
Evaluation of the Binding Kinetics of RHEB with mTORC1 by In-Cell and In Vitro Assays


Abstract
:1. Introduction
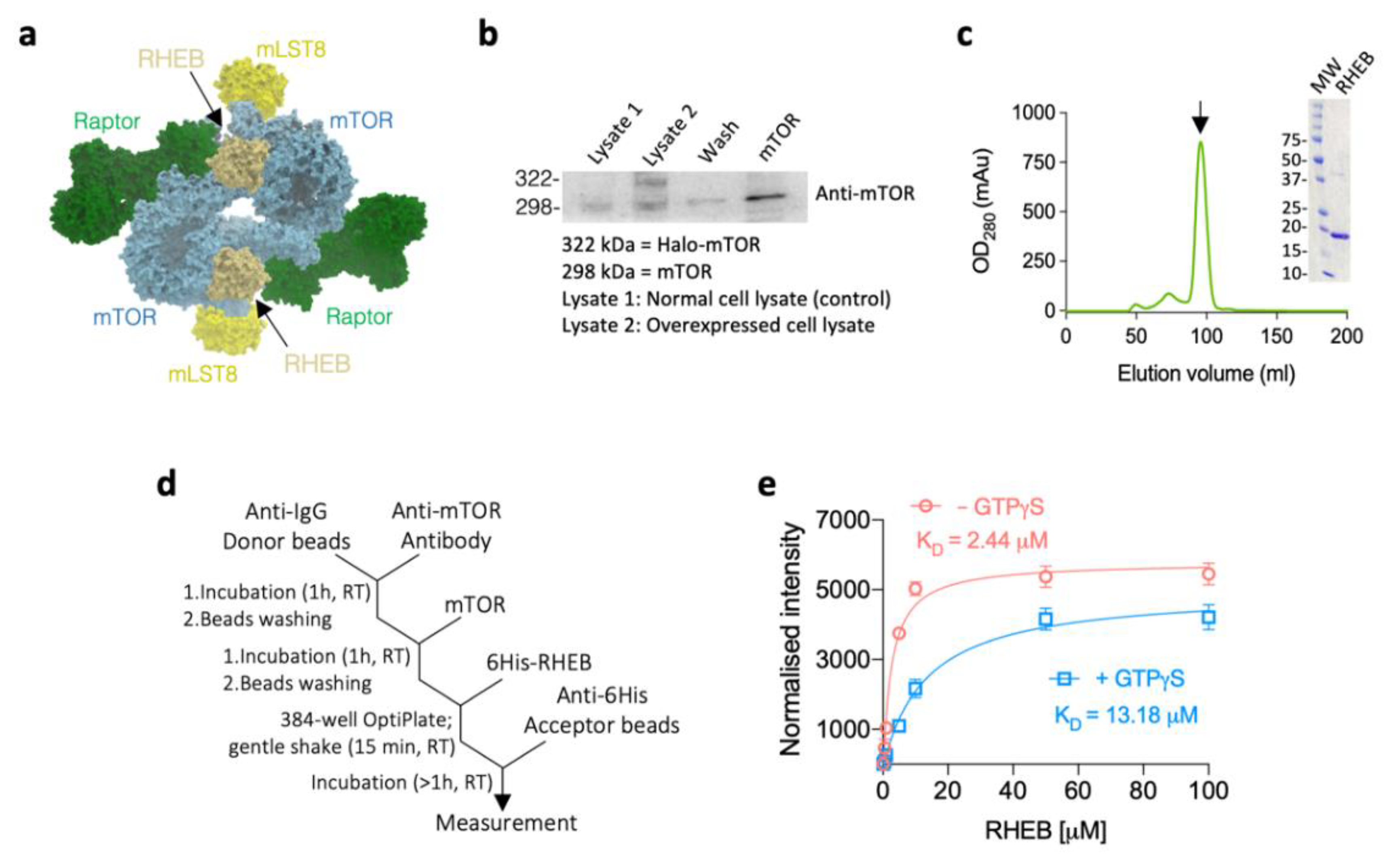
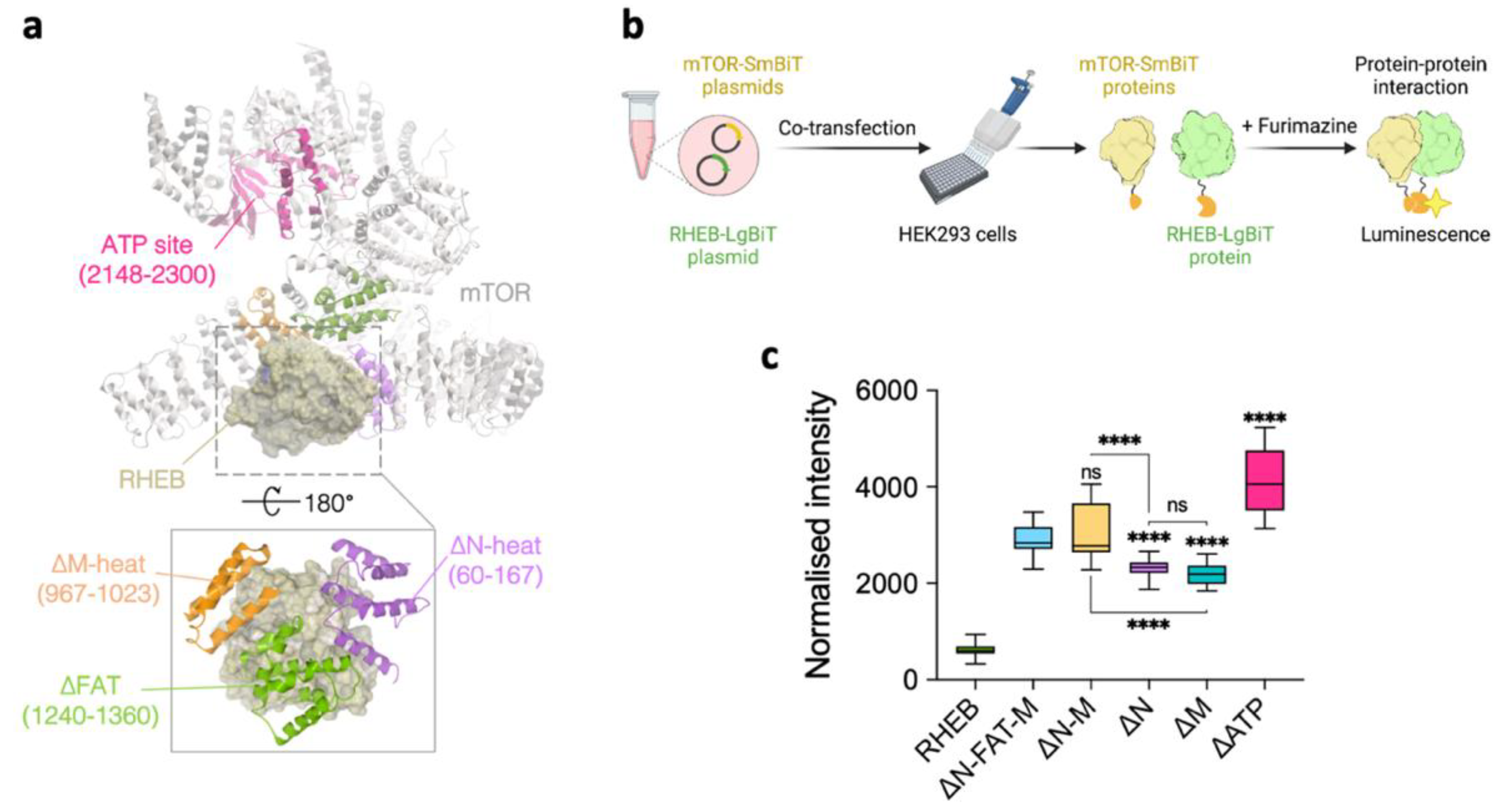
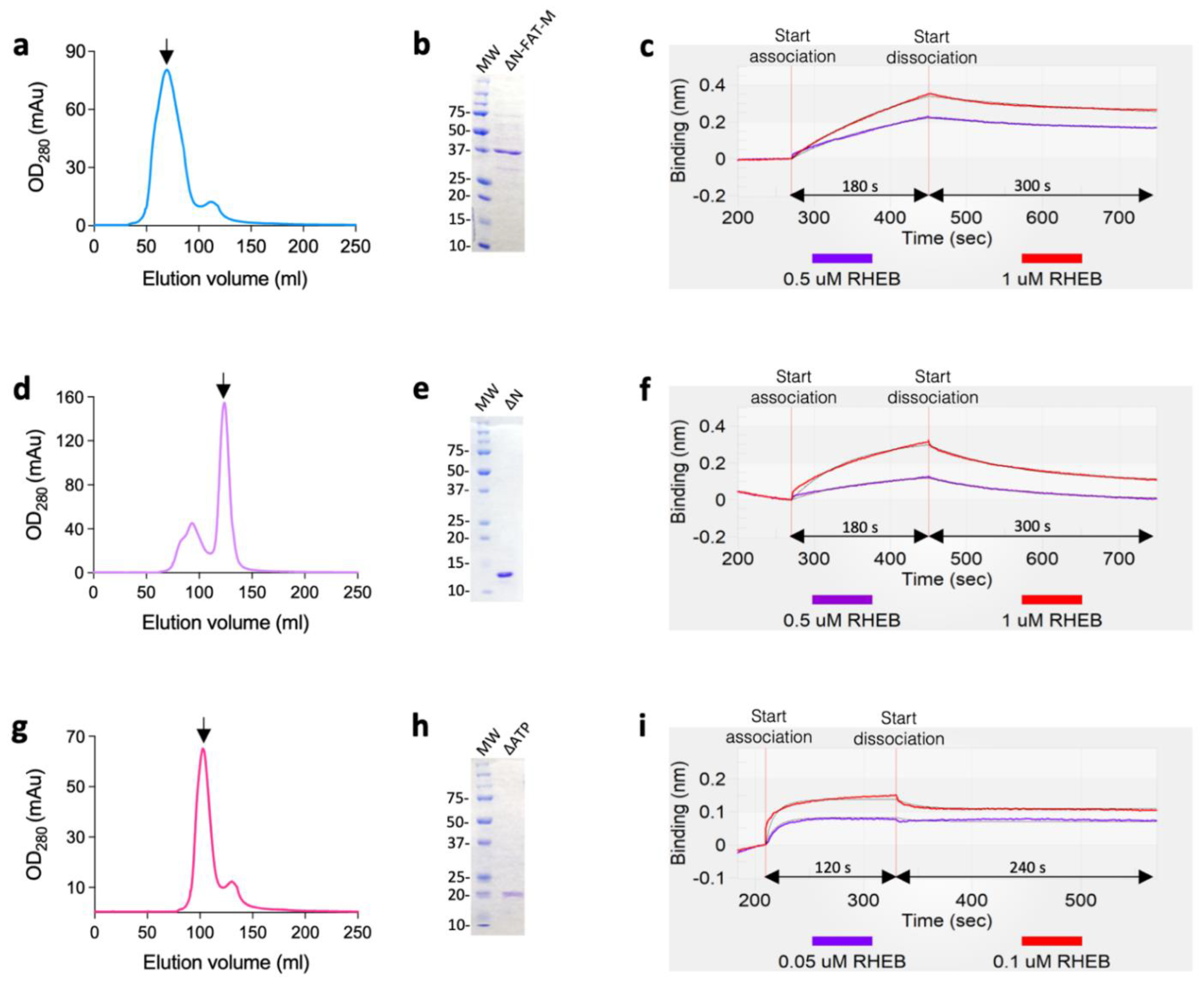
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. mTOR Expression and Purification
3.2.2. Western Blotting
3.2.3. Preparation of RHEB and Truncated mTOR Fragments
- Plasmid construction
- b.
- Protein expression
- c.
- Protein purification
3.2.4. His-Tag Cleavage from RHEB
3.2.5. RHEB Charging with GTPγS
3.2.6. AlphaLISA Assay for RHEB–mTOR Protein–Protein Interaction (PPI)
3.2.7. Preparation of Plasmids for In-Cell Protein–Protein Interaction (NanoBiT Assay)
3.2.8. In-Cell NanoBiT Assay
3.2.9. BLItz Measurements of RHEB Interactions with mTOR Truncates
3.2.10. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| ATP | Adenosine tri-phosphate |
| Cryo-EM | Cryo-Electron Micoroscopy |
| Da | Dalton |
| 4EB-P1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| FAT | Focal adhesion targeting domain |
| FBS | Fetal bovine serum |
| FKBP12 | FK506 binding protein of 12 kDa |
| FPLC | Fast protein liquid chromatography |
| GTPγS | Non-hydrolysable guanidine triphosphate |
| LgBiT | Large fragment of the NanoLuc binary technology (NanoBiT) |
| mTOR | Mammalian/mechanistic target of rapamycin |
| PI3K | Phosphoinositide 3-kinase |
| PPI | Protein–protein interaction |
| P/S | Penicillin/streptomycin |
| RT | Room temperature |
| SmBiT | Small fragment of the NanoLuc binary technology (NanoBiT) |
| RHEB | Ras homolog enriched in brain protein |
| S6K1 | Ribosomal protein S6 kinase 1 |
| TSC | Tuberous sclerosis complex |
| RAPTOR | Regulatory-associated protein of mTOR |
| DEPTOR | DEP domain containing protein 6 |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| MW-CO | Molecular weight-cut off |
| PRAS40 | 40 kDa proline-rich AKT substrate |
References
- Liu, G.Y.; Sabatini, D.M. mTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR Signalling and Cellular Metabolism are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Sabatini, D.M. Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9545–9550. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabatini, D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heard, J.J.; Fong, V.; Bathaie, S.Z.; Tamanoi, F. Recent progress in the study of the Rheb family GTPases. Cell Signal 2014, 26, 1950–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Ma, D.; Liu, A.; Shen, X.; Wang, Q.J.; Liu, Y.; Jiang, Y. RHEB Activates mTOR by Antagonizing its Endogenous Inhibitor, FKBP38. Science 2007, 318, 977–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.; Ortiz-Vega, S.; Lin, Y.; Avruch, J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 2005, 280, 23433–23436. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, S.J.; Narayan, S.; Molz, L.; Berstler, L.A.; Kang, S.A.; Vlasuk, G.P.; Saiah, E. A Small Molecule Inhibitor of Rheb Selectively Targets mTORC1 Signaling. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Castro, A.F.; Rebhun, J.F.; Clark, G.J.; Quilliam, L.A. RHEB Binds Tuberous Sclerosis Complex 2 (TSC2) and Promotes S6 Kinase Activation in a Rapamycin- and Farnesylation-Dependent Manner. J. Biol. Chem. 2003, 278, 32493–32496. [Google Scholar] [CrossRef] [Green Version]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi, L.F.; Bottino, A.M.S.; Dueholm, R.; Niehaus, A.; Overwijn, D.; Fils, A.P.; et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef]
- Anandapadamanaban, M.; Masson, G.R.; Perisic, O.; Berndt, A.; Kaufman, J.; Johnson, C.M.; Santhanam, B.; Rogala, K.B.; Sabatini, D.M.; Williams, R.L. Architecture of Human Rag GTPase Heterodimers and their Complex with mTORC1. Science 2019, 366, 203–210. [Google Scholar] [CrossRef]
- Ning, B.; Ren, X.; Hagiwara, K.; Takeoka, S.; Ito, Y.; Miyatake, H. Development of a Non-IgG PD-1/PD-L1 Inhibitor by in Silico Mutagenesis and an In-Cell Protein-Protein Interaction Assay. ACS Chem. Biol. 2021, 16, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Shams, R.; Matsukawa, A.; Ochi, Y.; Ito, Y.; Miyatake, H. In Silico and In Cell Hybrid Selection of Nonrapalog Ligands to Allosterically Inhibit the Kinase Activity of mTORC1. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Unni, N.; Arteaga, C.L. Is Dual mTORC1 and mTORC2 Therapeutic Blockade Clinically Feasible in Cancer? JAMA Oncol. 2019, 5, 1564–1565. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Li, C.; Khan, J.; Banerjee, N.S.; Chow, L.T.; Athar, M. Combined mTORC1/mTORC2 inhibition blocks growth and induces catastrophic macropinocytosis in cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 24583–24592. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.A.; O’Neill, D.J.; Machl, A.W.; Lumpkin, C.J.; Galda, S.N.; Sengupta, S.; Mahoney, S.J.; Howell, J.J.; Molz, L.; Hahm, S.; et al. Discovery of Small-Molecule Selective mTORC1 Inhibitors via Direct Inhibition of Glucose Transporters. Cell Chem. Biol. 2019, 26, 1203–1213. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, T.; Heilmeyer, L.M., Jr. Phosphatidylinositol 4-kinases. Eur. J. Biochem. 1998, 253, 357–370. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem. 2009, 284, 12783–12791. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Mendez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef]
- Ohana, R.F.; Hurst, R.; Vidugiriene, J.; Slater, M.R.; Wood, K.V.; Urh, M. HaloTag-based purification of functional human kinases from mammalian cells. Protein Expr. Purif. 2011, 76, 154–164. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Nokin, M.J.; Egorov, M.; Tome, M.; Bodineau, C.; Di Primo, C.; Minder, L.; Wdzieczak-Bakala, J.; Garcia-Alvarez, M.C.; Bignon, J.; et al. mTOR Inhibition via Displacement of Phosphatidic Acid Induces Enhanced Cytotoxicity Specifically in Cancer Cells. Cancer Res. 2018, 78, 5384–5397. [Google Scholar] [CrossRef] [Green Version]
- Dharmatti, R.; Miyatake, H.; Zhang, C.; Ren, X.; Yumoto, A.; Kiga, D.; Yamamura, M.; Ito, Y. Escherichia coli expression, purification, and refolding of human folate receptor alpha (hFRalpha) and beta (hFRbeta). Protein Expr. Purif. 2018, 149, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Yasgar, A.; Jadhav, A.; Simeonov, A.; Coussens, N.P. AlphaScreen-Based Assays: Ultra-High-Throughput Screening for Small-Molecule Inhibitors of Challenging Enzymes and Protein-Protein Interactions. Methods Mol. Biol. 2016, 1439, 77–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Protein | KD (M) 1,* | ka (M−1 s−1) 2,* | kd (s−1) 3,* | χ2 4 |
|---|---|---|---|---|
| ∆N-FAT-M | 1.26 ± 0.11 × 10−6 | 2.00 ± 0.12 × 103 | 2.40 ± 0.11 × 10−3 | 0.04 |
| ∆N | 6.47 ± 0.13 × 10−6 | 1.51 ± 0.30 × 103 | 9.77 ± 0.07 × 10−3 | 0.04 |
| ∆ATP | 2.91 ± 0.10 × 10−8 | 8.50 ± 0.13 × 105 | 2.47 ± 0.03 × 10−2 | 0.02 |
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit mAb anti-mTOR | Cell Signaling Technology | 2983; RRID: AB_2105622 |
| Goat anti-rabbit-HRP secondary antibody | Invitrogen, Thermo Fisher | A16104; RRID: AB_2534776 |
| Bacterial Strains | ||
| ECOSTM Competent E. coli DH5α | Nippon Gene | 316-06233 |
| ECOSTM Competent E. coli BL21(DE3) | Nippon Gene | 312-06534 |
| Chemicals and Recombinant Proteins | ||
| Dpn1 enzyme | Takara | 1235A |
| ExoSAP-IT enzyme | Bioscience, Thermo Fisher | 75001.1 |
| KOD one PCR enzyme mix | Toyobo | KMM-201 |
| DMEM (High-Glucose) media | FujiFilm Wako Pure Chemicals | 044-29765 |
| Opti-MEM media | Gibco, Thermo Fisher | 31985-070 |
| FuGENE HD | Promega | E2311 |
| Ampicillin, sodium salt | Nacalai Tesque | 02739-32 |
| Kanamycin | FujiFilm Wako Pure Chemicals | 113-00343 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Nacalai Tesque | 19742-94 |
| IGEPAL CA-630 | MP Biomedicals | 198596 |
| Luria–Bertani agar media | Sigma-Aldrich | 1002650948 |
| Luria–Bertani Broth media | Nacalai Tesque | 20068-75 |
| Modified Terrific Broth media | Sigma-Aldrich | 1002891164 |
| Antifoam SI | FujiFilm Wako Pure Chemicals | 018-17435 |
| Protease inhibitors | Roche (Merck) | 06538282001 |
| Thrombin | FujiFilm Wako Pure Chemicals | 206-18411 |
| Coomassie brilliant blue R-250 | FujiFilm Wako Pure Chemicals | 6104-59-2 |
| GTPγS | Millipore | 20-176 |
| Commercial kits | ||
| NucleoSpin® EasyPure kit | Macherey-Nagel | 740727.50 |
| NanoBiT® PPI Control Pair (FKBP/FRB) | Promega | N2016 |
| Nano-Glo® Live Cell Assay System | Promega | N2012 |
| Anti-Rabbit IgG Alpha Donor beads | PerkinElmer | AS105M |
| Anti-6xHis AlphaLISA Acceptor beads | PerkinElmer | AL178M |
| HaloTag protein purification system | Promega | G6270 |
| In-Fusion cloning kit | Takara | 639650 |
| HisTrapTM HP Ni column | Cytiva | 17524802 |
| HisTrapTM FF crude Ni column | Cytiva | 17528601 |
| Superdex-200 HiLoad 16/60 column | GE Healthcare | 28-9893-35 |
| His SpinTrapTM column | GE Healthcare | 28401353 |
| HiTrapTM Benzamidine FF column | GE Healthcare | 17-5143-02 |
| PD spintrap G-25 | GE Healthcare | 28918004 |
| Cell Lines | ||
| HEK293 | RIKEN Cell Bank | N/A |
| Oligonucleotides | ||
| Synthetic human RHEB gene (507 bp) UniportKB ID: Q15382 | Eurofins Genomics | GSY1601-1 |
| Human mTOR ORF/pFN21A UniportKB ID: P42345 | Kazusa Institute / Promega | FHC01207 |
| pET15b vector | Novagen | 69661 |
| Primers for RHEB fragmentation for pET15b: FOR: ′GTGCCGCGCGGGCAGCCAGTCCAAAAGCCGCAAAATC′ REV: ′ATCGATAAGCTTCTATTCCAACTTTTCCGCTTCCAG′ | Eurofins Genomics | N/A |
| Primers for pET15b linearization: FOR: ′CATATGGCTGCCGCGCGGCACCAGGCCGCTGCTG′ REV: ′TAGAAGCTTATCGATGATAAGCTGTCAAACATGAG′ | Eurofins Genomics | N/A |
| Primers for RHEB fragmentation for LgBiT: FOR: ′ATCGCCATGGTGGCCCAGTCCAAAAGCCGCAAAATC′ REV: ′ACTGCCTTGAGAAACTTCCAACTTTTCCGCTTCC′ | Eurofins Genomics | N/A |
| Primers for LgBiT vector linearization: FOR: ′GTTTCTCAAGGCAGTTCAGGTGGTGGCGGGAGCGG′ REV: ′GGCCACCATGGCGATCGCTAGCGGTGGCTTTACC′ | Eurofins Genomics | N/A |
| Primers for SmBiT vector linearization: FOR: ′TGGGCTAGCAGATCTTCTAGAGTCGGGGCGGCCGG′ REV: ′CATTCCACCGCTCGAGCCTCCACCTCCGCTCCCGC′ | Eurofins Genomics | N/A |
| Primers for mTORΔN Fragment for SmBiT: FOR: ′GGCTCGAGCGGTGGATCTACTCGCTTCTATGACC′ REV: ′AGAAGATCTGCTAGCACCCAGCCATTCCAGGGC′ | Eurofins Genomics | N/A |
| Primers for mTORΔM Fragment for SmBiT: FOR: ′GGCTCGAGCGGTGGACATCACACCATGGTTGTCC′ REV: ′AGAAGATCTGCTAGCCACAAAGGACACCAACATTC′ | Eurofins Genomics | N/A |
| Primers for mTORΔN-M Fragment for SmBiT: FOR: ′ACATGCACATCACACCATGGTTGTCCAGGCCATC′ REV: ′GTGTGATGTGCATGTCTCCGGCCCTCATTGCGG′ | Eurofins Genomics | N/A |
| Primers for mTORΔN-F-M Fragment for SmBiT: FOR: ′GGCCGGAGACATGCAGGCCAAGGGGATGCATTGG′ REV: ′AACCATGGTGTGATGCAAGTTTAAGAGGGTCTGTG′ | Eurofins Genomics | N/A |
| Primers for mTORΔATP Fragment for SmBiT: FOR: ′GCTCGAGCGGTGGACAGCCAATCATTCGCATTCAG′ REV: ′AGAAGATCTGCTAGCGGCCAGGTCGTCCCCAGCTG′ | Eurofins Genomics | N/A |
| Primers for mTORΔN Fragment for pET15b: FOR: ′GTGCCGCGCGGCAGCTCTACTCGCTTCTATGACC′ REV: ′ATCGATAAGCTTCTAACCCAGCCATTCCAGGGCTC′ | Eurofins Genomics | N/A |
| Primers for mTORΔN-F-M Fragment for pET15b: FOR: ′GATAACGCGATCGCCTCTACTCGCTTCTATGACC′ REV: ′CGAATTCGTTTAAACCACAAAGGACACCAACATTC′ | Eurofins Genomics | N/A |
| Primers for mTORΔATP Fragment for pET15b: FOR: ′GATAACGCGATCGCCCTGCCTCAGCTCACATCC′ REV: ′CGAATTCGTTTAAACGCATGTGATTCTGTAGTTGC′ | Eurofins Genomics | N/A |
| Primers for colony PCR of SmBiT/LgBiT: FOR: ′GAAGTCGAACACGCAGATGCAGTCG′ REV: ′CACTGCATTCTAGTTGTGGTTTGTCCAAACTC′ | Eurofins Genomics | N/A |
| Primers for colony PCR of pET15b: FOR: ′CGATCCCGCGAAATTAATACGACTCACTATAG′ REV: ′GACATTAACCTATAAAAATAGGCGTATCACGAGG′ | Eurofins Genomics | N/A |
| Software | ||
| ICM-Pro 3.9 software | Molsoft L.L.C. | https://www.molsoft.com/products.html |
| SnapGene 5.1.7 software | GSL Biotech L.L.C. | https://www.snapgene.com/ |
| BLItzPro 1.2 software | FortéBio (Sartorius) | https://www.sartorius.com/ |
| Prism 8.4.3 software | GraphPad | https://www.graphpad.com/ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shams, R.; Ito, Y.; Miyatake, H. Evaluation of the Binding Kinetics of RHEB with mTORC1 by In-Cell and In Vitro Assays. Int. J. Mol. Sci. 2021, 22, 8766. https://doi.org/10.3390/ijms22168766
Shams R, Ito Y, Miyatake H. Evaluation of the Binding Kinetics of RHEB with mTORC1 by In-Cell and In Vitro Assays. International Journal of Molecular Sciences. 2021; 22(16):8766. https://doi.org/10.3390/ijms22168766
Chicago/Turabian StyleShams, Raef, Yoshihiro Ito, and Hideyuki Miyatake. 2021. "Evaluation of the Binding Kinetics of RHEB with mTORC1 by In-Cell and In Vitro Assays" International Journal of Molecular Sciences 22, no. 16: 8766. https://doi.org/10.3390/ijms22168766
APA StyleShams, R., Ito, Y., & Miyatake, H. (2021). Evaluation of the Binding Kinetics of RHEB with mTORC1 by In-Cell and In Vitro Assays. International Journal of Molecular Sciences, 22(16), 8766. https://doi.org/10.3390/ijms22168766