
Hearing Impairment in a Mouse Model of Diabetes Is Associated with Mitochondrial Dysfunction, Synaptopathy, and Activation of the Intrinsic Apoptosis Pathway

 and
and 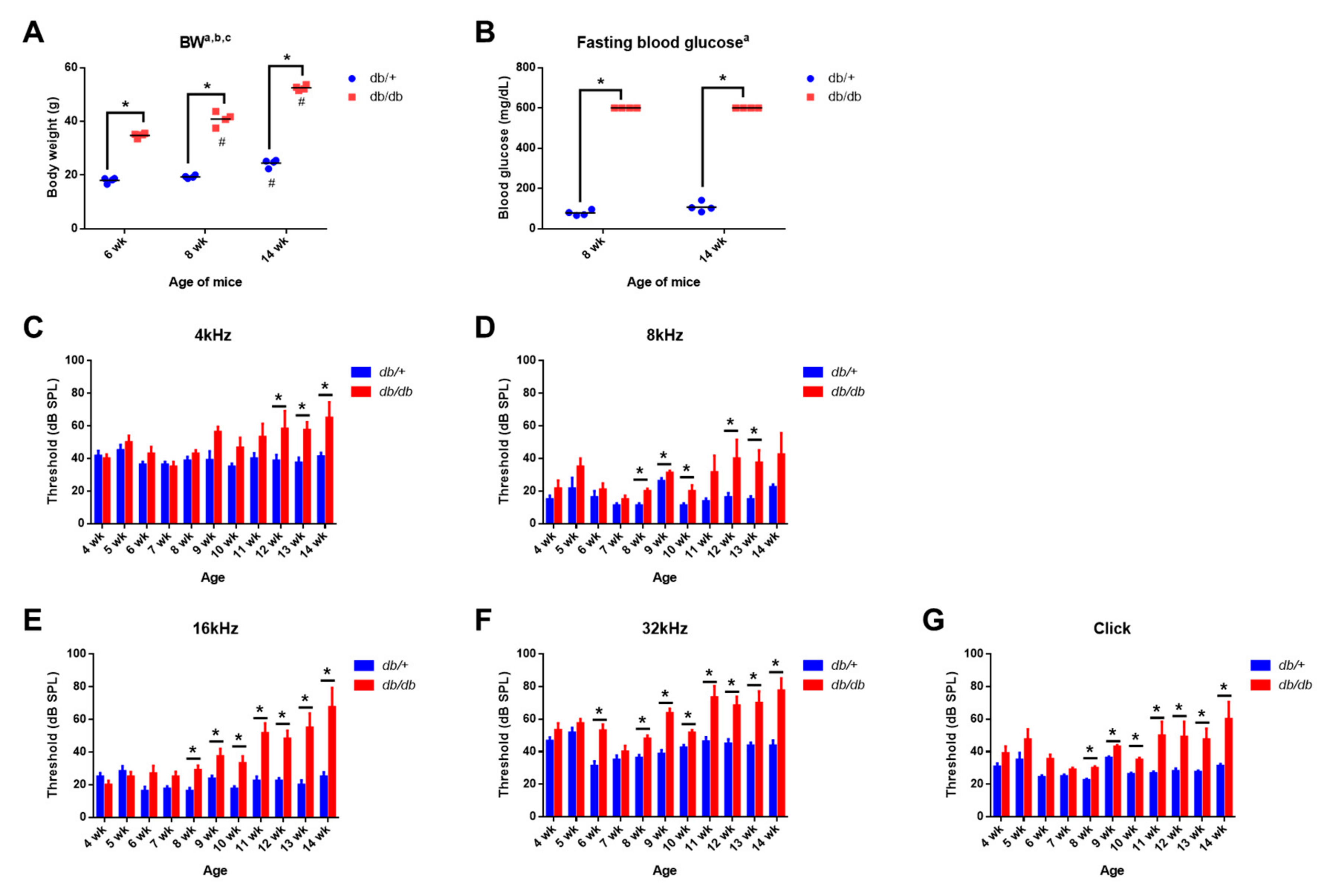{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
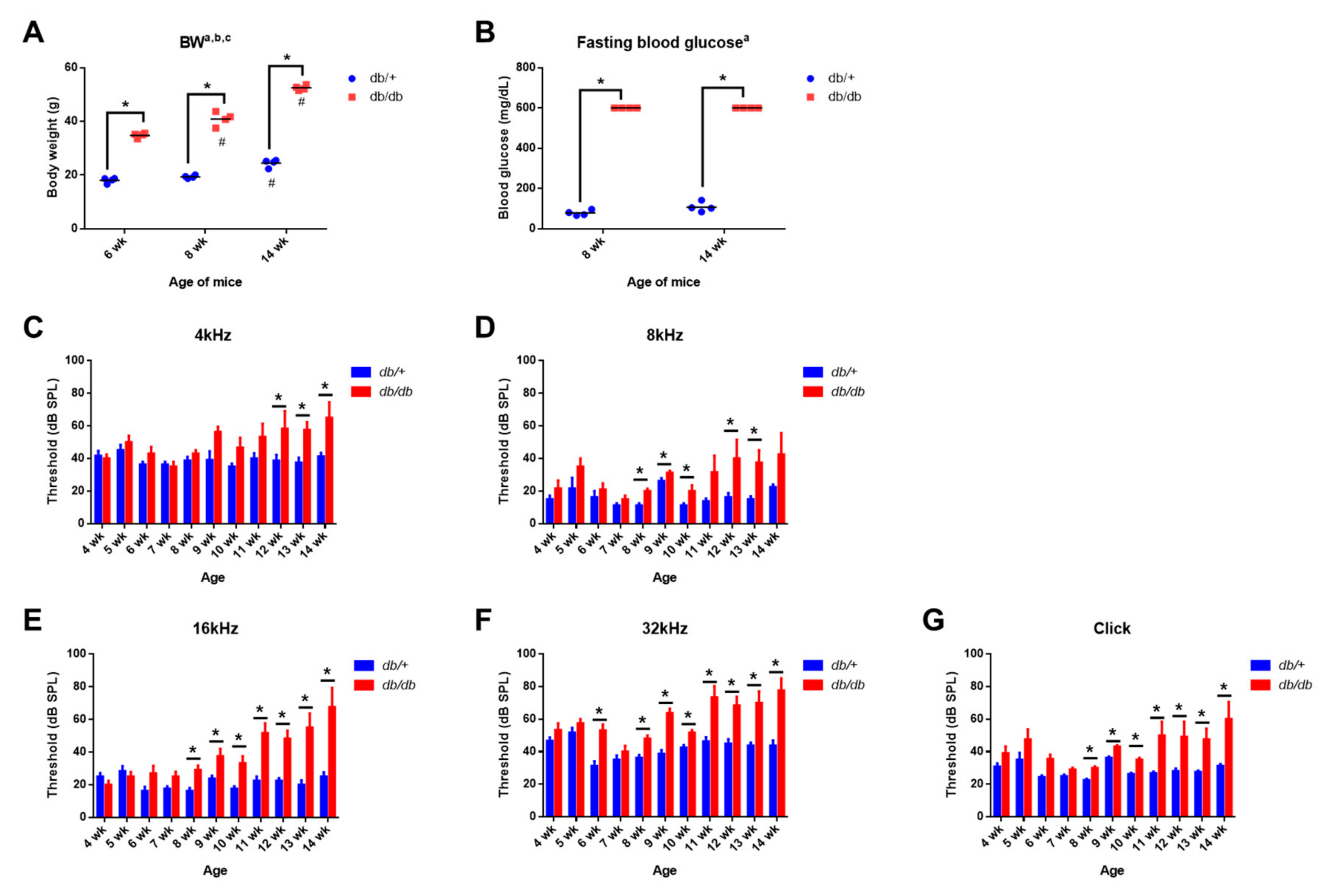
2.1. Body Weight and Blood Glucose Levels in Control and Diabetic Mice
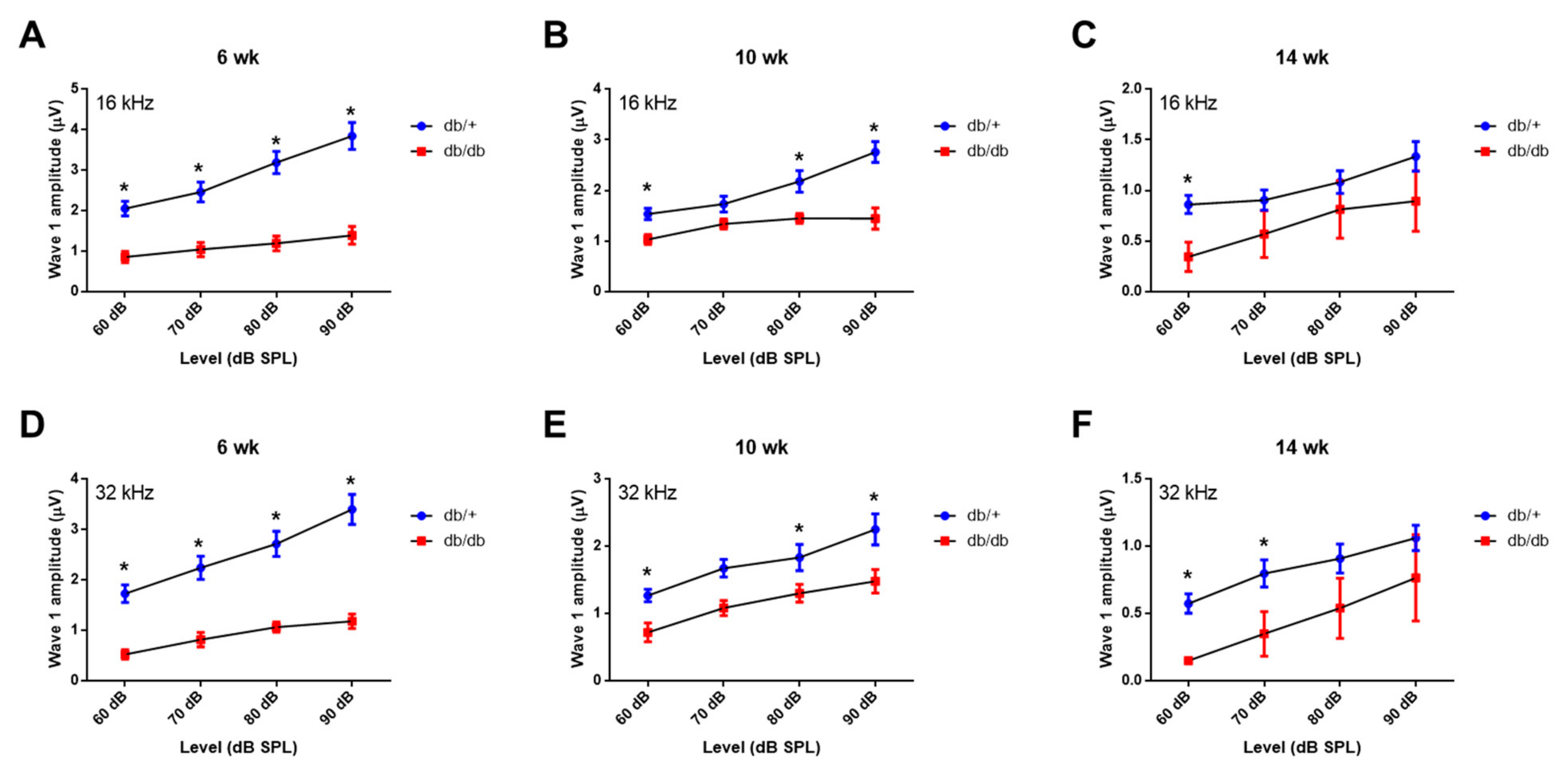
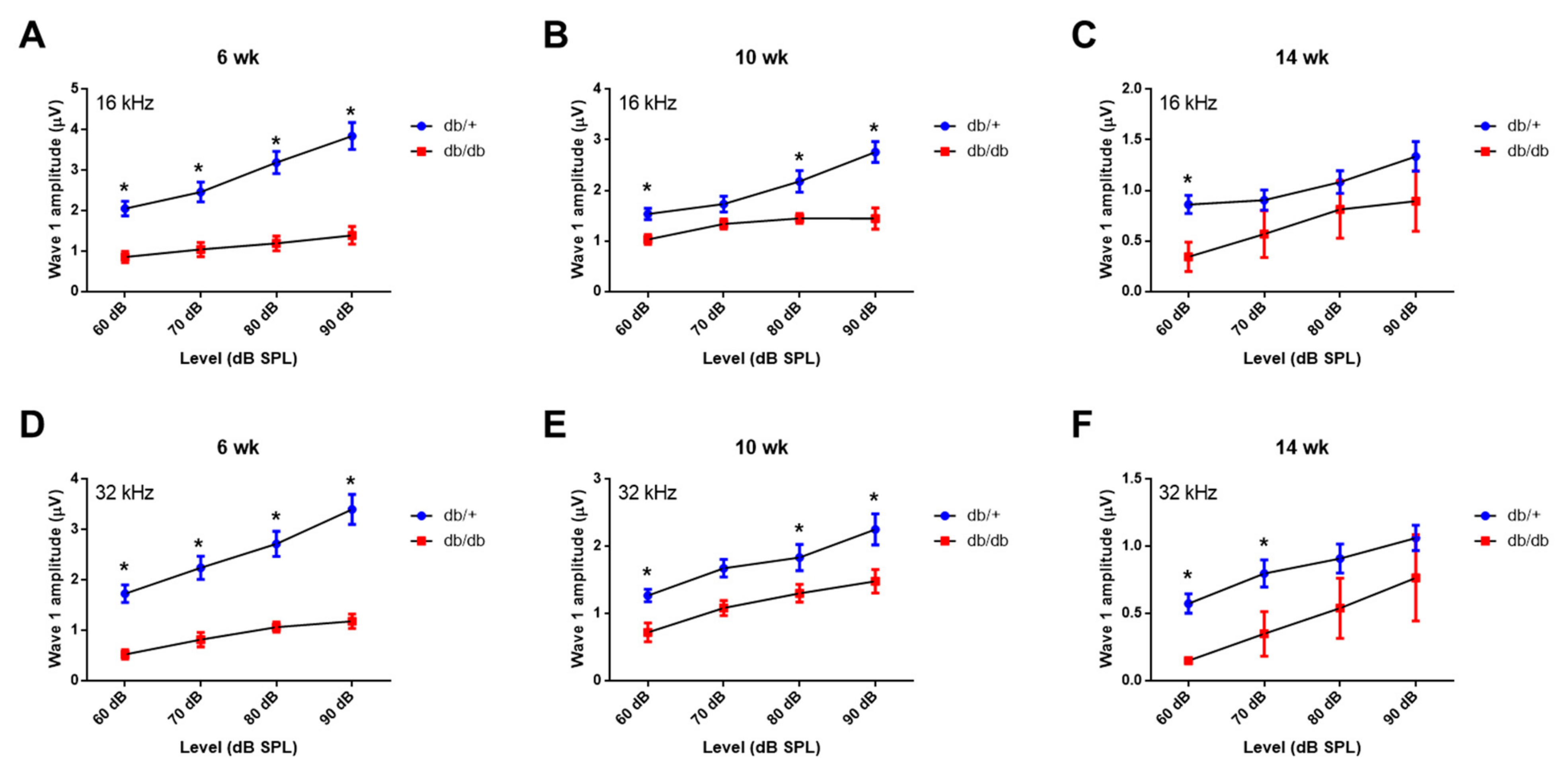
2.2. Weekly ABR Threshold Measurements in Wild-Type and Diabetic Mice
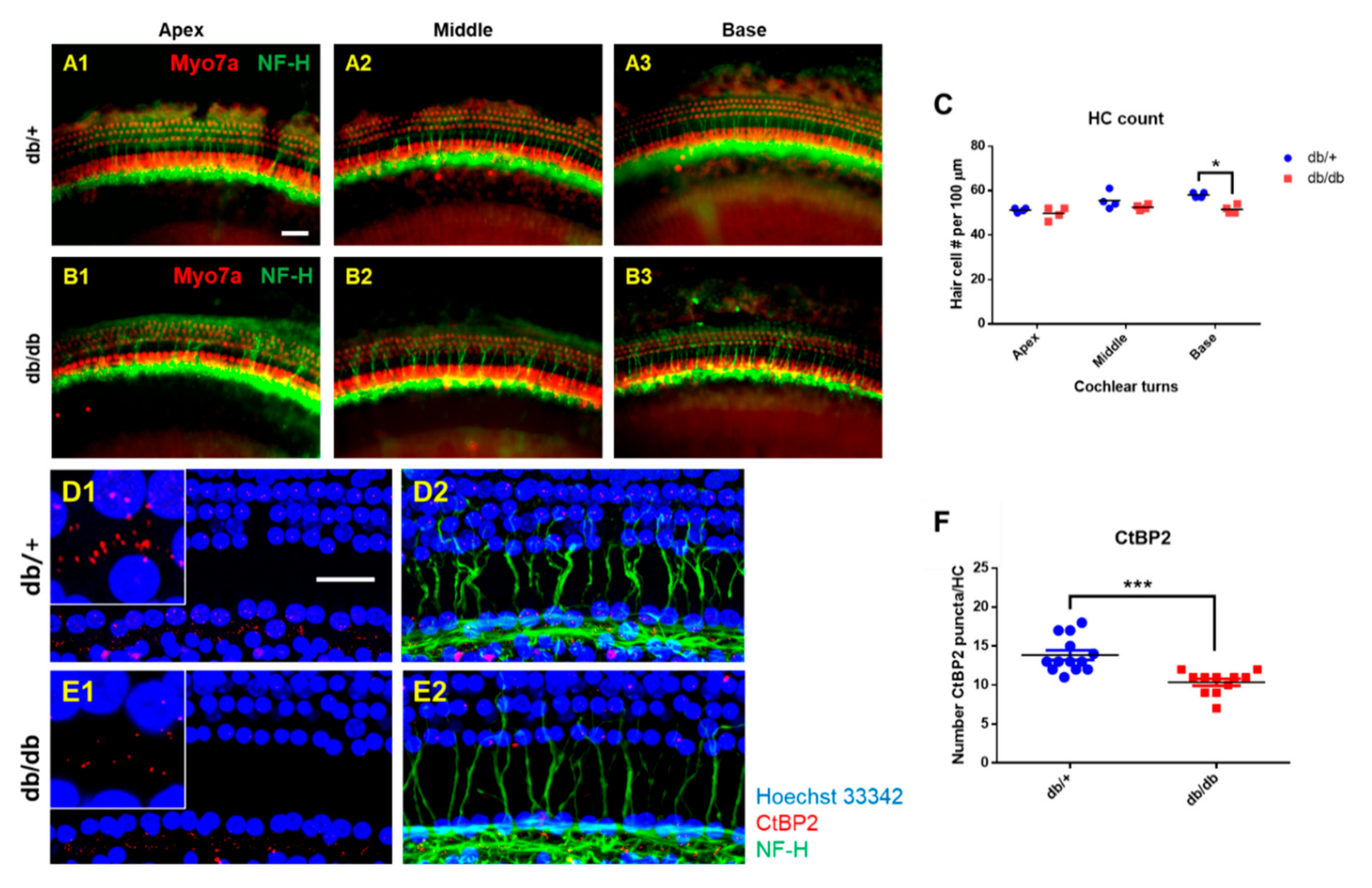
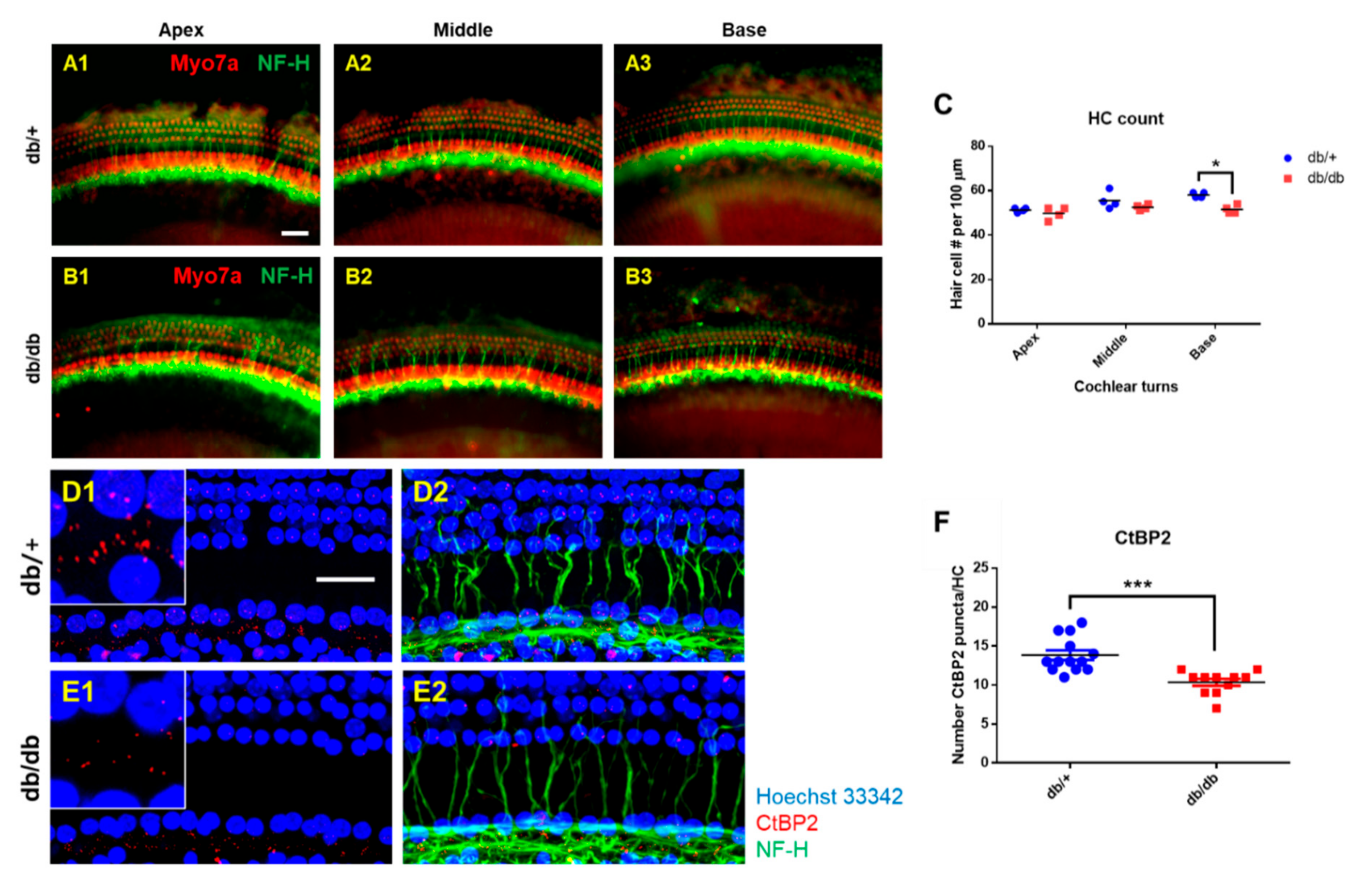
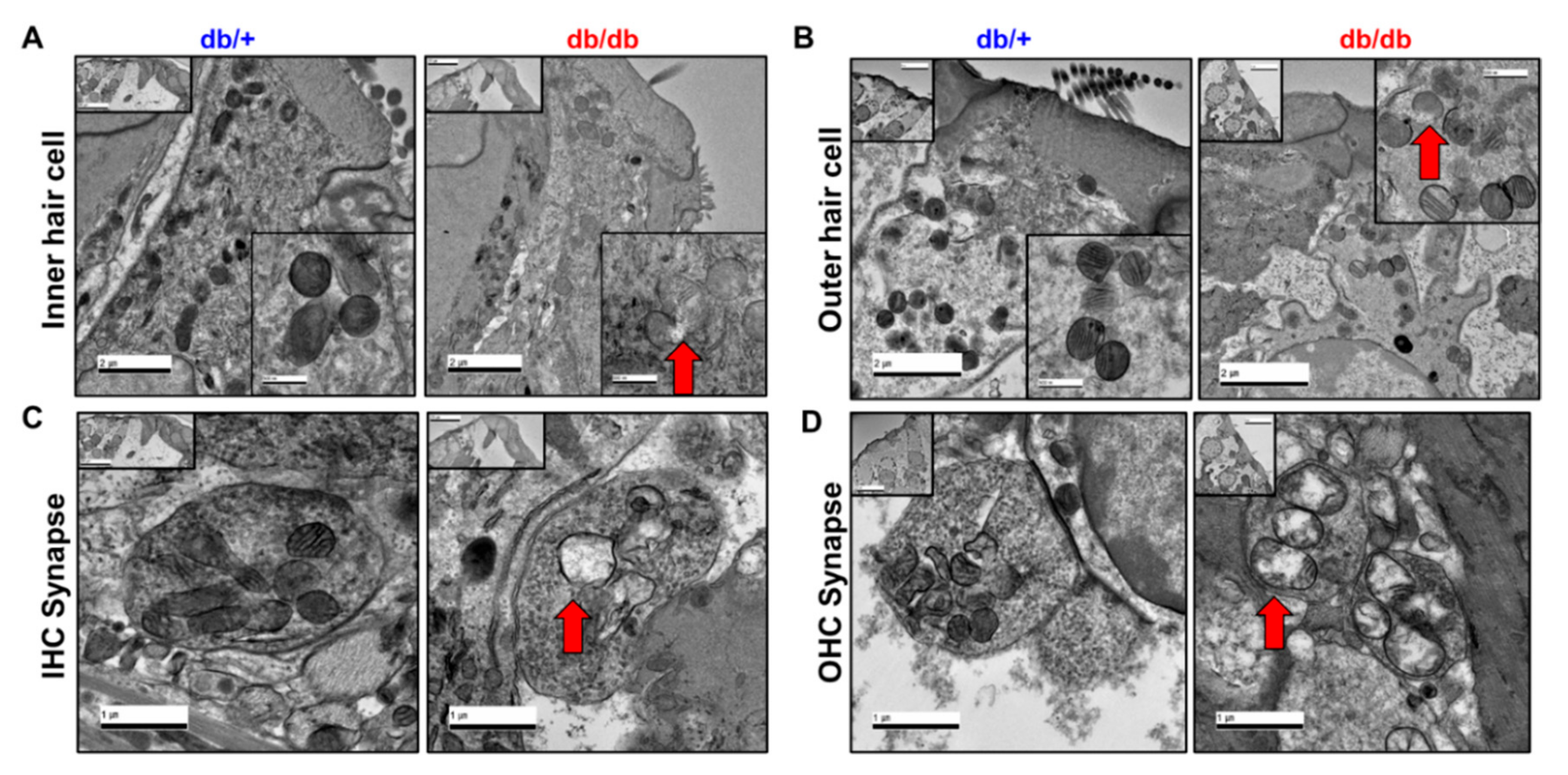
2.3. Hair Cell Loss and Synaptopathy in the Cochlea of Diabetic Mice
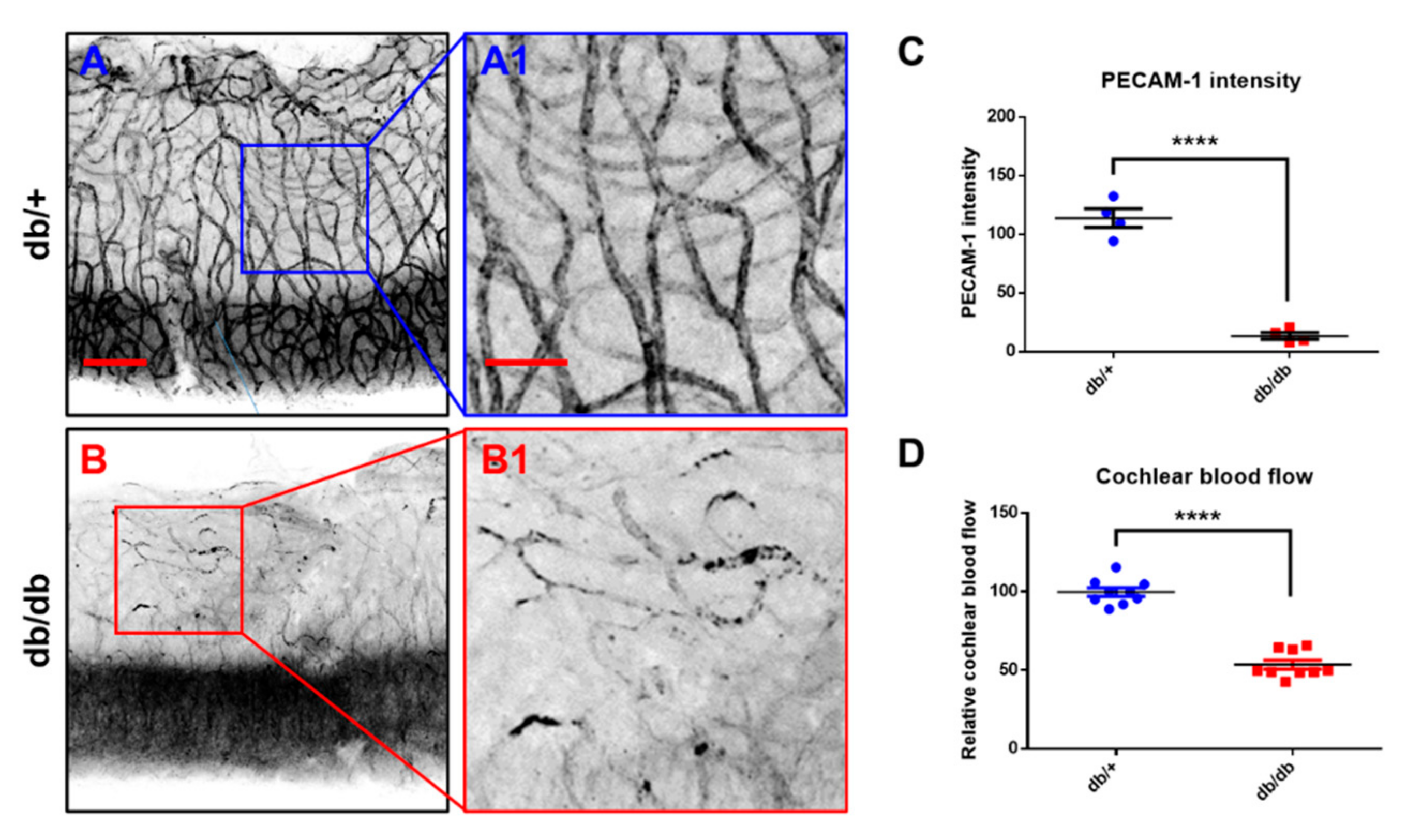
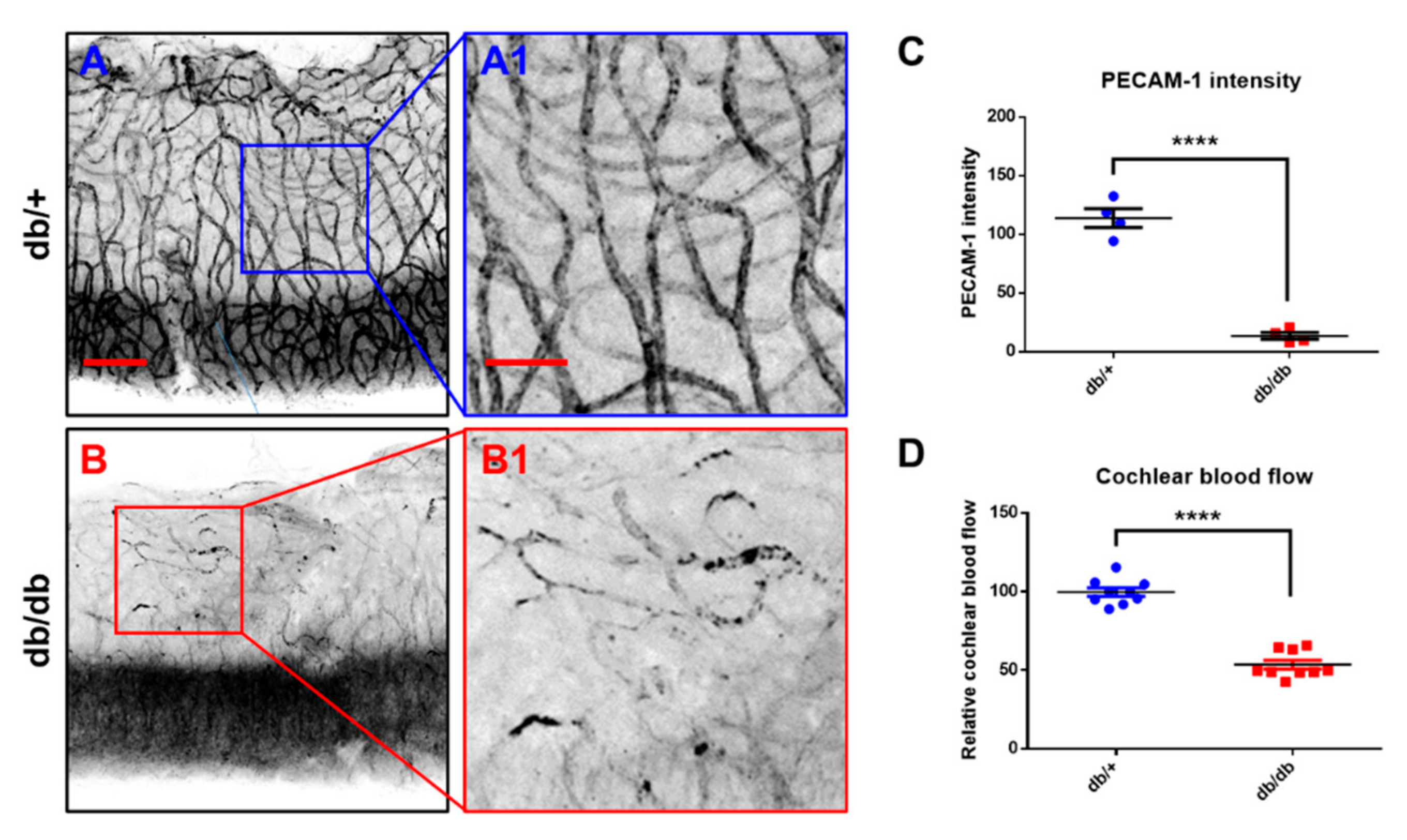
2.4. Microvasculature and Cochlear Blood Flow
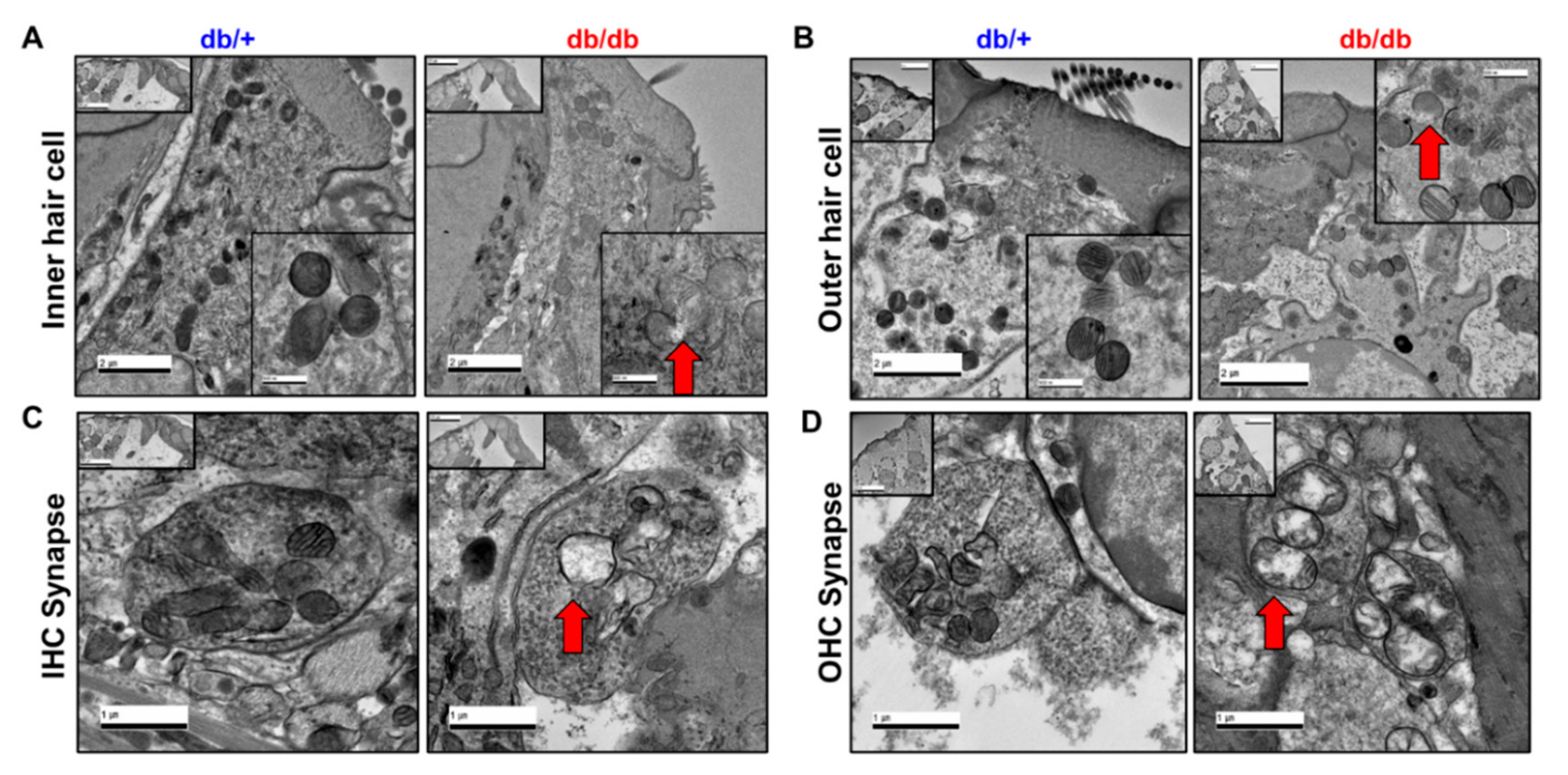
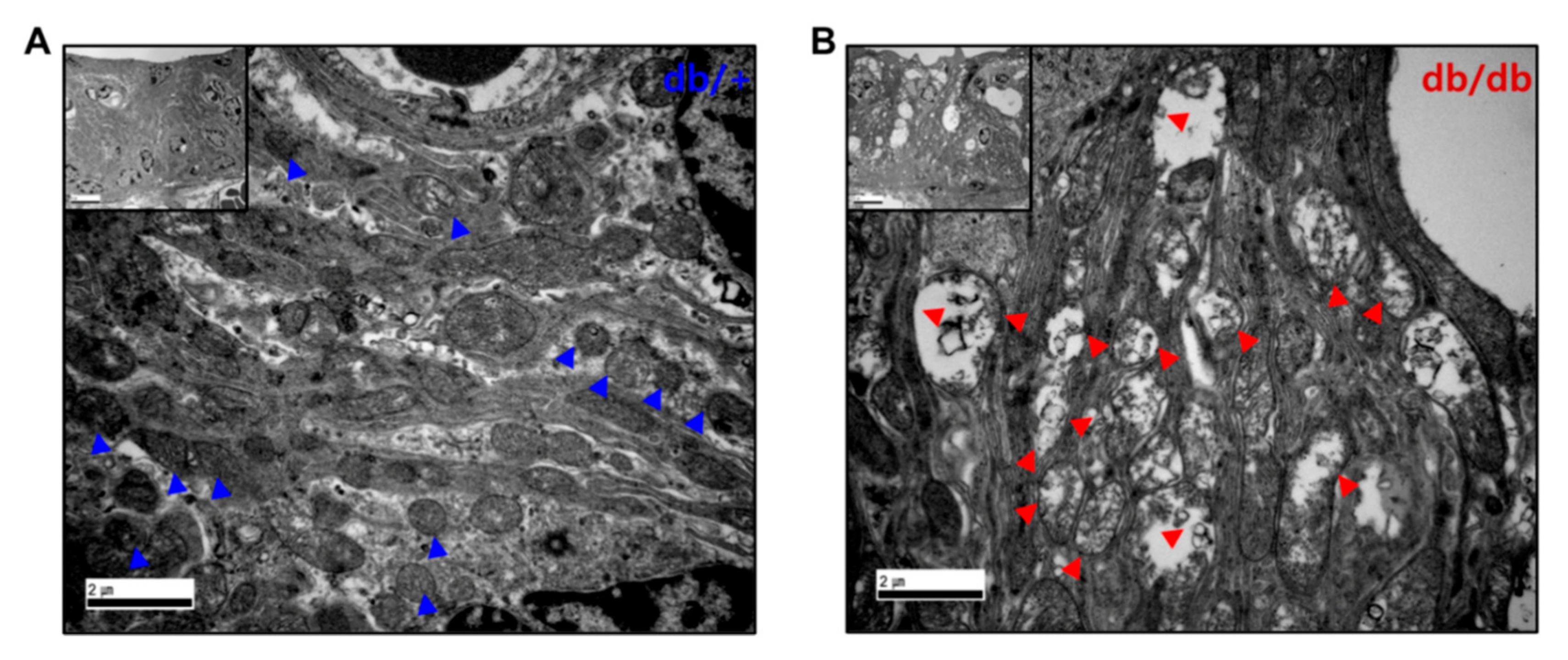
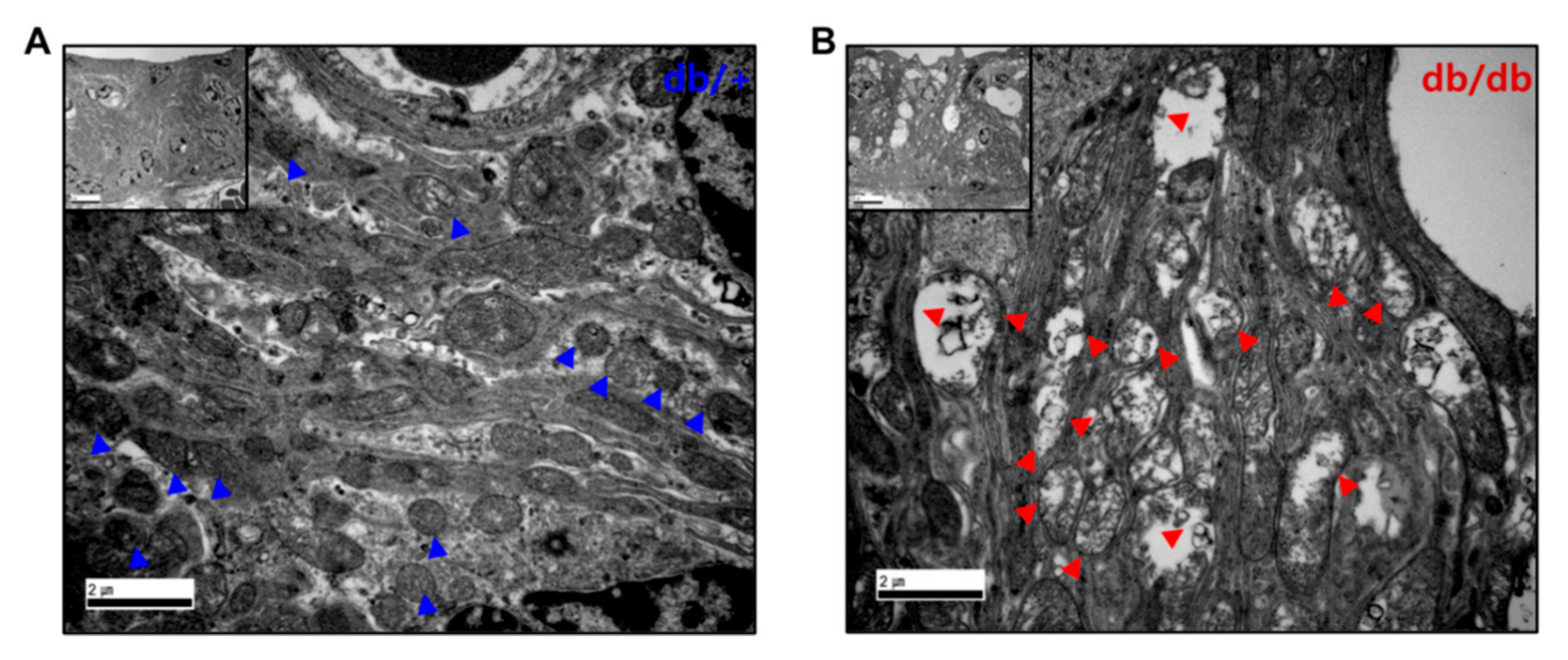
2.5. Mitochondrial Damage in the Cochlea of db/db Mice
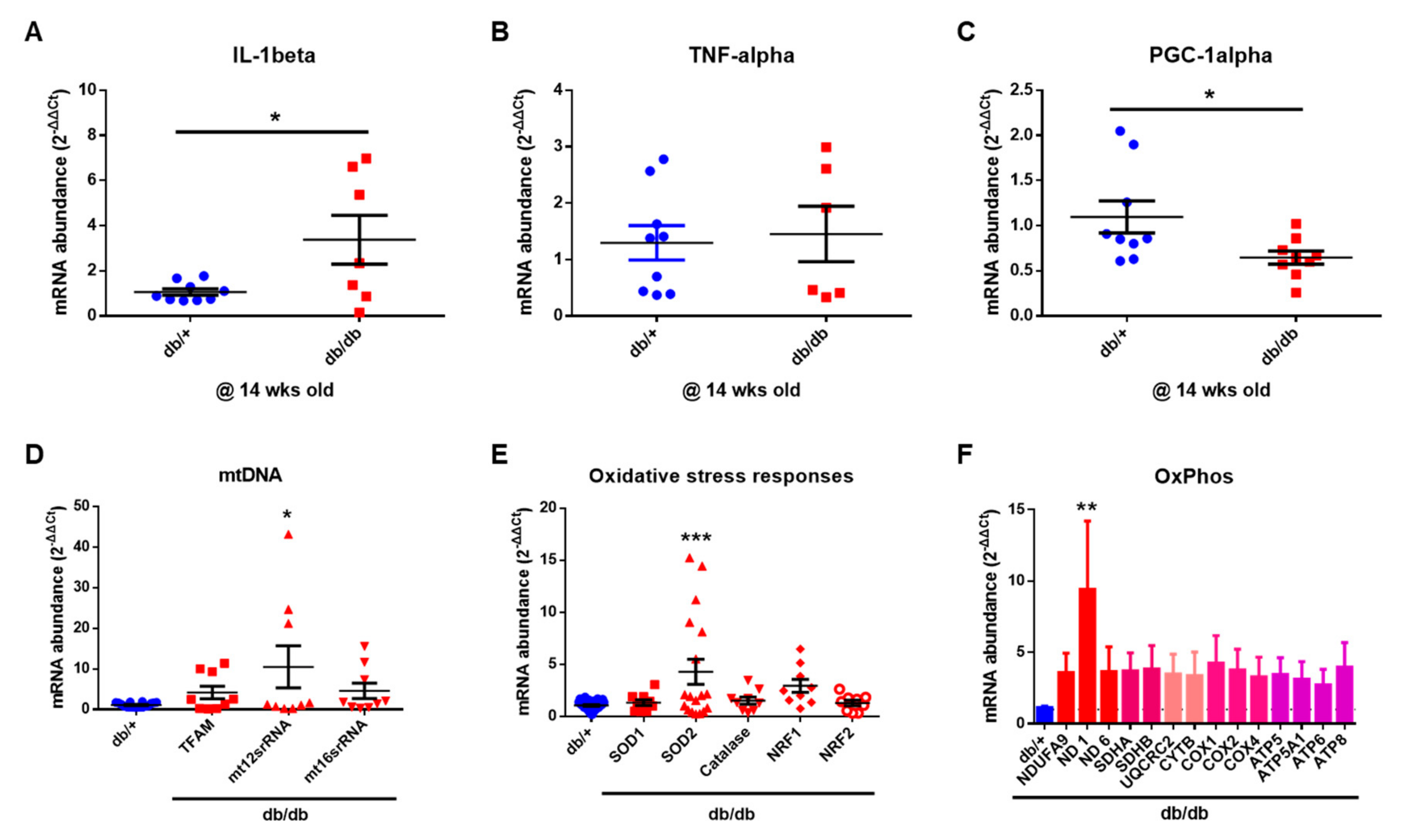
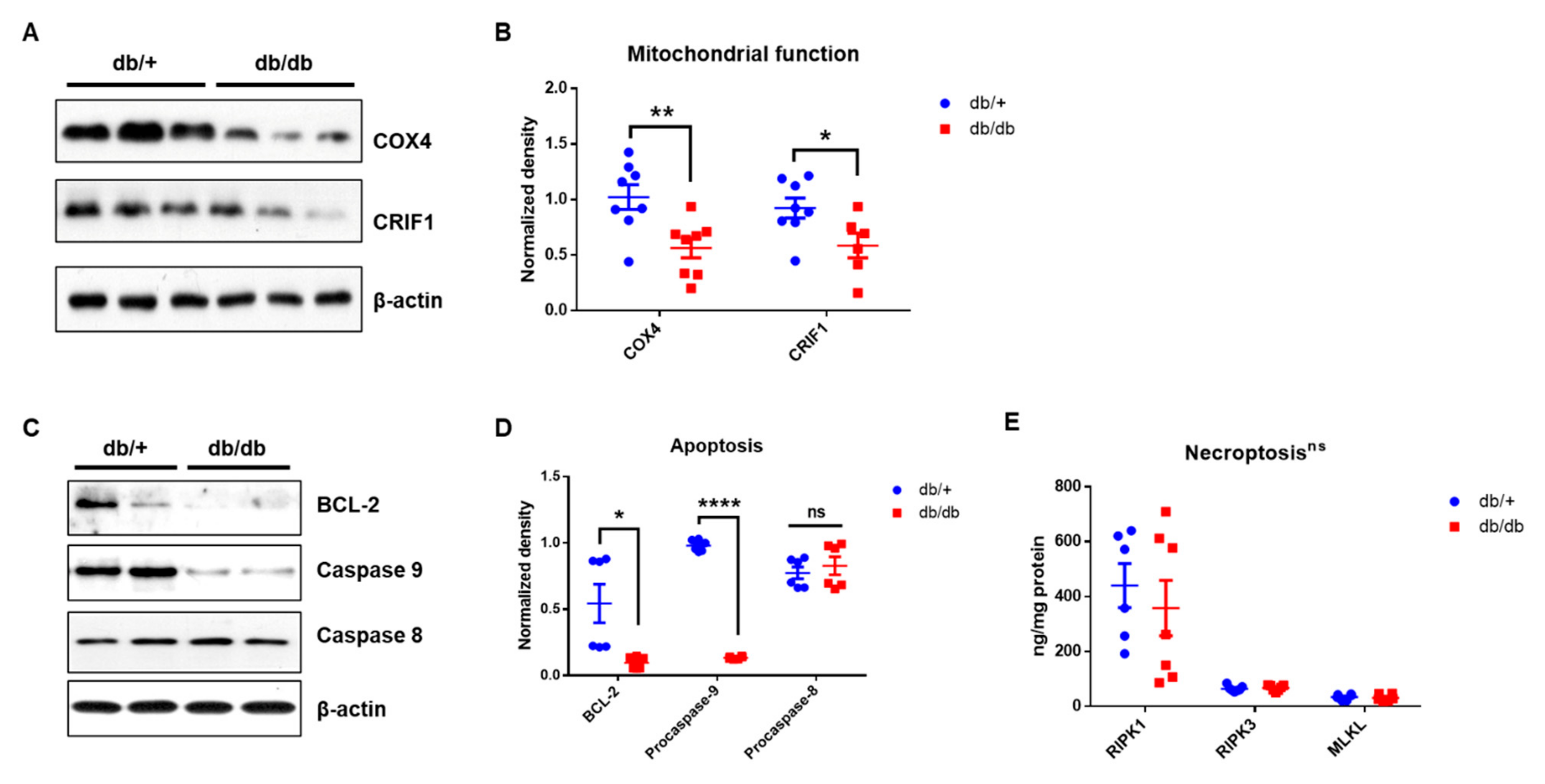
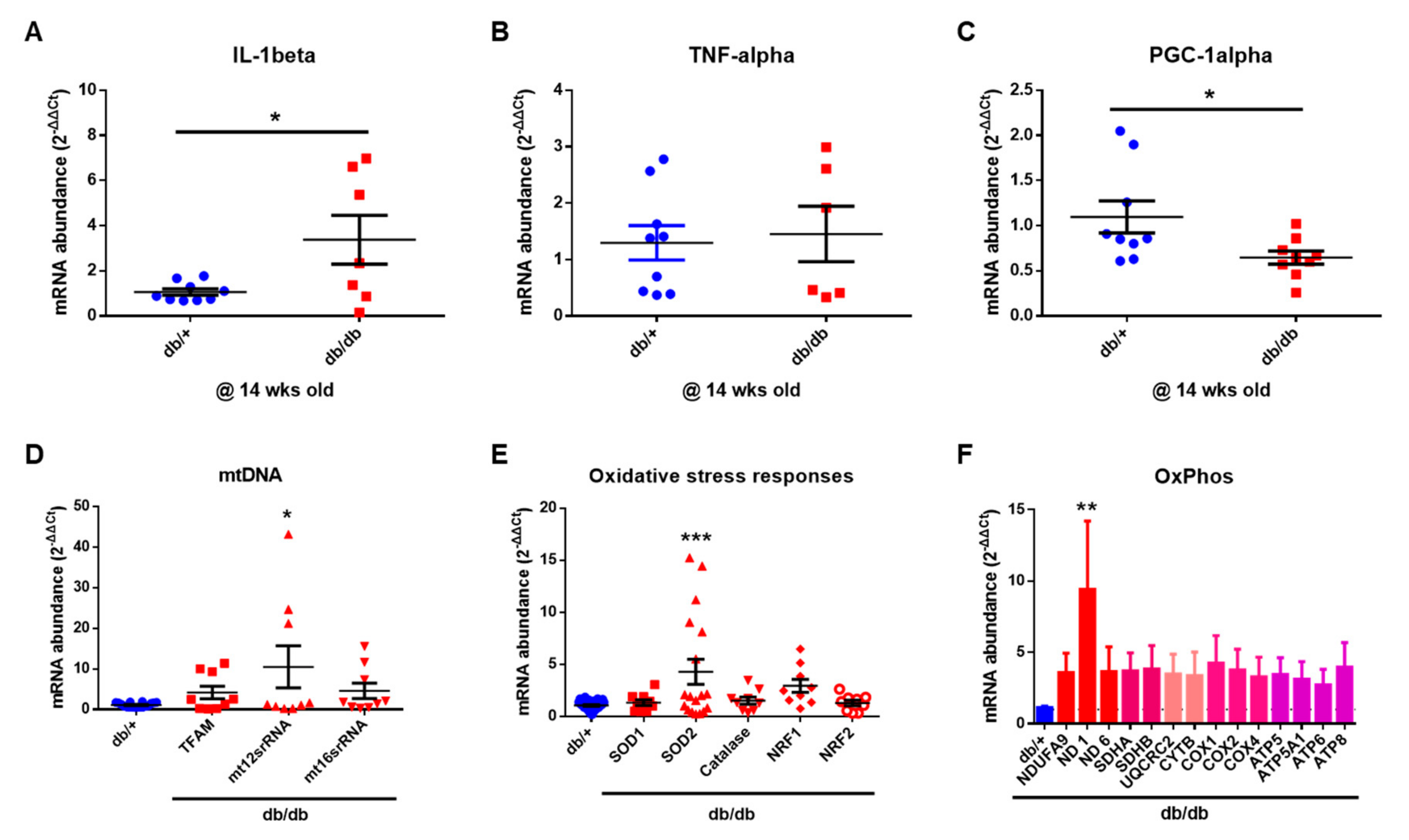
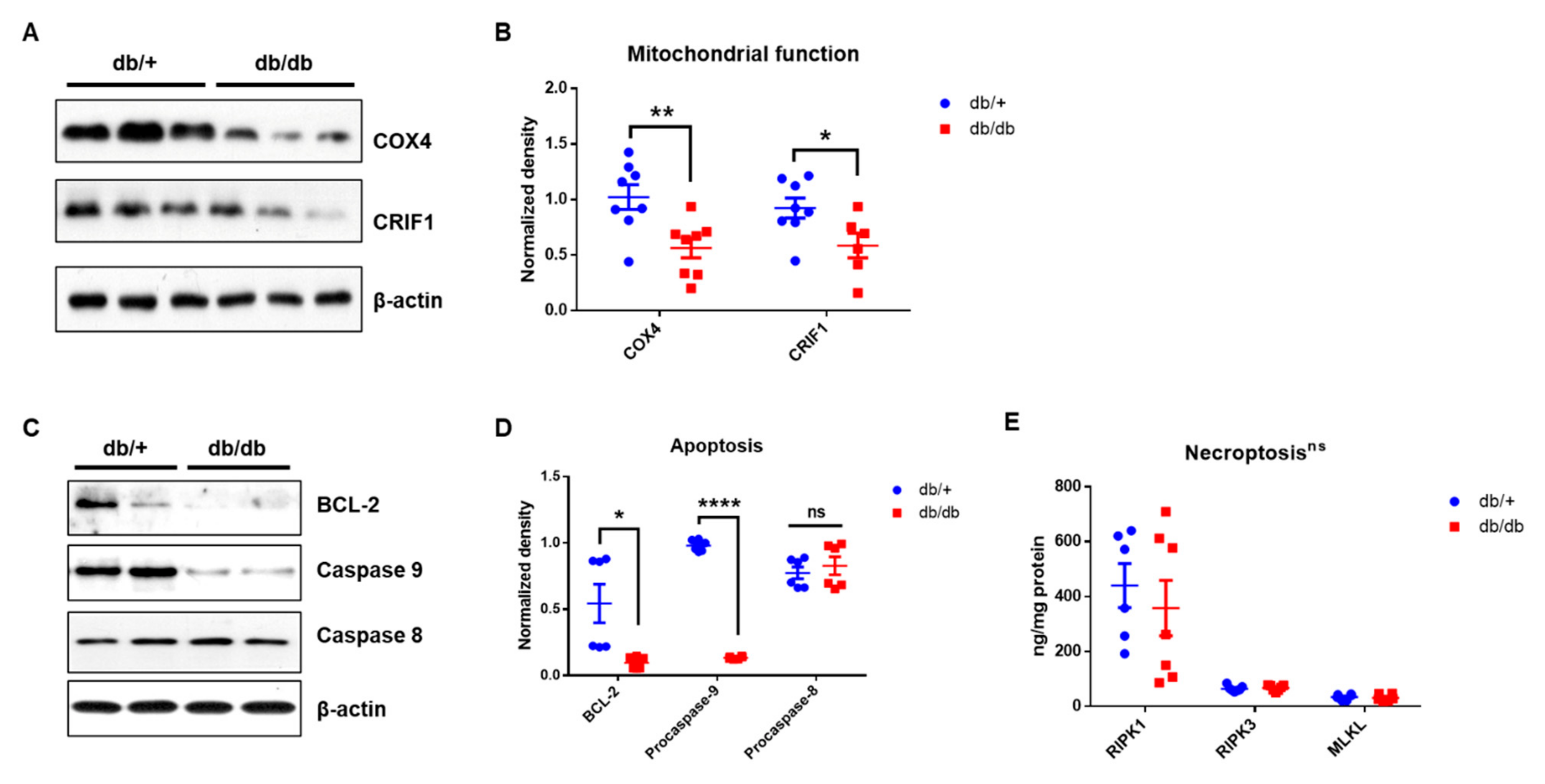
2.6. Mitochondrial Dysfunction in the Cochlea of db/db Mice
2.7. Apoptotic Cell Death, However, Not Necroptosis, in Diabetic Cochlea
3. Discussion
4. Materials and Methods
4.1. Research Design and Experimental Animals
4.2. Data and Resource Availability
4.3. Auditory Brainstem Response
4.4. Immunostaining
4.5. Transmission Electron Microscope (TEM)
4.6. Measurement of Cochlear Blood Flow
4.7. Quantitative Real Time Polymerase Chain Reaction (qRT-PCR)
4.8. Western Blotting
4.9. Measurement of Tissue Levels of RIPK1, RIPK3 and MLKL
4.10. Image Processing and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Aimoni, C.; Bianchini, C.; Borin, M.; Ciorba, A.; Fellin, R.; Martini, A.; Scanelli, G.; Volpato, S. Diabetes, cardiovascular risk factors and idiopathic sudden sensorineural hearing loss: A case-control study. Audiol. Neurotol. 2010, 15, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, K.E.; Hoffman, H.J.; Cowie, C.C. Diabetes and hearing impairment in the United States: Audiometric evidence from the National Health and Nutrition Examination Survey, 1999 to 2004. Ann. Intern. Med. 2008, 149, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, C.; Kodama, S.; Tanaka, S.; Fujihara, K.; Hirasawa, R.; Yachi, Y.; Shimano, H.; Yamada, N.; Saito, K.; Sone, H. Diabetes and risk of hearing impairment in adults: A meta-analysis. J. Clin. Endocrinol. Metab. 2013, 98, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Wang, Z.; Mao, Z.; Zhang, P.; Wang, C.; Liu, A.; Yuan, G. Hearing Loss in Type 2 Diabetes in Association with Diabetic Neuropathy. Arch. Med. Res. 2017, 48, 631–637. [Google Scholar] [CrossRef]
- Lerman-Garber, I.; Cuevas-Ramos, D.; Valdes, S.; Enriquez, L.; Lobato, M.; Osornio, M.; Escobedo, A.R.; Pascual-Ramos, V.; Mehta, R.; Ramirez-Anguiano, J.; et al. Sensorineural hearing loss—A common finding in early-onset type 2 diabetes mellitus. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2012, 18, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Elamin, A.; Fadlallah, M.; Tuevmo, T. Hearing loss in children with type 1 diabetes. Indian Pediatr. 2005, 42, 15–21. [Google Scholar] [PubMed]
- Ferrer, J.P.; Biurrun, O.; Lorente, J.; Conget, J.I.; de Espana, R.; Esmatjes, E.; Gomis, R. Auditory function in young patients with type 1 diabetes mellitus. Diabetes Res. Clin. Pract. 1991, 11, 17–22. [Google Scholar] [CrossRef]
- Gao, C.; Sun, W.; Christofidou-Solomidou, M.; Sawada, M.; Newman, D.K.; Bergom, C.; Albelda, S.M.; Matsuyama, S.; Newman, P.J. PECAM-1 functions as a specific and potent inhibitor of mitochondrial-dependent apoptosis. Blood 2003, 102, 169–179. [Google Scholar] [CrossRef]
- Falati, S.; Patil, S.; Gross, P.L.; Stapleton, M.; Merrill-Skoloff, G.; Barrett, N.E.; Pixton, K.L.; Weiler, H.; Cooley, B.; Newman, D.K.; et al. Platelet PECAM-1 inhibits thrombus formation in vivo. Blood 2006, 107, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Byun, J.; Son, S.; Cha, M.; Shong, M.; Hwang, Y.; Kim, Y.; Ryu, H.; Moon, M.; Kim, K.; Mook-Jung, I. CR6-interacting factor 1 is a key regulator in A β-induced mitochondrial disruption and pathogenesis of Alzheimer’s disease. Cell Death Differ. 2015, 22, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Joung, K.H.; Ryu, M.J.; Kim, S.J.; Kim, H.; Chung, H.K.; Lee, M.H.; Lee, S.E.; Choi, M.J.; Chang, J.Y.; et al. Disruption of CR6-interacting factor-1 (CRIF1) in mouse islet beta cells leads to mitochondrial diabetes with progressive beta cell failure. Diabetologia 2015, 58, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kwon, M.-C.; Ryu, M.J.; Chung, H.K.; Tadi, S.; Kim, Y.K.; Kim, J.M.; Lee, S.H.; Park, J.H.; Kweon, G.R. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab. 2012, 16, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, M.J.; Kim, S.J.; Kim, Y.K.; Choi, M.J.; Tadi, S.; Lee, M.H.; Lee, S.E.; Chung, H.K.; Jung, S.B.; Kim, H.J.; et al. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet. 2013, 9, e1003356. [Google Scholar] [CrossRef]
- Furman, A.C.; Kujawa, S.G.; Liberman, M.C. Noise-induced cochlear neuropathy is selective for fibers with low spontaneous rates. J. Neurophysiol. 2013, 110, 577–586. [Google Scholar] [CrossRef]
- Lin, H.W.; Furman, A.C.; Kujawa, S.G.; Liberman, M.C. Primary neural degeneration in the Guinea pig cochlea after reversible noise-induced threshold shift. J. Assoc. Res. Otolaryngol. 2011, 12, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: Cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberman, M.C.; Kujawa, S.G. Cochlear synaptopathy in acquired sensorineural hearing loss: Manifestations and mechanisms. Hear. Res. 2017, 349, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Liberman, L.D.; Suzuki, J.; Liberman, M.C. Dynamics of cochlear synaptopathy after acoustic overexposure. J. Assoc. Res. Otolaryngol. 2015, 16, 205–219. [Google Scholar] [CrossRef]
- Kujawa, S.G.; Liberman, M.C. Synaptopathy in the noise-exposed and aging cochlea: Primary neural degeneration in acquired sensorineural hearing loss. Hear. Res. 2015, 330, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Deng, X.; Shi, Y.; Su, Y.; Wei, J.; Duan, H. PGC-1α, glucose metabolism and type 2 diabetes mellitus. J. Endocrinol. 2016, 229, R99–R115. [Google Scholar] [CrossRef]
- Zhang, L.N.; Zhou, H.Y.; Fu, Y.Y.; Li, Y.Y.; Wu, F.; Gu, M.; Wu, L.Y.; Xia, C.M.; Dong, T.C.; Li, J.Y.; et al. Novel small-molecule PGC-1alpha transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes 2013, 62, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Park, J.-S.; Deng, J.-H.; Bai, Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 2006, 38, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontanesi, F.; Soto, I.C.; Barrientos, A. Cytochrome c oxidase biogenesis: New levels of regulation. IUBMB Life 2008, 60, 557–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pålbrink, A.-K.; Kopietz, F.; Morén, B.; In’t Zandt, R.; Kalinec, F.; Stenkula, K.; Göransson, O.; Holm, C.; Magnusson, M.; Degerman, E. Inner ear is a target for insulin signaling and insulin resistance: Evidence from mice and auditory HEI-OC1 cells. BMJ Open Diabetes Res. Care 2020, 8, e000820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Chang, Y.; Li, X.; Aiken, S.; Liu, L.; Wang, J. Cochlear Synaptopathy and Noise-Induced Hidden Hearing Loss. Neural Plast. 2016, 2016, 6143164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobel, M.; Le Prell, C.G.; Liu, J.; Hawks, J.W.; Bao, J. Noise-induced cochlear synaptopathy: Past findings and future studies. Hear. Res. 2017, 349, 148–154. [Google Scholar] [CrossRef]
- Sergeyenko, Y.; Lall, K.; Liberman, M.C.; Kujawa, S.G. Age-related cochlear synaptopathy: An early-onset contributor to auditory functional decline. J. Neurosci. 2013, 33, 13686–13694. [Google Scholar] [CrossRef] [PubMed]
- Valero, M.D.; Burton, J.A.; Hauser, S.N.; Hackett, T.A.; Ramachandran, R.; Liberman, M.C. Noise-induced cochlear synaptopathy in rhesus monkeys (Macaca mulatta). Hear. Res. 2017, 353, 213–223. [Google Scholar] [CrossRef]
- Han, W.K.; Kim, E.H.; Shin, S.A.; Shin, D.S.; Kim, B.J.; Lyu, A.R.; Park, Y.H. Susceptibility of Diabetic Mice to Noise Trauma. BioMed Res. Int. 2018, 2018, 7601232. [Google Scholar] [CrossRef] [Green Version]
- Lyu, A.R.; Kim, T.H.; Park, S.J.; Shin, S.A.; Jeong, S.H.; Yu, Y.; Huh, Y.H.; Je, A.R.; Park, M.J.; Park, Y.H. Mitochondrial Damage and Necroptosis in Aging Cochlea. Int. J. Mol. Sci. 2020, 21, 2505. [Google Scholar] [CrossRef] [Green Version]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of PECAM-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K.; Fung, M.K.; Lo, A.C.; Lam, T.T.; So, K.F.; Chung, S.S.; Chung, S.K. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes 2005, 54, 3119–3125. [Google Scholar] [CrossRef] [Green Version]
- Eshaq, R.S.; Harris, N.R. Loss of Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1) in the Diabetic Retina: Role of Matrix Metalloproteinases. Investig. Ophthalmol. Vis. Sci. 2019, 60, 748–760. [Google Scholar] [CrossRef]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R. FLICE, a novel FADD-homologous ICE/CED-3–like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8–FLIP L complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Maiese, K. New Insights for Oxidative Stress and Diabetes Mellitus. Oxidative Med. Cell. Longev. 2015, 2015, 875961. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Scott, N.A.; Fynch, S.; Elkerbout, L.; Wong, W.W.-L.; Mason, K.D.; Strasser, A.; Huang, D.C.; Kay, T.W.; Thomas, H.E. Autoreactive T cells induce necrosis and not BCL-2-regulated or death receptor-mediated apoptosis or RIPK3-dependent necroptosis of transplanted islets in a mouse model of type 1 diabetes. Diabetologia 2015, 58, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Lyu, A.R.; Kim, D.H.; Lee, S.H.; Shin, D.S.; Shin, S.A.; Park, Y.H. Effects of dexamethasone on intracochlear inflammation and residual hearing after cochleostomy: A comparison of administration routes. BioMed Res. Int. 2018, 13, e0195230. [Google Scholar] [CrossRef] [PubMed]
- Mehraei, G.; Hickox, A.E.; Bharadwaj, H.M.; Goldberg, H.; Verhulst, S.; Liberman, M.C.; Shinn-Cunningham, B.G. Auditory brainstem response latency in noise as a marker of cochlear synaptopathy. J. Neurosci. 2016, 36, 3755–3764. [Google Scholar] [CrossRef]
- Spurr, A.R. A low-viscosity epoxy resin embedding medium for electron microscopy. J. Ultrastruct. Res. 1969, 26, 31–43. [Google Scholar] [CrossRef]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, A.-R.; Kim, T.-H.; Shin, S.-A.; Kim, E.-H.; Yu, Y.; Gajbhiye, A.; Kwon, H.-C.; Je, A.R.; Huh, Y.H.; Park, M.J.; et al. Hearing Impairment in a Mouse Model of Diabetes Is Associated with Mitochondrial Dysfunction, Synaptopathy, and Activation of the Intrinsic Apoptosis Pathway. Int. J. Mol. Sci. 2021, 22, 8807. https://doi.org/10.3390/ijms22168807
Lyu A-R, Kim T-H, Shin S-A, Kim E-H, Yu Y, Gajbhiye A, Kwon H-C, Je AR, Huh YH, Park MJ, et al. Hearing Impairment in a Mouse Model of Diabetes Is Associated with Mitochondrial Dysfunction, Synaptopathy, and Activation of the Intrinsic Apoptosis Pathway. International Journal of Molecular Sciences. 2021; 22(16):8807. https://doi.org/10.3390/ijms22168807
Chicago/Turabian StyleLyu, Ah-Ra, Tae-Hwan Kim, Sun-Ae Shin, Eung-Hyub Kim, Yang Yu, Akanksha Gajbhiye, Hyuk-Chan Kwon, A Reum Je, Yang Hoon Huh, Min Jung Park, and et al. 2021. "Hearing Impairment in a Mouse Model of Diabetes Is Associated with Mitochondrial Dysfunction, Synaptopathy, and Activation of the Intrinsic Apoptosis Pathway" International Journal of Molecular Sciences 22, no. 16: 8807. https://doi.org/10.3390/ijms22168807
APA StyleLyu, A.-R., Kim, T.-H., Shin, S.-A., Kim, E.-H., Yu, Y., Gajbhiye, A., Kwon, H.-C., Je, A. R., Huh, Y. H., Park, M. J., & Park, Y.-H. (2021). Hearing Impairment in a Mouse Model of Diabetes Is Associated with Mitochondrial Dysfunction, Synaptopathy, and Activation of the Intrinsic Apoptosis Pathway. International Journal of Molecular Sciences, 22(16), 8807. https://doi.org/10.3390/ijms22168807