
Disruption of Cytosolic Folate Integrity Aggravates Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors and Modulates Metastatic Properties in Non-Small-Cell Lung Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
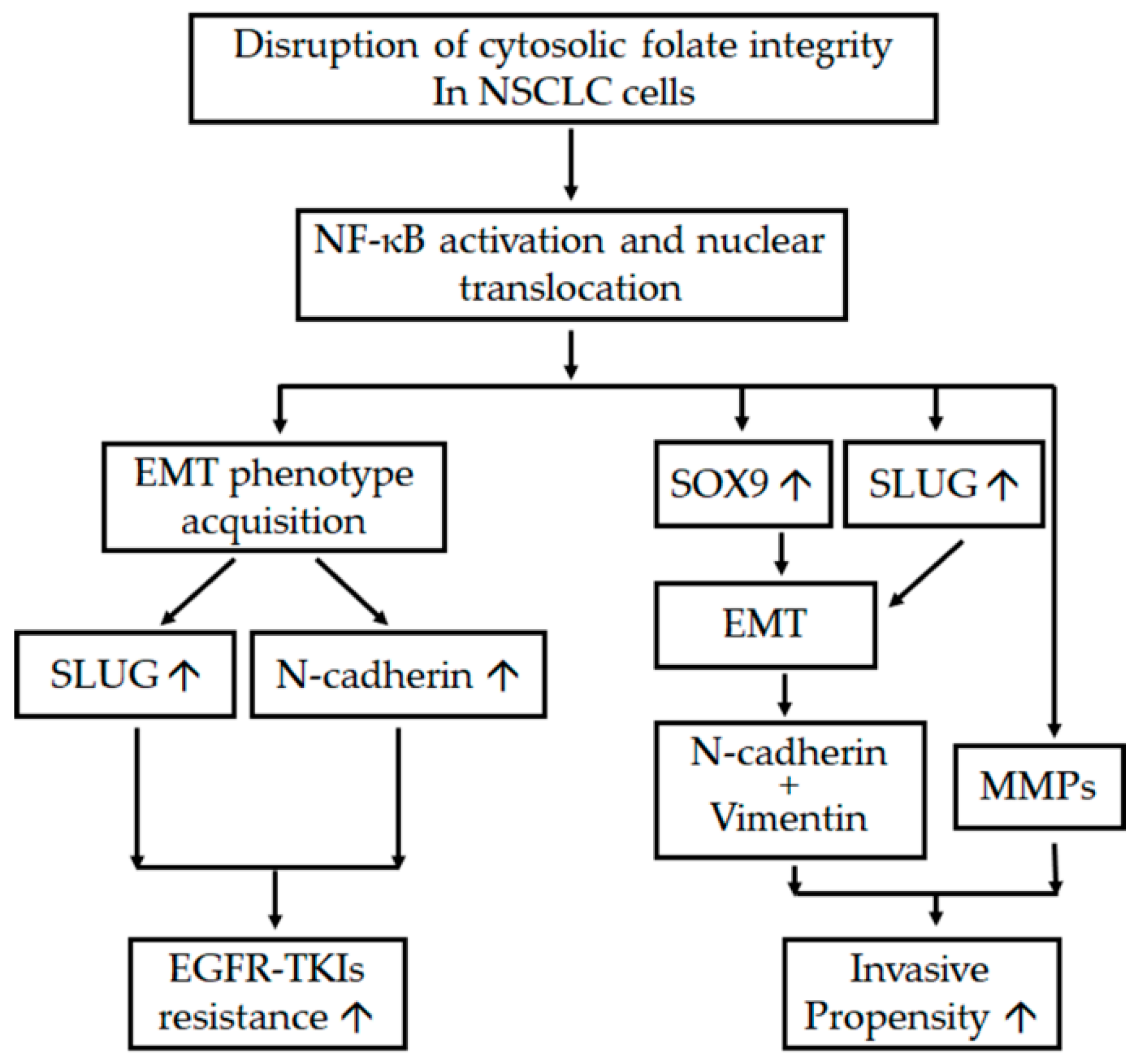
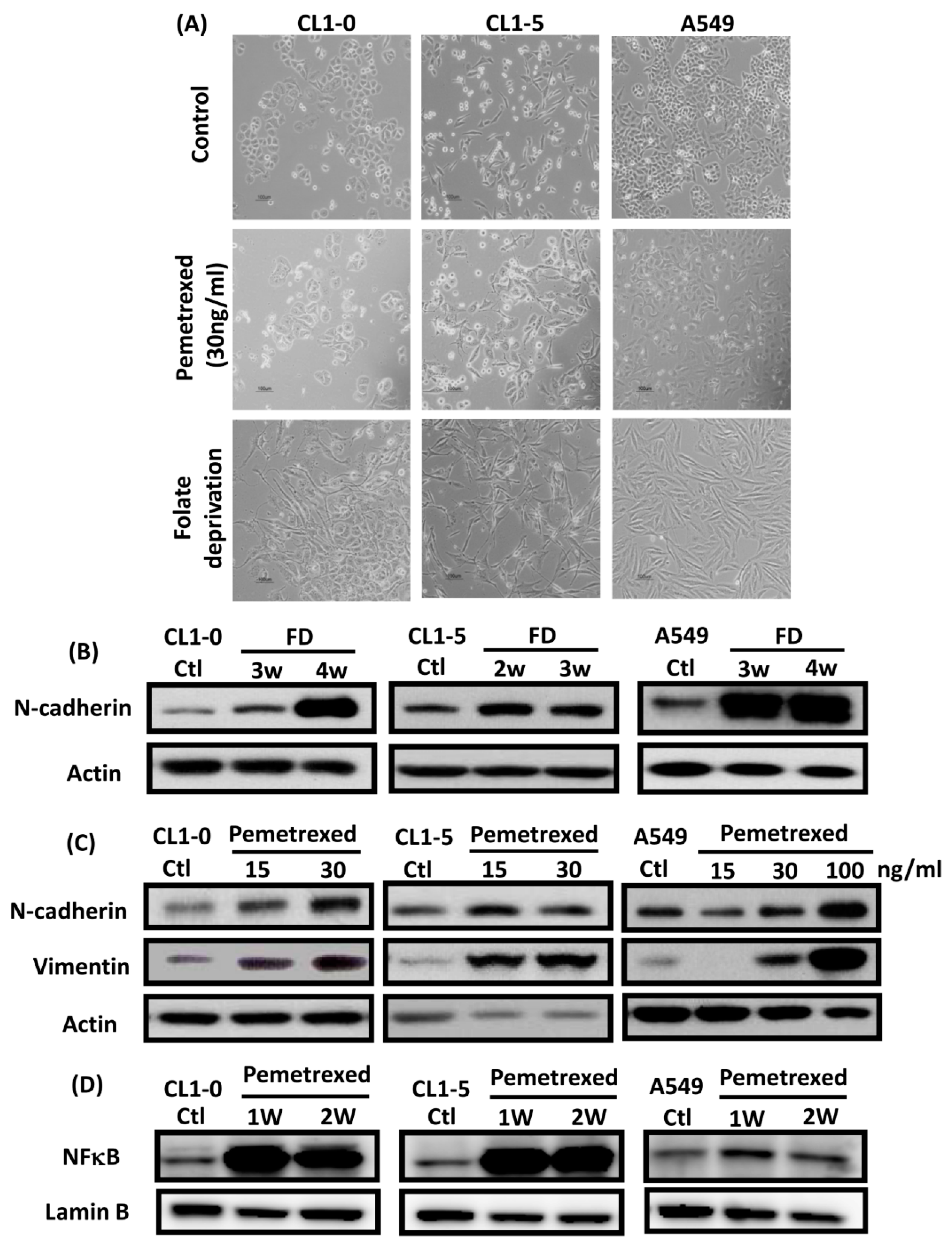
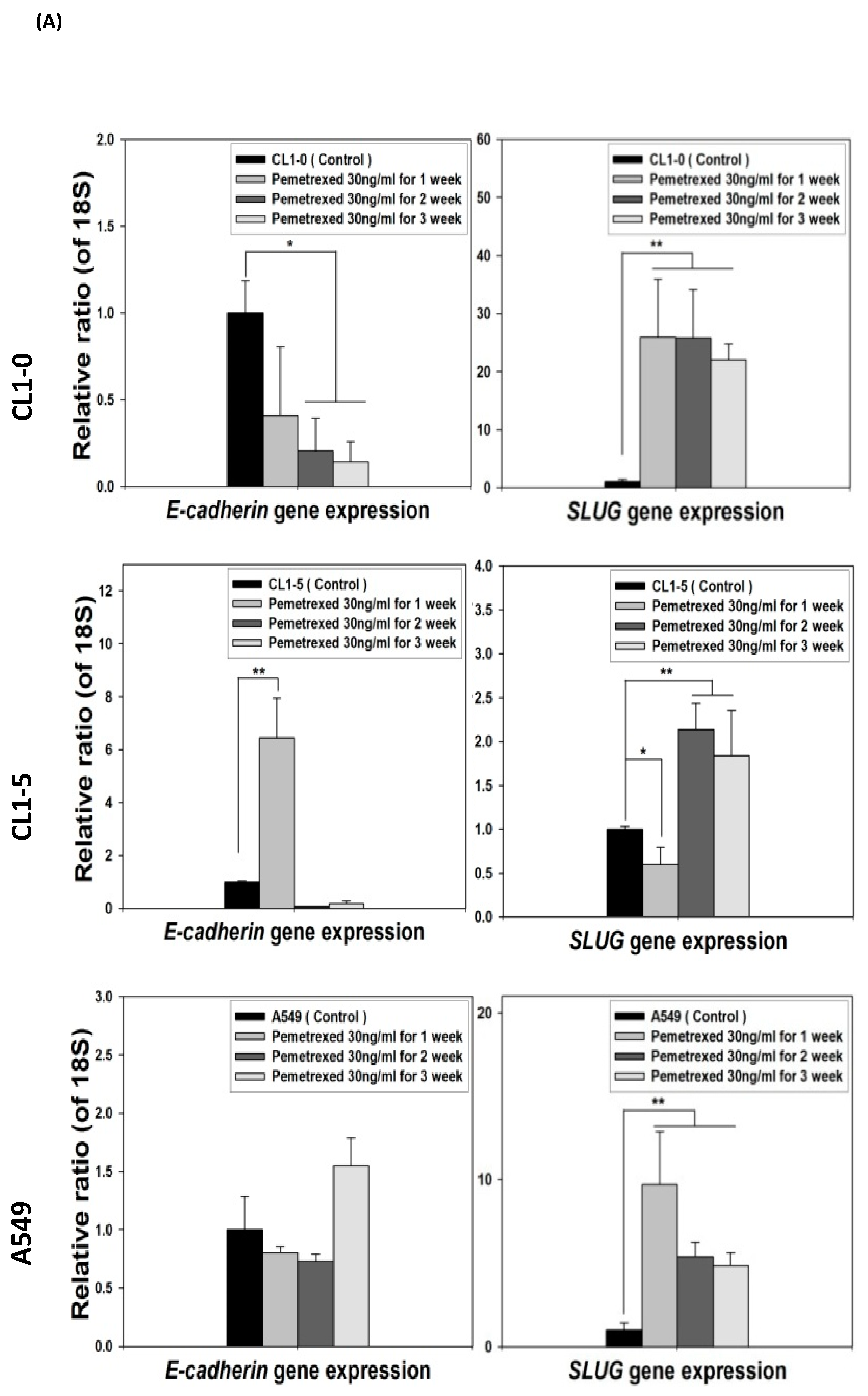
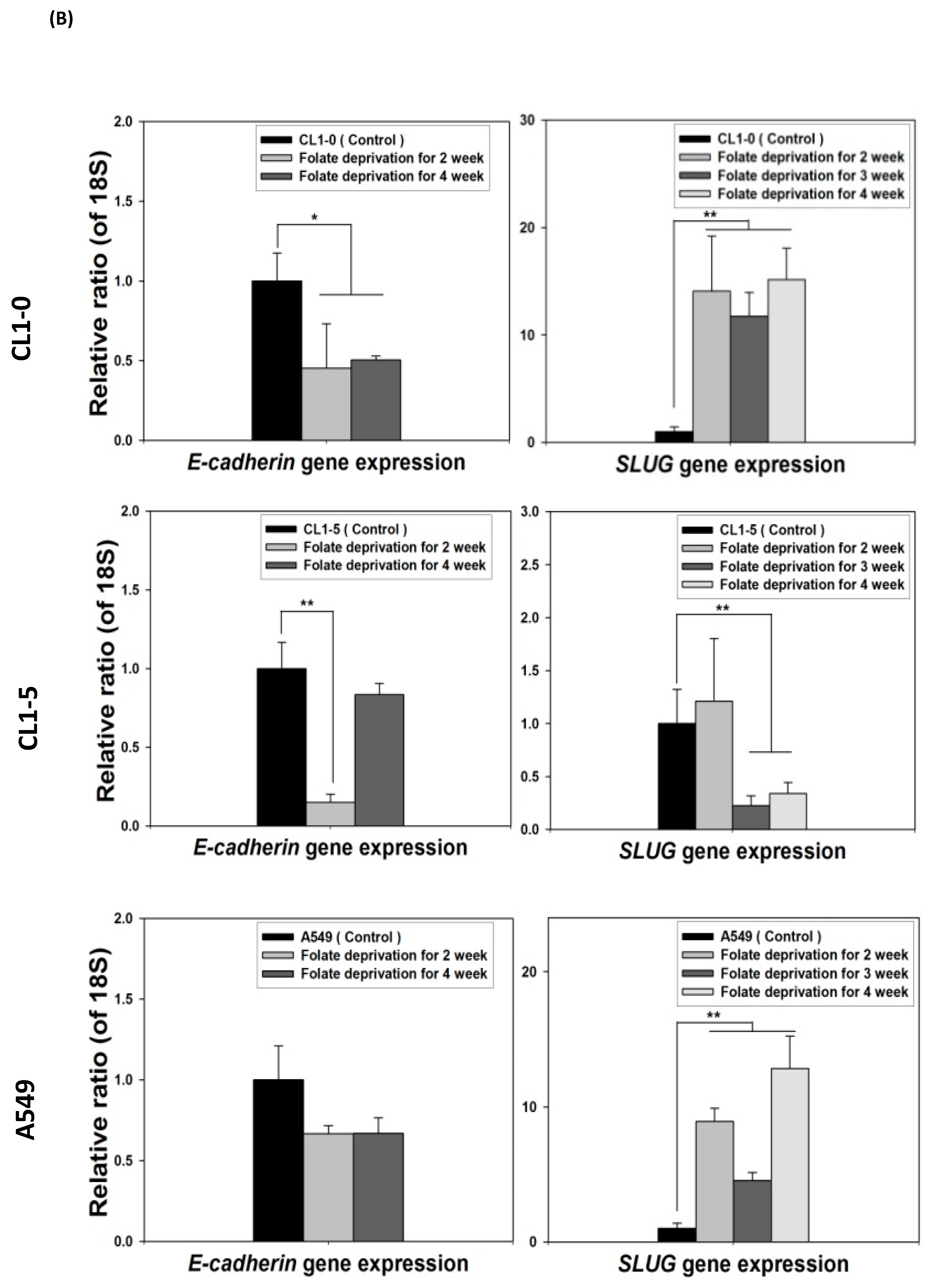
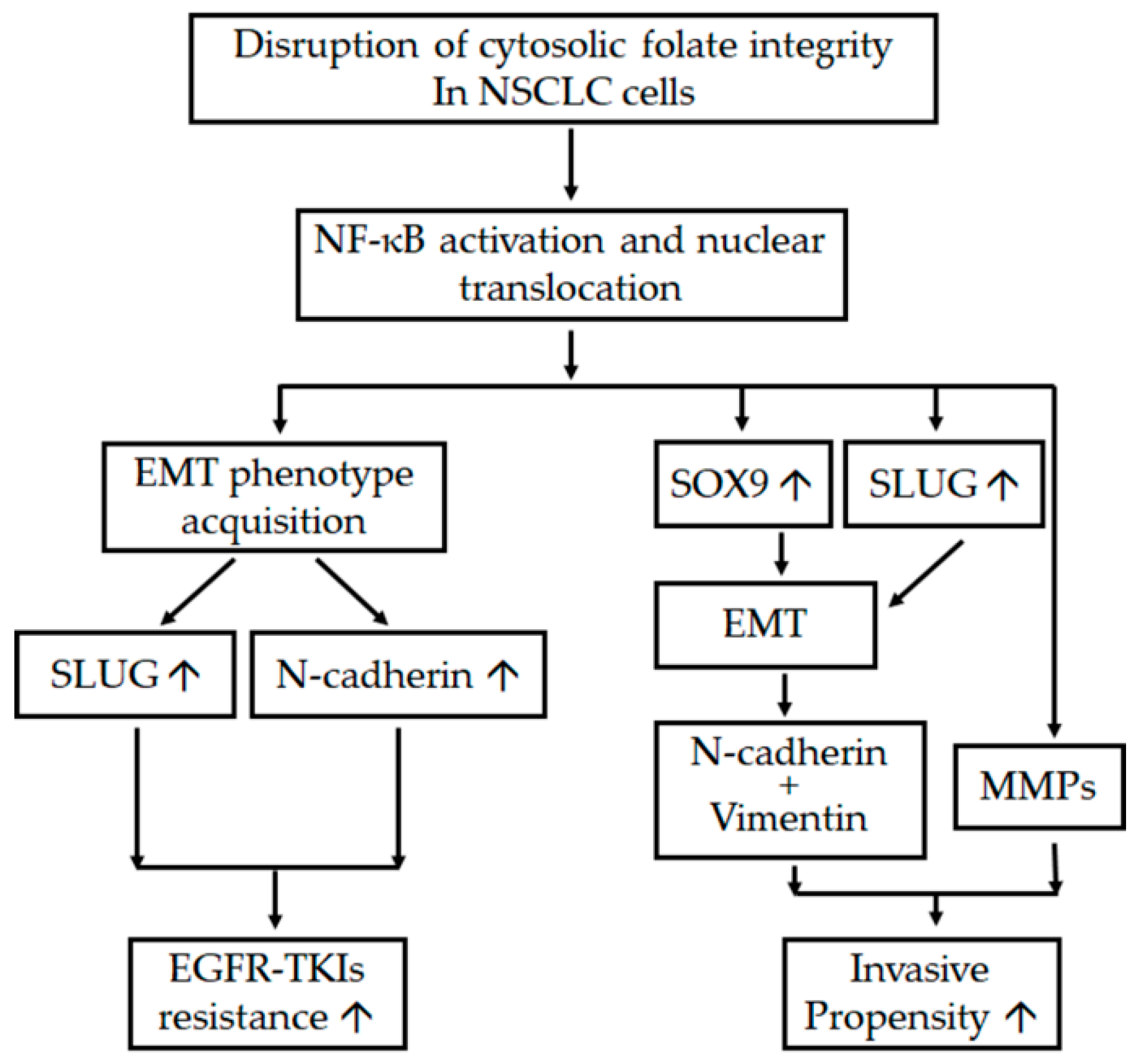
2.1. Disruption of Cytosolic Folate Integrity Induces EMT Transformation in NSCLC Cells
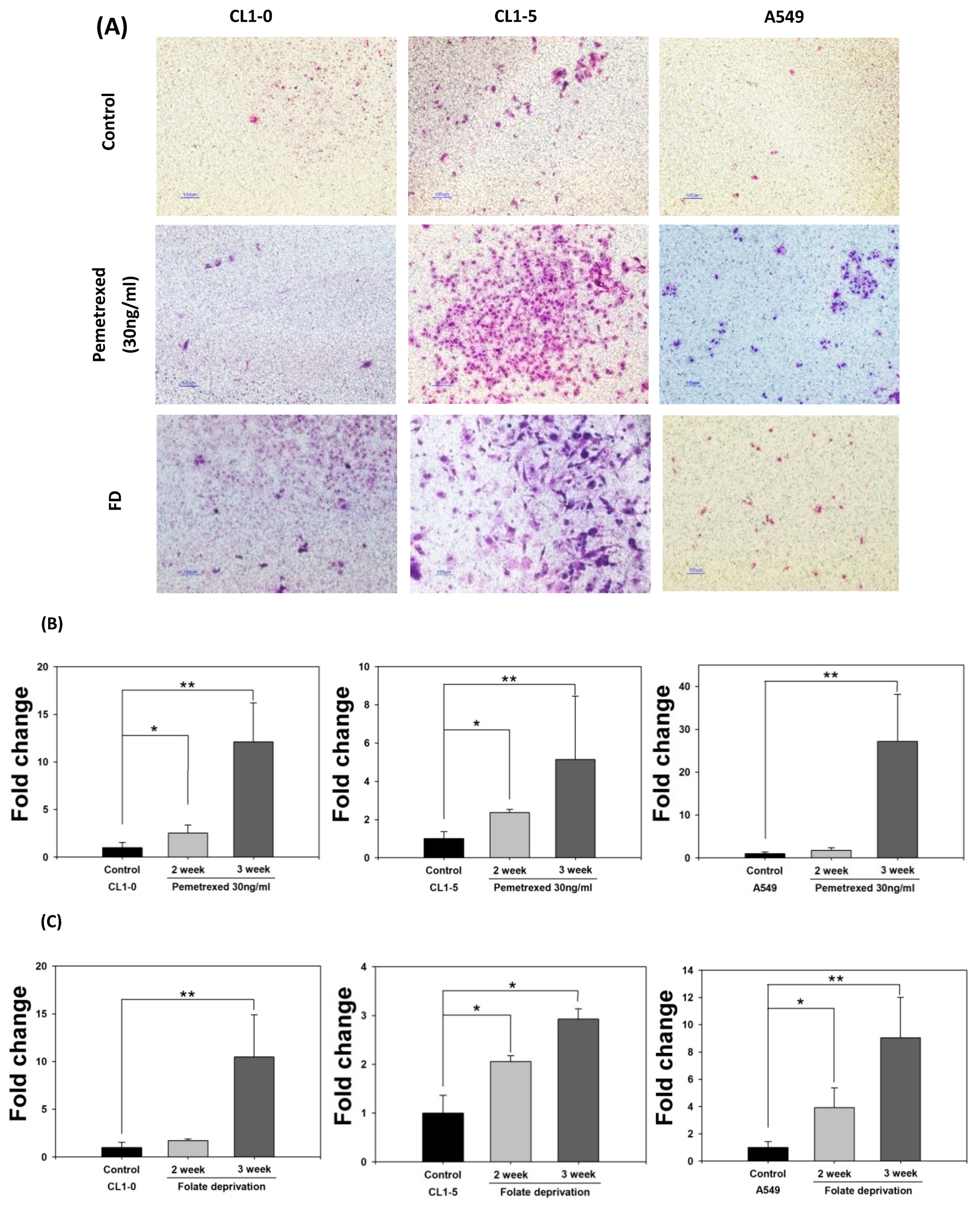
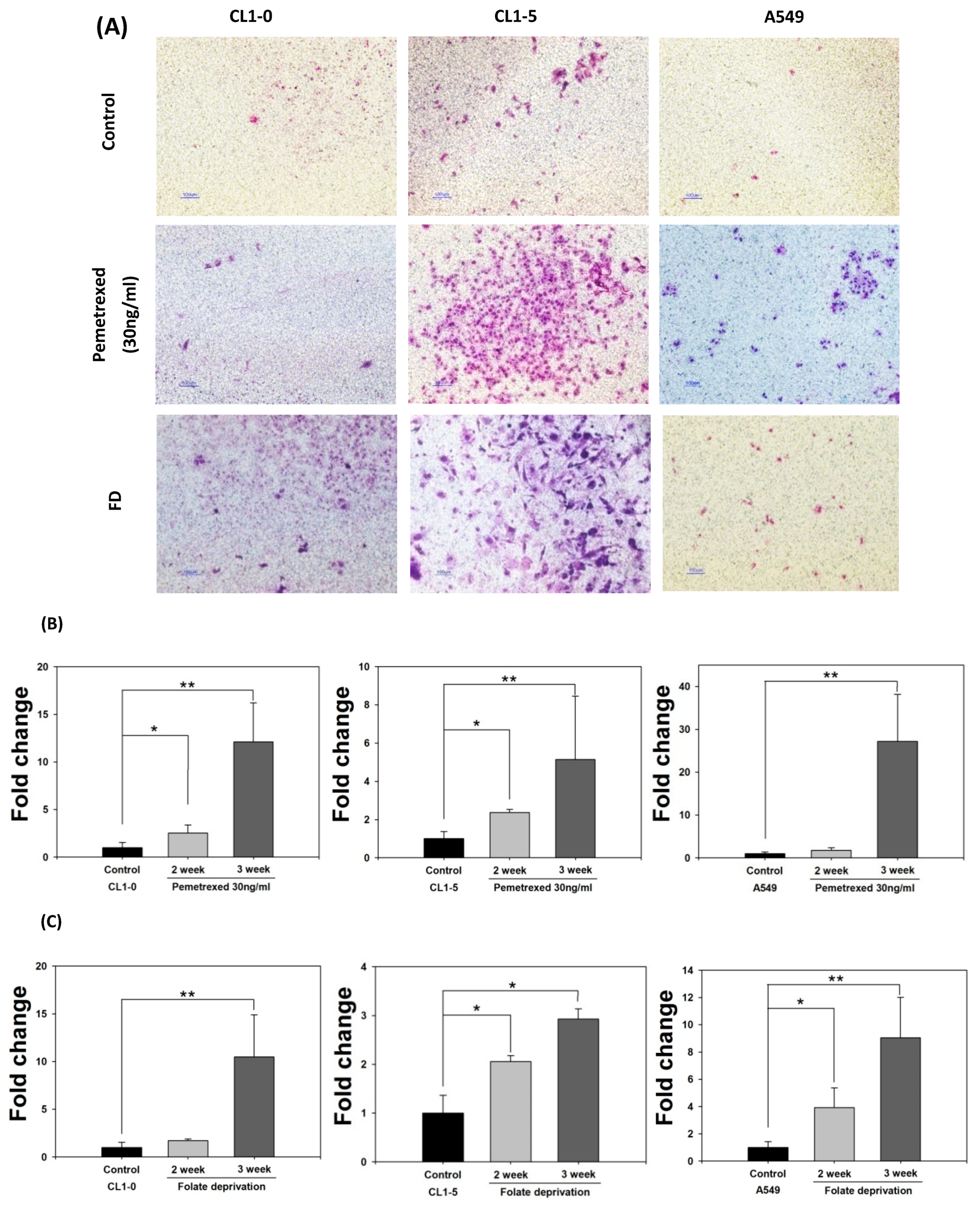
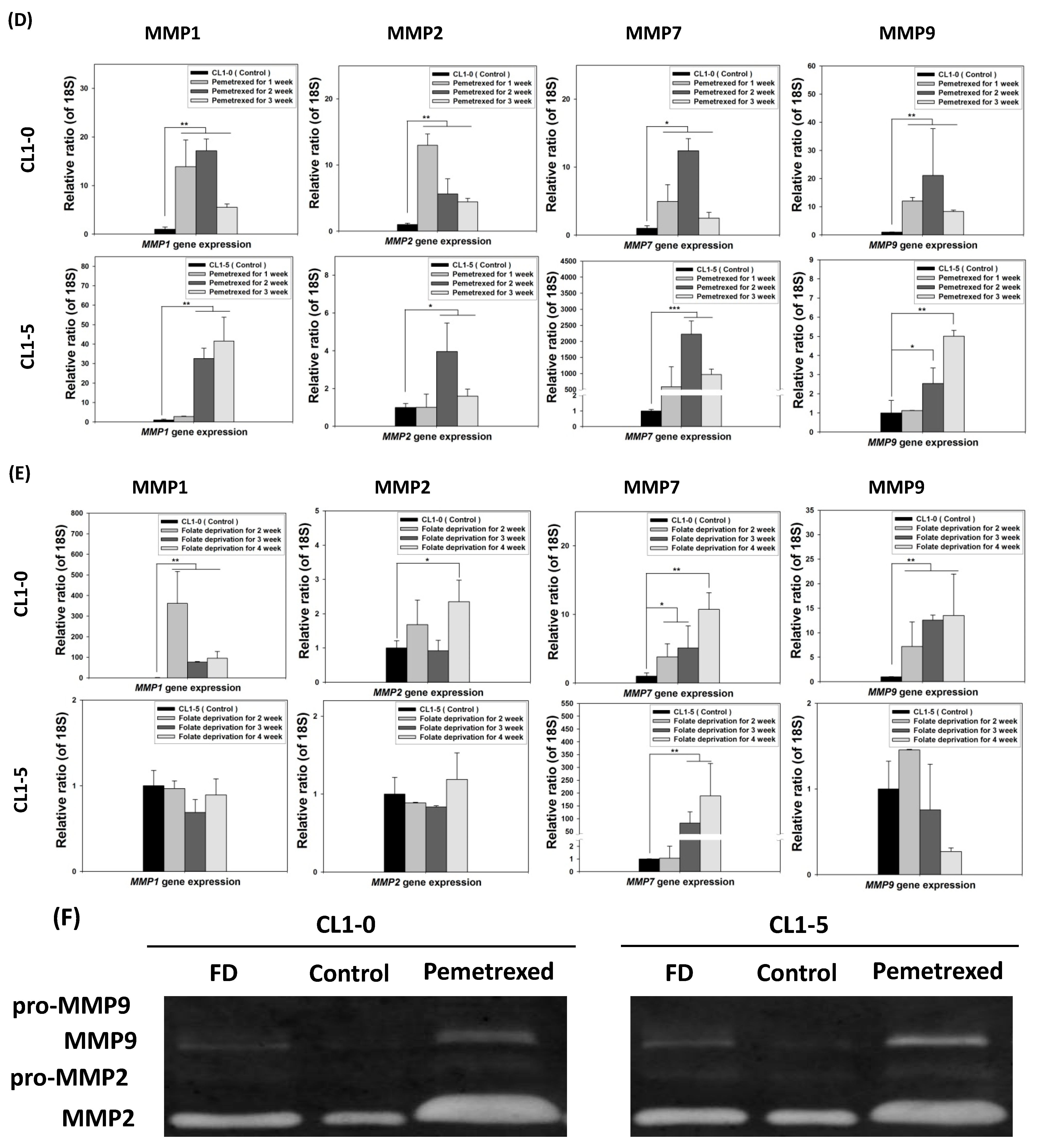
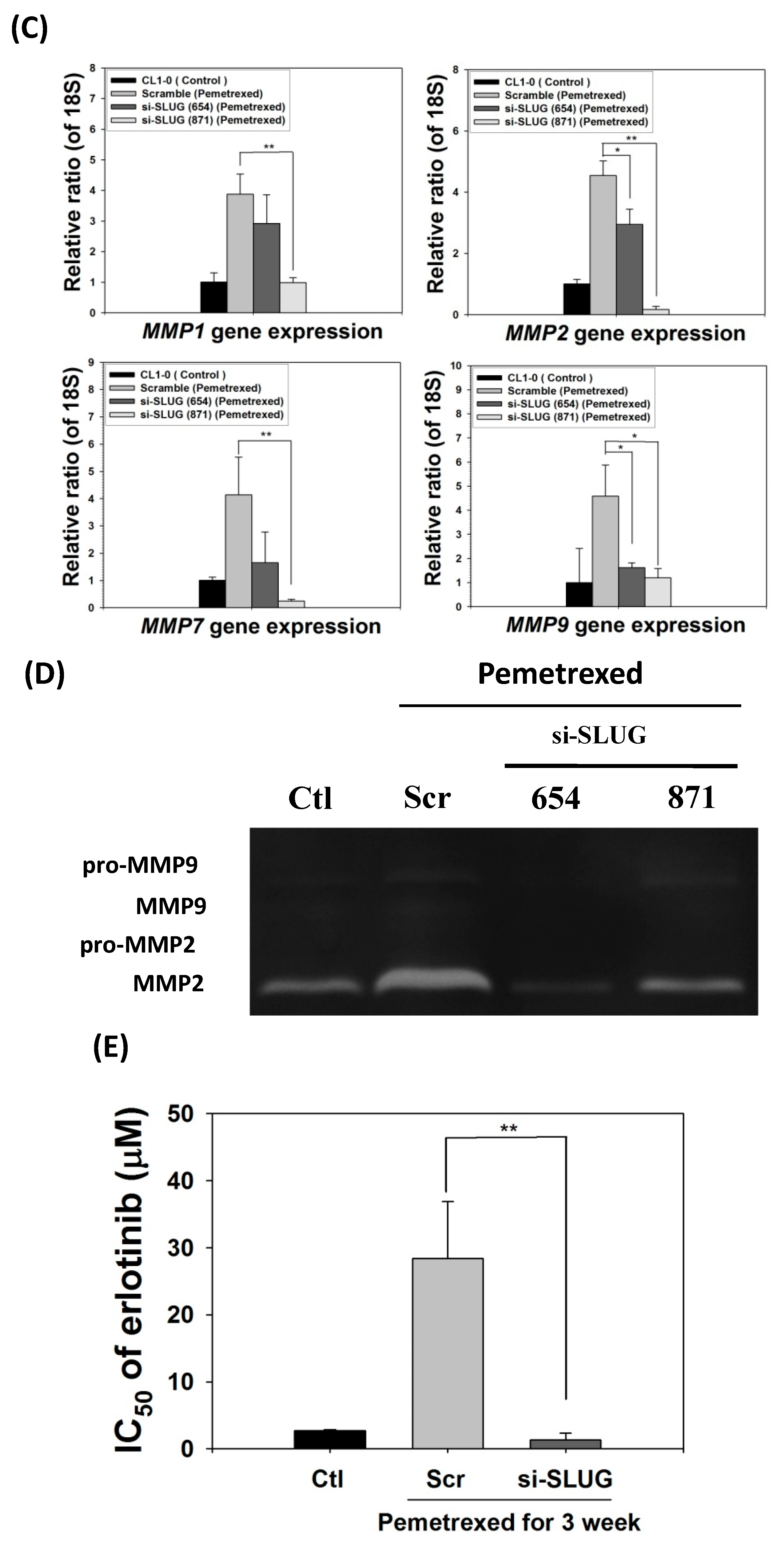
2.2. Disruption of Cytosolic Folate Integrity Promotes Increased NSCLC Cell Invasiveness by Upregulating Matrix Metalloproteinase Activities
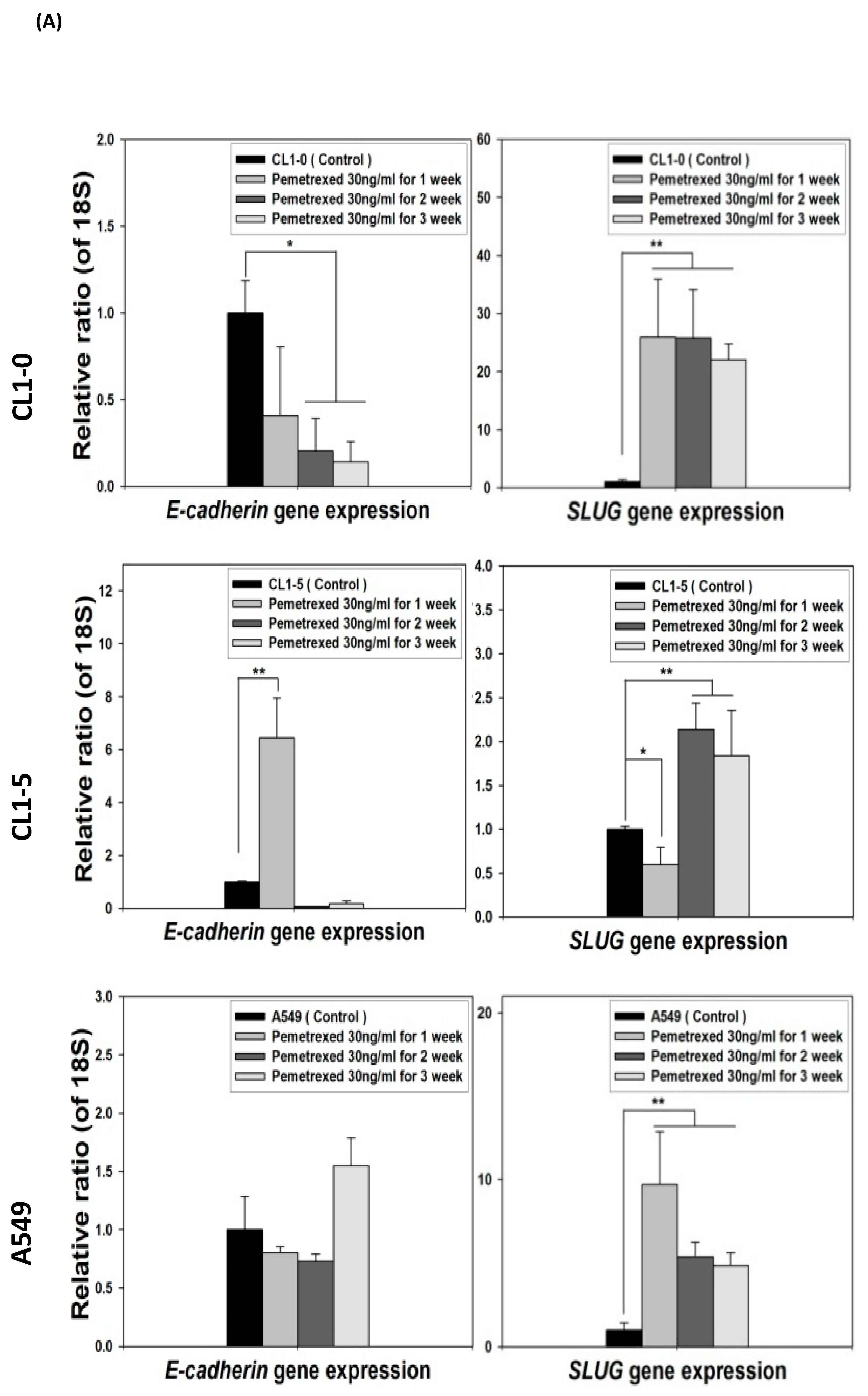
2.3. Folate Antagonist Enhances EGFR-TKI Resistance in NSCLC Cells
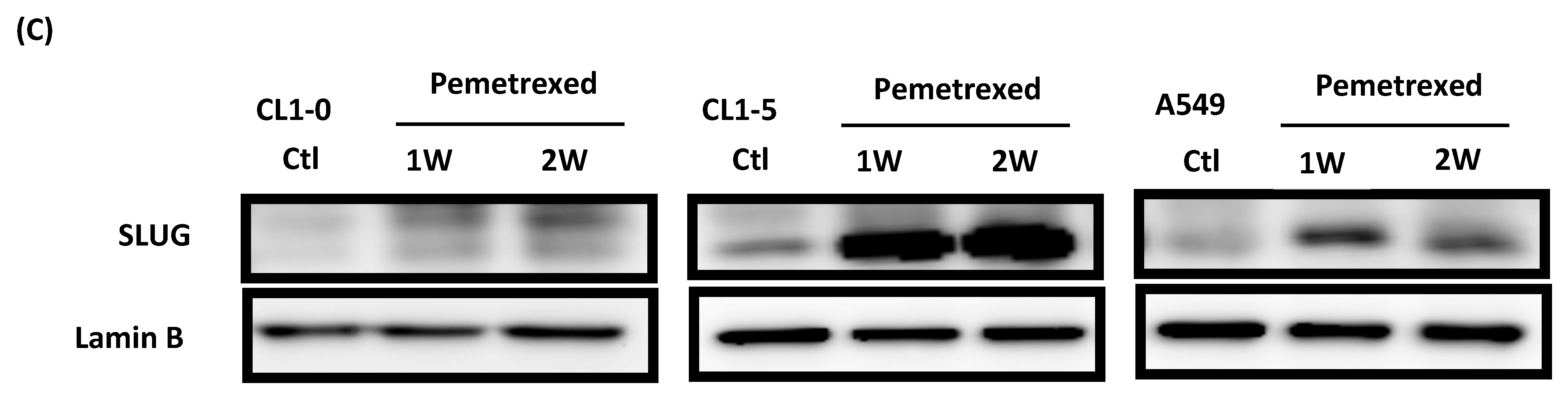
2.4. Acquisition of FD-Mediated EGFR-TKI Resistance in NSCLC Cells Is Mechanistically Linked to SLUG and N-Cadherin Upregulation
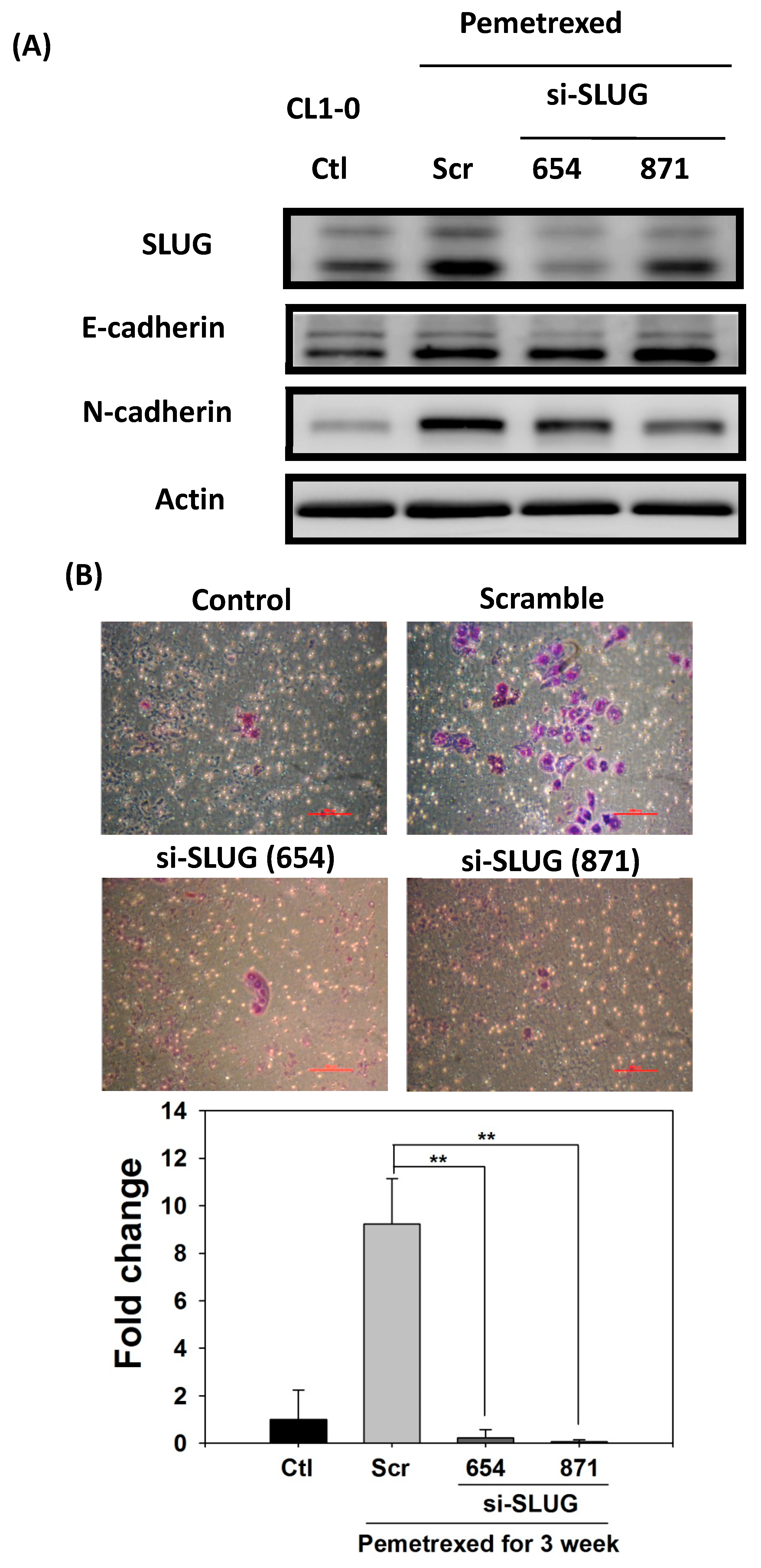
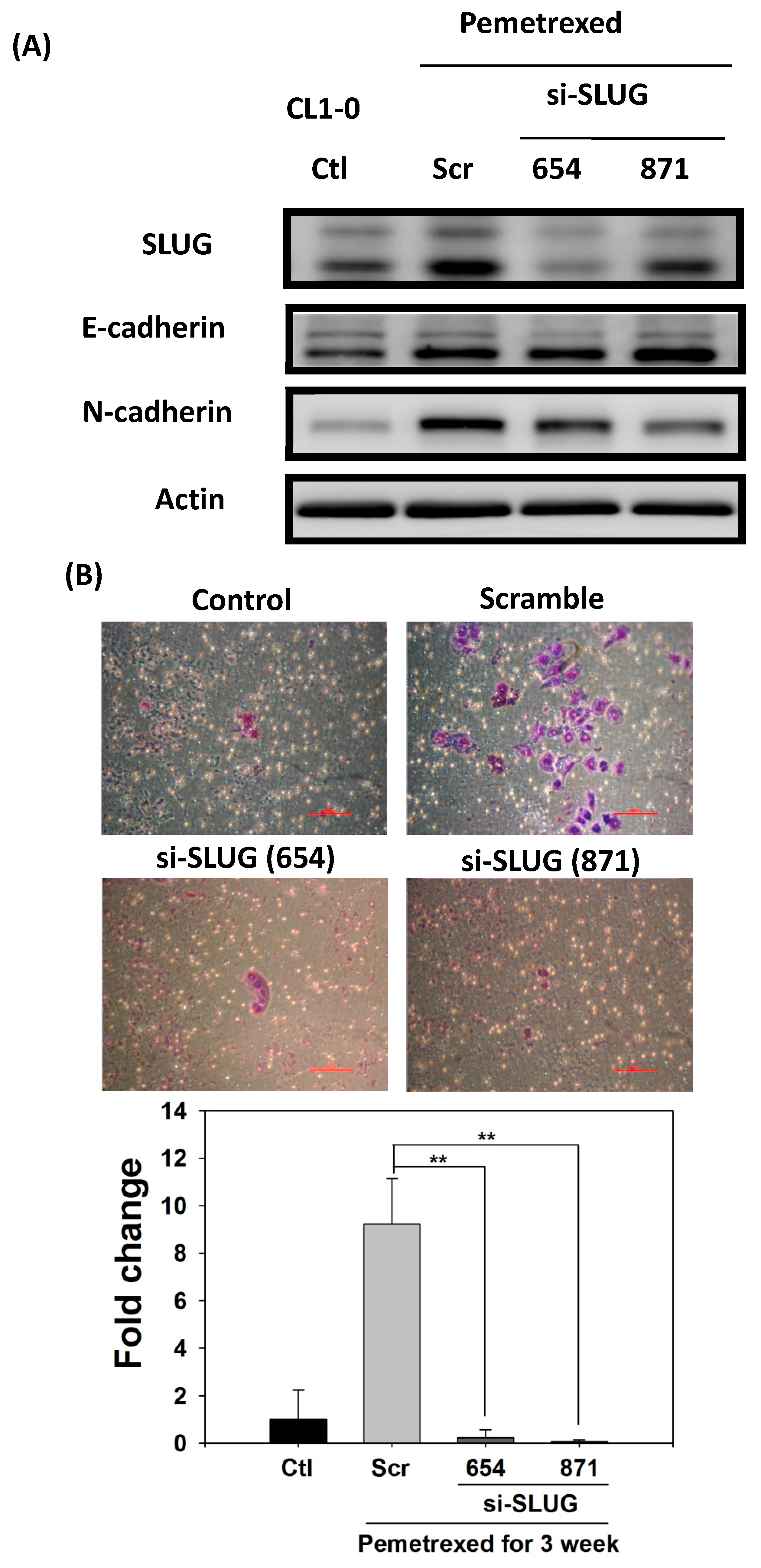
2.5. SLUG Silencing Suppresses Invasiveness and Mitigates Erlotinib Resistance Acquisition in NSCLC Cells
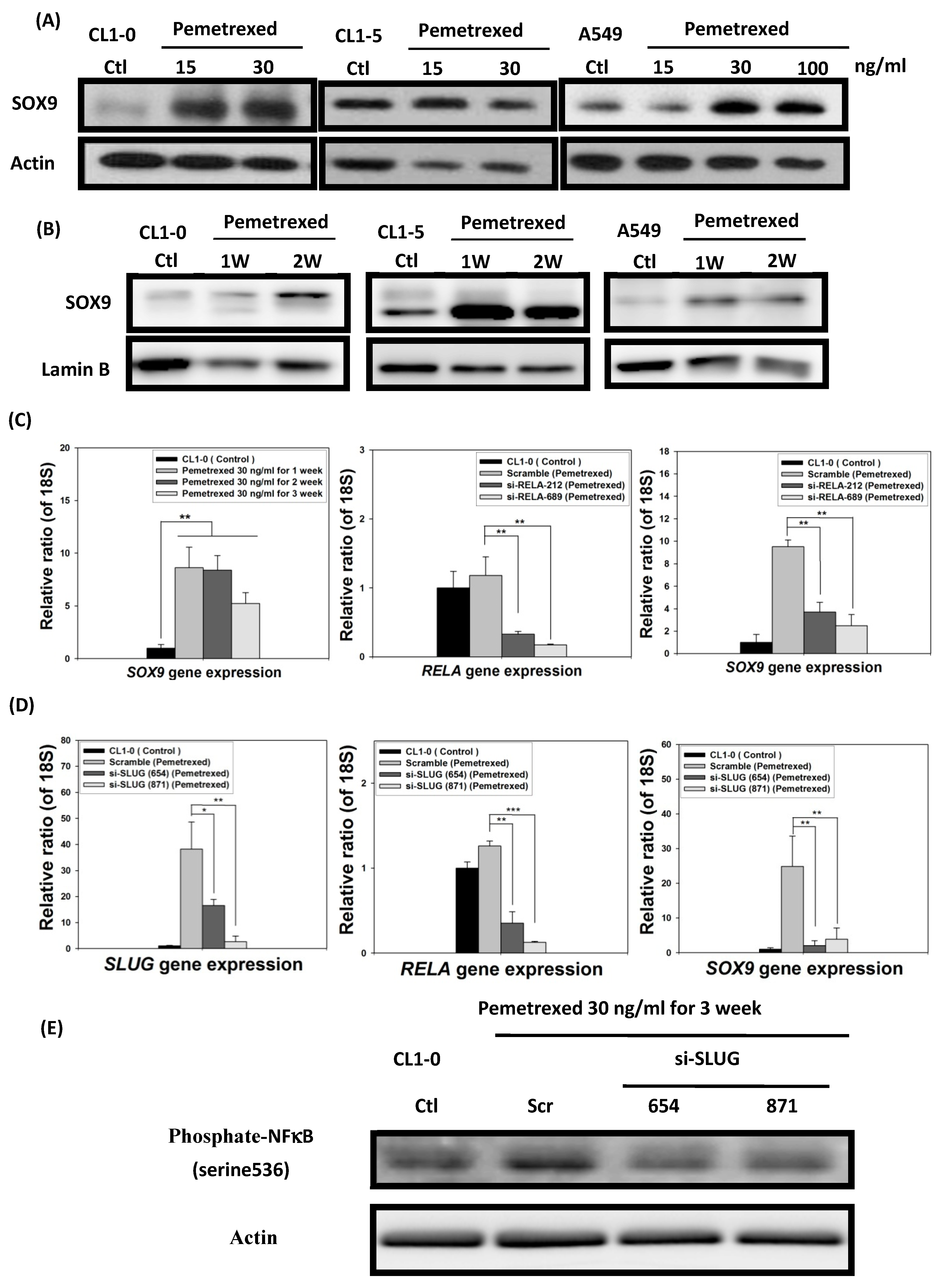
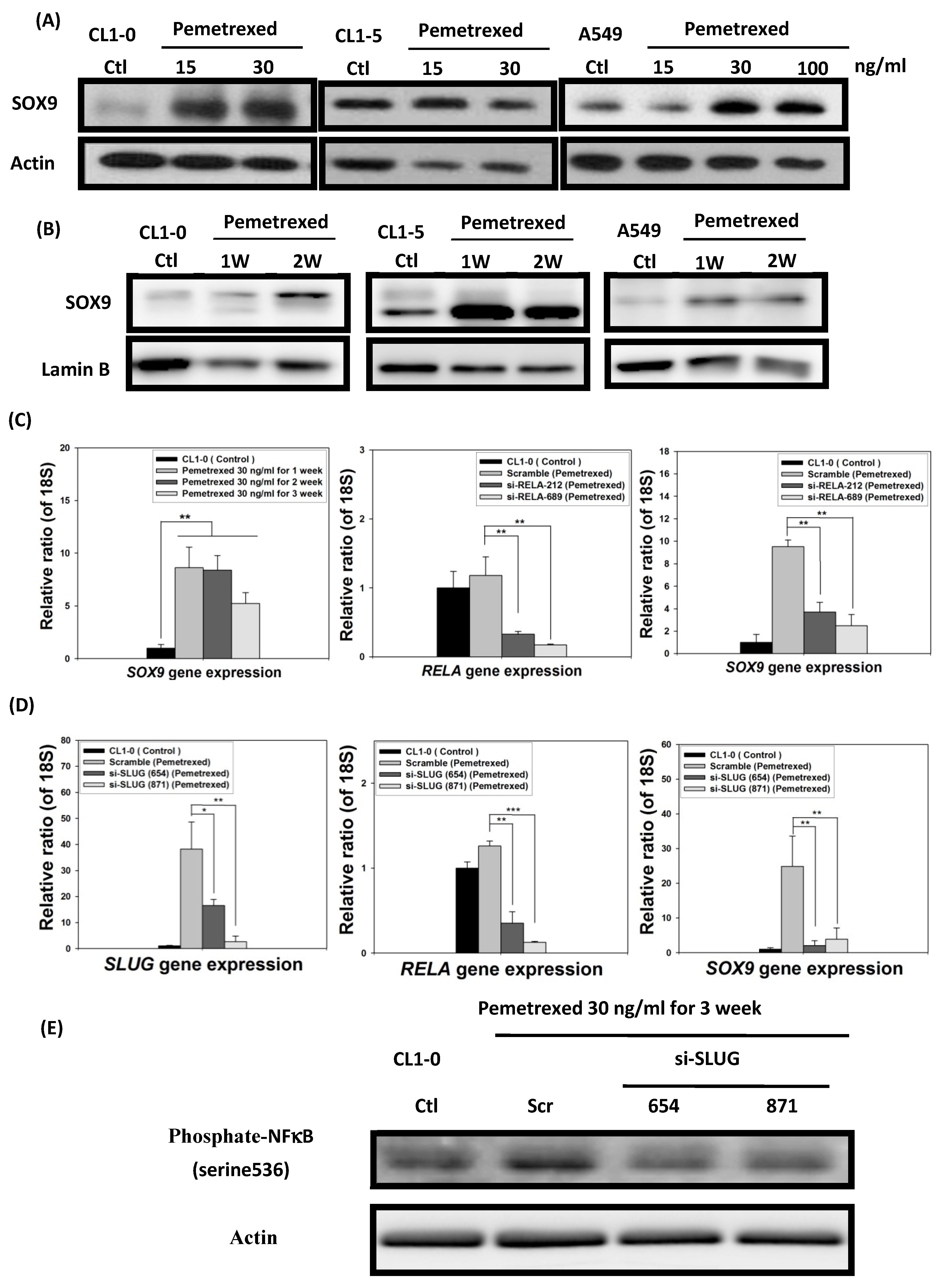
2.6. EMT in Disrupted Folate Metabolism Is Mechanistically Linked to NF-κB/Sox9 Pathway Activation
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture Mediums, Drugs, and Antibodies
4.2. Cell Viability Assay
4.3. Western Blot Analysis
4.4. RT-qPCR
4.5. Lentivirus-Mediated Knockdown of NF-κB (RELA) or SLUG Expression
4.6. Invasion Assay and Gelatin Zymography
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Bacha, S.; Mejdoub El Fehri, S.; Habibech, S.; Cheikhrouhou, S.; Racil, H.; Chaouch, N.; Sghaier, A.; Chabbou, A. Impact of malnutrition in advanced non-small cell lung cancer. Tunis. Med. 2018, 96, 59–63. [Google Scholar]
- Bittner, N.; Ostoros, G.; Geczi, L. New treatment options for lung adenocarcinoma—In view of molecular background. Pathol. Oncol. Res. 2014, 20, 11–25. [Google Scholar] [CrossRef]
- Zarogoulidis, K.; Zarogoulidis, P.; Darwiche, K.; Boutsikou, E.; Machairiotis, N.; Tsakiridis, K.; Katsikogiannis, N.; Kougioumtzi, I.; Karapantzos, I.; Huang, H.; et al. Treatment of non-small cell lung cancer (NSCLC). J. Thorac. Dis. 2013, 5, S389–S396. [Google Scholar] [PubMed]
- Leighl, N.B. Treatment paradigms for patients with metastatic non-small-cell lung cancer: First-, second-, and third-line. Curr. Oncol. 2012, 19, S52–S58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, H.; Solomon, B. The current status of targeted therapy for non-small cell lung cancer. Intern. Med. J. 2010, 40, 611–618. [Google Scholar] [CrossRef]
- Al-Farsi, A.; Ellis, P.M. Treatment paradigms for patients with metastatic non-small cell lung cancer, squamous lung cancer: First, second, and third-line. Front. Oncol. 2014, 4, 157. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.C.; Chooback, N.; Ho, C.; Melosky, B. Outcome Differences Between First- and Second-generation EGFR Inhibitors in Advanced EGFR Mutated NSCLC in a Large Population-based Cohort. Clin. Lung Cancer 2019, 20, e576–e583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Lee, B.; Shim, J.H.; Lee, S.H.; Park, W.Y.; Choi, Y.L.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K. Concurrent Genetic Alterations Predict the Progression to Target Therapy in EGFR-Mutated Advanced NSCLC. J. Thorac. Oncol. 2019, 14, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, R.; Nadal, E.; Peiro, I.; Masuet-Aumatell, C.; Macia, I.; Rivas, F.; Rosado, G.; Rodriguez, P.; Urena, A.; Padrones, S.; et al. Preoperative nutritional status assessment predicts postoperative outcomes in patients with surgically resected non-small cell lung cancer. Eur. J. Surg. Oncol. 2018, 44, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Lin, T.; Yang, J.; Wang, M. Nutritional status and related factors of patients with advanced lung cancer in northern China: A retrospective study. Cancer Manag. Res. 2019, 11, 2225–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Baldi, M.; Kaur, J.; Muthu, V.; Prasad, K.T.; Behera, D.; Bal, A.; Gupta, N.; Kapoor, R. Timing of folic acid/vitamin B12 supplementation and hematologic toxicity during first-line treatment of patients with nonsquamous non-small cell lung cancer using L01BA04-based chemotherapy: The PEMVITASTART randomized trial. Cancer 2019, 125, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Durda, K.; Kaklewski, K.; Gupta, S.; Szydlowski, M.; Baszuk, P.; Jaworska-Bieniek, K.; Sukiennicki, G.; Kaczmarek, K.; Waloszczyk, P.; Narod, S.; et al. Serum folate concentration and the incidence of lung cancer. PLoS ONE 2017, 12, e0177441. [Google Scholar] [CrossRef]
- Fang, J.Y.; Zhu, S.S.; Xiao, S.D.; Jiang, S.J.; Shi, Y.; Chen, X.Y.; Zhou, X.M.; Qian, L.F. Studies on the hypomethylation of c-myc, c-Ha-ras oncogenes and histopathological changes in human gastric carcinoma. J. Gastroenterol. Hepatol. 1996, 11, 1079–1082. [Google Scholar] [CrossRef]
- Han, J.; Hankinson, S.E.; Zhang, S.M.; De Vivo, I.; Hunter, D.J. Interaction between genetic variations in DNA repair genes and plasma folate on breast cancer risk. Cancer Epidemiol. Biomark. Prev. 2004, 13, 520–524. [Google Scholar]
- Reidy, J.A. Role of deoxyuridine incorporation and DNA repair in the expression of human chromosomal fragile sites. Mutat. Res. 1988, 200, 215–220. [Google Scholar] [CrossRef]
- Duthie, S.J.; Narayanan, S.; Brand, G.M.; Pirie, L.; Grant, G. Impact of folate deficiency on DNA stability. J. Nutr. 2002, 132, 2444S–2449S. [Google Scholar] [CrossRef]
- Linhart, H.G.; Troen, A.; Bell, G.W.; Cantu, E.; Chao, W.H.; Moran, E.; Steine, E.; He, T.; Jaenisch, R. Folate deficiency induces genomic uracil misincorporation and hypomethylation but does not increase DNA point mutations. Gastroenterology 2009, 136, 227–235 e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hines, P.J. Folate manages cell shape during neurulation. Science 2017, 356, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.P.; Hsu, S.H.; Feng, H.C.; Huang, R.F. Folate deprivation enhances invasiveness of human colon cancer cells mediated by activation of sonic hedgehog signaling through promoter hypomethylation and cross action with transcription nuclear factor-kappa B pathway. Carcinogenesis 2012, 33, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.H.; Huang, W.C.; Huang, T.H.; Huang, Y.J.; Sue, Y.K.; Huynh, T.T.; Hsiao, M.; Liu, T.Z.; Wu, A.T.; Lin, C.M. Folate deficient tumor microenvironment promotes epithelial-to-mesenchymal transition and cancer stem-like phenotypes. Oncotarget 2016, 7, 33246–33256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.T.; Shang, H.S.; Chang, J.B.; Liu, J.J.; Liu, T.Z. Folate deficiency-triggered redox pathways confer drug resistance in hepatocellular carcinoma. Oncotarget 2015, 6, 26104–26118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Lin, T.Y.; Lee, G.; Paddock, M.N.; Momb, J.; Cheng, Z.; Li, Q.; Fei, D.L.; Stein, B.D.; Ramsamooj, S.; et al. Mitochondrial One-Carbon Pathway Supports Cytosolic Folate Integrity in Cancer Cells. Cell 2018, 175, 1546–1560.e17. [Google Scholar] [CrossRef] [Green Version]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jimenez-Sanchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.H.; Tsai, M.F.; Su, K.Y.; Wu, S.G.; Huang, C.P.; Yu, S.L.; Yu, Y.L.; Lan, C.C.; Yang, C.H.; Lin, S.B.; et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am. J. Respir. Crit. Care Med. 2011, 183, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-kappaB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef]
- Izumchenko, E.; Chang, X.; Michailidi, C.; Kagohara, L.; Ravi, R.; Paz, K.; Brait, M.; Hoque, M.O.; Ling, S.; Bedi, A.; et al. The TGFbeta-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res. 2014, 74, 3995–4005. [Google Scholar] [CrossRef] [Green Version]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, M.; Yoshino, I.; Yamaguchi, R.; Shimamura, T.; Nagasaki, M.; Imoto, S.; Niida, A.; Koizumi, F.; Kohno, T.; Yokota, J.; et al. N-cadherin expression is a potential survival mechanism of gefitinib-resistant lung cancer cells. Am. J. Cancer Res. 2011, 1, 823–833. [Google Scholar] [PubMed]
- Mendelsohn, J. Targeting the epidermal growth factor receptor for cancer therapy. J. Clin. Oncol. 2002, 20, 1S–13S. [Google Scholar] [PubMed]
- Vidal, V.P.; Ortonne, N.; Schedl, A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. J. Cutan. Pathol. 2008, 35, 373–379. [Google Scholar] [CrossRef]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Adjei, A.A. The role of L01BA04 (Alimta, LY231514) in lung cancer therapy. Clin. Lung Cancer 2003, 5, 21–27. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.F.; Ho, Y.H.; Lin, H.L.; Wei, J.S.; Liu, T.Z. Folate deficiency induces a cell cycle-specific apoptosis in HepG2 cells. J. Nutr. 1999, 129, 25–31. [Google Scholar] [CrossRef]
- Chern, C.L.; Huang, R.F.; Chen, Y.H.; Cheng, J.T.; Liu, T.Z. Folate deficiency-induced oxidative stress and apoptosis are mediated via homocysteine-dependent overproduction of hydrogen peroxide and enhanced activation of NF-kappaB in human Hep G2 cells. Biomed. Pharmacother. 2001, 55, 434–442. [Google Scholar] [CrossRef]
- Huang, R.F.; Huang, S.M.; Lin, B.S.; Wei, J.S.; Liu, T.Z. Homocysteine thiolactone induces apoptotic DNA damage mediated by increased intracellular hydrogen peroxide and caspase 3 activation in HL-60 cells. Life Sci. 2001, 68, 2799–2811. [Google Scholar] [CrossRef]
- Hsu, H.C.; Chiou, J.F.; Wang, Y.H.; Chen, C.H.; Mau, S.Y.; Ho, C.T.; Chang, P.J.; Liu, T.Z.; Chen, C.H. Folate deficiency triggers an oxidative-nitrosative stress-mediated apoptotic cell death and impedes insulin biosynthesis in RINm5F pancreatic islet beta-cells: Relevant to the pathogenesis of diabetes. PLoS ONE 2013, 8, e77931. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.W.; Chen, S.C.; Cheng, E.C.; Ko, Y.P.; Lin, Y.C.; Kao, Y.R.; Tsay, Y.G.; Yang, P.C.; Wu, C.W.; Roffler, S.R. CD13 (aminopeptidase N) can associate with tumor-associated antigen L6 and enhance the motility of human lung cancer cells. Int. J. Cancer 2005, 116, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.Y.; Chen, Y.C.; Chang, H.Y.; Jeng, J.C.; Lin, E.H.; Pan, C.M.; Chang, Y.W.; Wang, M.L.; Chou, Y.T.; Shih, H.M.; et al. The PML isoform IV is a negative regulator of nuclear EGFR’s transcriptional activity in lung cancer. Carcinogenesis 2013, 34, 1708–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.W.; Peck, K.; Hong, T.-M.; Yang, S.-C.; Sher, Y.-P.; Shih, J.-Y.; Wu, R.; Cheng, J.-L.; Roffler, S.R.; Wu, C.-W.; et al. Global analysis of gene expression in invasion by a lung cancer model. Cancer Res. 2001, 61, 5223–5230. [Google Scholar]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, P.-W.; Ho, C.-T.; Hsiao, S.-H.; Chou, Y.-T.; Chang, Y.-C.; Liu, J.-J. Disruption of Cytosolic Folate Integrity Aggravates Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors and Modulates Metastatic Properties in Non-Small-Cell Lung Cancer Cells. Int. J. Mol. Sci. 2021, 22, 8838. https://doi.org/10.3390/ijms22168838
Shen P-W, Ho C-T, Hsiao S-H, Chou Y-T, Chang Y-C, Liu J-J. Disruption of Cytosolic Folate Integrity Aggravates Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors and Modulates Metastatic Properties in Non-Small-Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2021; 22(16):8838. https://doi.org/10.3390/ijms22168838
Chicago/Turabian StyleShen, Po-Wen, Chun-Te Ho, Shih-Hsin Hsiao, Yu-Ting Chou, Yi-Cheng Chang, and Jun-Jen Liu. 2021. "Disruption of Cytosolic Folate Integrity Aggravates Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors and Modulates Metastatic Properties in Non-Small-Cell Lung Cancer Cells" International Journal of Molecular Sciences 22, no. 16: 8838. https://doi.org/10.3390/ijms22168838
APA StyleShen, P.-W., Ho, C.-T., Hsiao, S.-H., Chou, Y.-T., Chang, Y.-C., & Liu, J.-J. (2021). Disruption of Cytosolic Folate Integrity Aggravates Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors and Modulates Metastatic Properties in Non-Small-Cell Lung Cancer Cells. International Journal of Molecular Sciences, 22(16), 8838. https://doi.org/10.3390/ijms22168838