | Fluorapatite Glass-Ceramic | Alumina Ceramic | Zirconium Oxide Ceramic | |
| Flexural strength (MPa) | 60–110 | 120–160 | 350–400 | 90–110 | 350–600 | 840–1200 |
| Hardness by Vickers (GPa) | >6.5 | 6.67 | 5.3 | 4.5 | 11.5 | 13.7 |
| Young’s modulus (GPa) | - | 65 | 103 | 70 | 380 | 210 |
| Density (g/cm3) | 2.1 | 2.5 | 2.47 | 2.56 | 3.96 | 6.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarna-Boś, K.; Skic, K.; Sobieszczański, J.; Boguta, P.; Chałas, R. Contemporary Approach to the Porosity of Dental Materials and Methods of Its Measurement. Int. J. Mol. Sci. 2021, 22, 8903. https://doi.org/10.3390/ijms22168903
Sarna-Boś K, Skic K, Sobieszczański J, Boguta P, Chałas R. Contemporary Approach to the Porosity of Dental Materials and Methods of Its Measurement. International Journal of Molecular Sciences. 2021; 22(16):8903. https://doi.org/10.3390/ijms22168903
Chicago/Turabian StyleSarna-Boś, Katarzyna, Kamil Skic, Jarosław Sobieszczański, Patrycja Boguta, and Renata Chałas. 2021. "Contemporary Approach to the Porosity of Dental Materials and Methods of Its Measurement" International Journal of Molecular Sciences 22, no. 16: 8903. https://doi.org/10.3390/ijms22168903
APA StyleSarna-Boś, K., Skic, K., Sobieszczański, J., Boguta, P., & Chałas, R. (2021). Contemporary Approach to the Porosity of Dental Materials and Methods of Its Measurement. International Journal of Molecular Sciences, 22(16), 8903. https://doi.org/10.3390/ijms22168903