
Role of SSD1 in Phenotypic Variation of Saccharomyces cerevisiae Strains Lacking DEG1-Dependent Pseudouridylation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
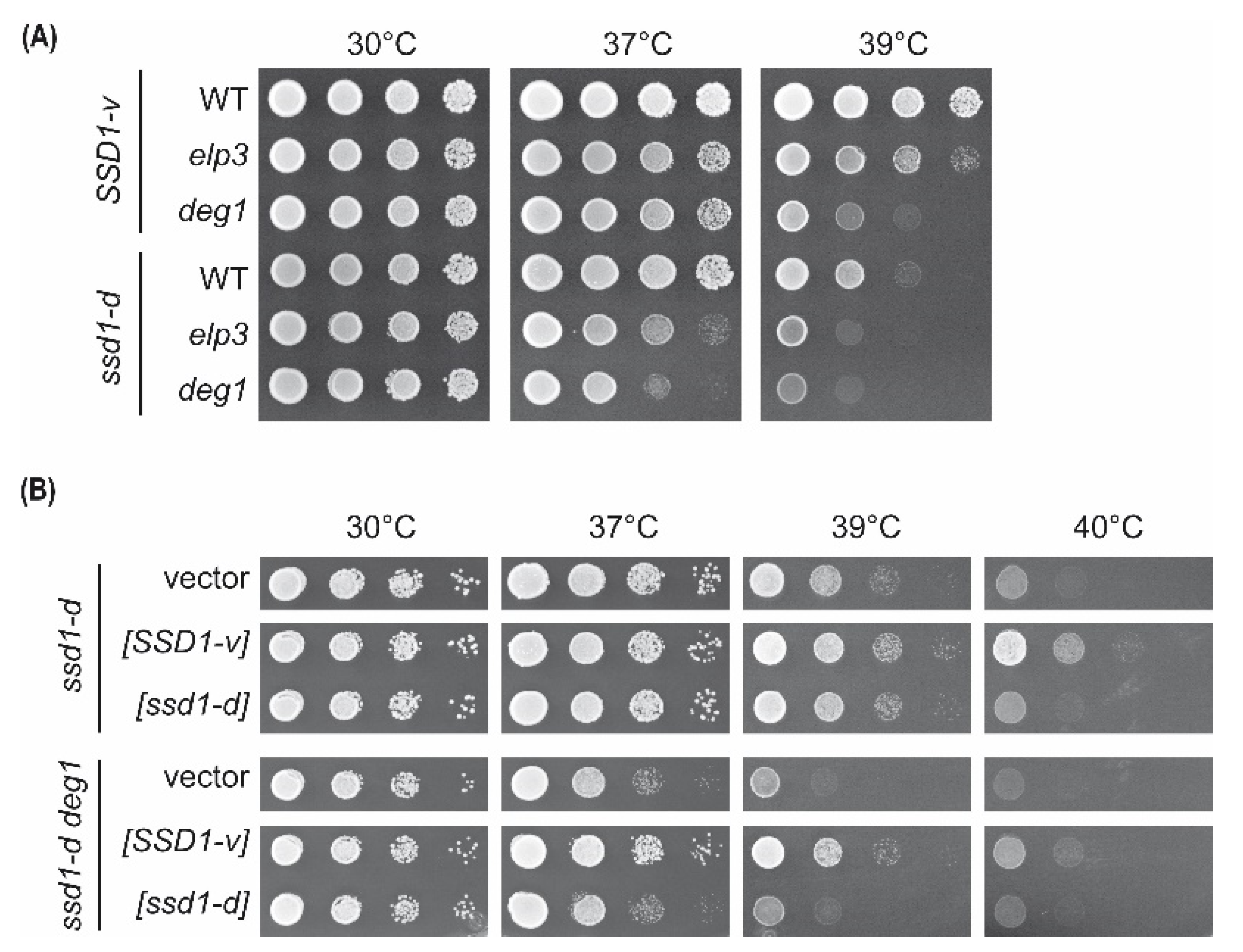
2.1. Comparison of elp3 and deg1 Mutant Phenotypes in ssd1-d and SSD1-v Strains
2.2. Impact of SSD1 on Genetic Interaction of DEG1 with mcm5s2U34-Relevant Genes
2.3. Expression of the Gln-Rich Protein Rnq1 in deg1 Mutants
2.4. Role of SSD1 Status Variation in Protein Aggregation
2.5. Chronological Aging in deg1 Mutants
2.6. Phenotypic Diversity of Other tRNA Modification Defects in ssd1-d and SSD1-v Strains
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Strains and General Methods
5.2. Plasmid Construction and Shuffling
5.3. Protein Isolation and Western Blotting
5.4. Isolation of Protein Aggregates
5.5. Chronological Aging Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Motorin, Y.; Helm, M. tRNA stabilization by modified nucleotides. Biochemistry 2010, 49, 4934–4944. [Google Scholar] [CrossRef]
- Phizicky, E.M.; Hopper, A.K. tRNA biology charges to the front. Genes Dev. 2010, 24, 1832–1860. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lünse, C.E.; Mörl, M. tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules 2017, 7, 35. [Google Scholar] [CrossRef]
- Sokołowski, M.; Klassen, R.; Bruch, A.; Schaffrath, R.; Glatt, S. Cooperativity between different tRNA modifications and their modification pathways. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2018, 1861, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Accornero, F.; Ross, R.L.; Alfonzo, J.D. From canonical to modified nucleotides: Balancing translation and metabolism. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 525–540. [Google Scholar] [CrossRef] [PubMed]
- De Zoysa, T.; Phizicky, E.M. Hypomodified tRNA in evolutionarily distant yeasts can trigger rapid tRNA decay to activate the general amino acid control response, but with different consequences. PLoS Genet. 2020, 16, e1008893. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Ciftci, A.; Funk, J.; Bruch, A.; Butter, F.; Schaffrath, R. tRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucleic Acids Res. 2016, 44, 10946–10959. [Google Scholar] [CrossRef]
- Han, L.; Kon, Y.; Phizicky, E.M. Functional importance of Ψ38 and Ψ39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA 2015, 21, 188–201. [Google Scholar] [CrossRef]
- Motorin, Y.; Helm, M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes 2019, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Rintala-Dempsey, A.C.; Kothe, U. Eukaryotic stand-alone pseudouridine synthases–RNA modifying enzymes and emerging regulators of gene expression? RNA Biol. 2017, 14, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Spenkuch, F.; Motorin, Y.; Helm, M. Pseudouridine: Still mysterious, but never a fake (uridine)! RNA Biol. 2014, 11, 1540–1554. [Google Scholar] [CrossRef]
- Borchardt, E.K.; Martinez, N.M.; Gilbert, W.V. Regulation and Function of RNA Pseudouridylation in Human Cells. Annu. Rev. Genet. 2020, 54, 309–336. [Google Scholar] [CrossRef]
- Hawer, H.; Hammermeister, A.; Ravichandran, K.E.; Glatt, S.; Schaffrath, R.; Klassen, R. Roles of Elongator Dependent tRNA Modification Pathways in Neurodegeneration and Cancer. Genes 2018, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Han, L.; Faqeih, E.; Ewida, N.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 2016, 135, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Bozaquel-Morais, B.L.; Vogt, L.; D’Angelo, V.; Schaffrath, R.; Klassen, R.; Montero-Lomelí, M. Protein Phosphatase Sit4 Affects Lipid Droplet Synthesis and Soraphen A Resistance Independent of Its Role in Regulating Elongator Dependent tRNA Modification. Biomolecules 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Schaffrath, R. Role of Pseudouridine Formation by Deg1 for Functionality of Two Glutamine Isoacceptor tRNAs. Biomolecules 2017, 7, 8. [Google Scholar] [CrossRef]
- Lecointe, F.; Simos, G.; Sauer, A.; Hurt, E.C.; Motorin, Y.; Grosjean, H. Characterization of yeast protein Deg1 as pseudouridine synthase (Pus3) catalyzing the formation of Ψ38 and Ψ39 in tRNA anticodon loop. J. Biol. Chem. 1998, 273, 1316–1323. [Google Scholar] [CrossRef]
- Mülleder, M.; Calvani, E.; Alam, M.T.; Wang, R.K.; Eckerstorfer, F.; Zelezniak, A.; Ralser, M. Functional Metabolomics Describes the Yeast Biosynthetic Regulome. Cell 2016, 167, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lu, J.; Byström, A.S. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA 2008, 14, 2183–2194. [Google Scholar] [CrossRef]
- Leidel, S.; Pedrioli, P.G.A.; Bucher, T.; Brost, R.; Costanzo, M.; Schmidt, A.; Aebersold, R.; Boone, C.; Hofmann, K.; Peter, M. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 2009, 458, 228–232. [Google Scholar] [CrossRef]
- Schaffrath, R.; Leidel, S.A. Wobble uridine modifications—A reason to live, a reason to die?! RNA Biol. 2017, 14, 1209–1222. [Google Scholar] [CrossRef]
- Abbassi, N.-E.-H.; Biela, A.; Glatt, S.; Lin, T.-Y. How Elongator Acetylates tRNA Bases. Int. J. Mol. Sci. 2020, 21, 8209. [Google Scholar] [CrossRef]
- Versées, W.; De Groeve, S.; van Lijsebettens, M. Elongator, a conserved multitasking complex? Mol. Microbiol. 2010, 76, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Schaffrath, R. Collaboration of tRNA modifications and elongation factor eEF1A in decoding and nonsense suppression. Sci. Rep. 2018, 8, 12749. [Google Scholar] [CrossRef] [PubMed]
- Pollo-Oliveira, L.; Klassen, R.; Davis, N.; Ciftci, A.; Bacusmo, J.M.; Martinelli, M.; DeMott, M.S.; Begley, T.J.; Dedon, P.C.; Schaffrath, R.; et al. Loss of Elongator- and KEOPS-Dependent tRNA Modifications Leads to Severe Growth Phenotypes and Protein Aggregation in Yeast. Biomolecules 2020, 10, 322. [Google Scholar] [CrossRef] [PubMed]
- Björk, G.R.; Huang, B.; Persson, O.P.; Byström, A.S. A conserved modified wobble nucleoside (mcm5s2U) in lysyl-tRNA is required for viability in yeast. RNA 2007, 13, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Grunewald, P.; Thüring, K.L.; Eichler, C.; Helm, M.; Schaffrath, R. Loss of anticodon wobble uridine modifications affects tRNALys function and protein levels in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0119261. [Google Scholar] [CrossRef] [PubMed]
- Nedialkova, D.D.; Leidel, S.A. Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef]
- Bruch, A.; Laguna, T.; Butter, F.; Schaffrath, R.; Klassen, R. Misactivation of multiple starvation responses in yeast by loss of tRNA modifications. Nucleic Acids Res. 2020, 48, 7307–7320. [Google Scholar] [CrossRef]
- Xu, F.; Byström, A.S.; Johansson, M.J.O. SSD1 suppresses phenotypes induced by the lack of Elongator-dependent tRNA modifications. PLoS Genet. 2019, 15, e1008117. [Google Scholar] [CrossRef] [PubMed]
- Hogan, D.J.; Riordan, D.P.; Gerber, A.P.; Herschlag, D.; Brown, P.O. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008, 6, e255. [Google Scholar] [CrossRef]
- Jansen, J.M.; Wanless, A.G.; Seidel, C.W.; Weiss, E.L. Cbk1 regulation of the RNA-binding protein Ssd1 integrates cell fate with translational control. Curr. Biol. 2009, 19, 2114–2120. [Google Scholar] [CrossRef]
- Kurischko, C.; Broach, J.R. Phosphorylation and nuclear transit modulate the balance between normal function and terminal aggregation of the yeast RNA-binding protein Ssd1. Mol. Biol. Cell 2017, 28, 3057–3069. [Google Scholar] [CrossRef][Green Version]
- Fiorentini, P.; Huang, K.N.; Tishkoff, D.X.; Kolodner, R.D.; Symington, L.S. Exonuclease I of Saccharomyces cerevisiae functions in mitotic recombination in vivo and in vitro. Mol. Cell. Biol. 1997, 17, 2764–2773. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.; Lin, F.; Sarabia, M.; Arndt, K.T. The SIT4 protein phosphatase is required in late G1 for progression into S phase. Cold Spring Harb. Symp. Quant. Biol. 1991, 56, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, Y.; Qin, L.-X.; Bar-Joseph, Z.; Werner-Washburne, M.; Breeden, L.L. Budding yeast SSD1-V regulates transcript levels of many longevity genes and extends chronological life span in purified quiescent cells. Mol. Biol. Cell 2009, 20, 3851–3864. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Andalis, A.A.; Liszt, G.B.; Fink, G.R.; Guarente, L. Saccharomyces cerevisiae SSD1-V confers longevity by a Sir2p-independent mechanism. Genetics 2004, 166, 1661–1672. [Google Scholar] [CrossRef]
- Frohloff, F.; Fichtner, L.; Jablonowski, D.; Breunig, K.D.; Schaffrath, R. Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J. 2001, 20, 1993–2003. [Google Scholar] [CrossRef]
- Sondheimer, N.; Lindquist, S. Rnq1: An Epigenetic Modifier of Protein Function in Yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Gustavsson, M.; Ronne, H. Evidence that tRNA modifying enzymes are important in vivo targets for 5-fluorouracil in yeast. RNA 2008, 14, 666–674. [Google Scholar] [CrossRef]
- Khonsari, B.; Klassen, R. Impact of Pus1 Pseudouridine Synthase on Specific Decoding Events in Saccharomyces cerevisiae. Biomolecules 2020, 10, 729. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Roig, C.; Viéitez, C.; Posas, F.; De Nadal, E. The Rpd3L HDAC complex is essential for the heat stress response in yeast. Mol. Microbiol. 2010, 76, 1049–1062. [Google Scholar] [CrossRef]
- Alexandrov, A.; Grayhack, E.J.; Phizicky, E.M. tRNA m7G methyltransferase Trm8p/Trm82p: Evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA 2005, 11, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, A.; Chernyakov, I.; Gu, W.; Hiley, S.L.; Hughes, T.R.; Grayhack, E.J.; Phizicky, E.M. Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 2006, 21, 87–96. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Guarente, L. Saccharomyces cerevisiae MPT5 and SSD1 function in parallel pathways to promote cell wall integrity. Genetics 2002, 160, 83–95. [Google Scholar] [CrossRef] [PubMed]
- López-García, B.; Gandía, M.; Muñoz, A.; Carmona, L.; Marcos, J.F. A genomic approach highlights common and diverse effects and determinants of susceptibility on the yeast Saccharomyces cerevisiae exposed to distinct antimicrobial peptides. BMC Microbiol. 2010, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Esberg, A.; Huang, B.; Johansson, M.J.O.; Byström, A.S. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell 2006, 24, 139–148. [Google Scholar] [CrossRef]
- Nuñez, I.; Pino, M.R.; Wiley, D.J.; Das, M.E.; Chen, C.; Goshima, T.; Kume, K.; Hirata, D.; Toda, T.; Verde, F. Spatial control of translation repression and polarized growth by conserved NDR kinase Orb6 and RNA-binding protein Sts5. eLife 2016, 5, e14216. [Google Scholar] [CrossRef]
- Ballou, E.R.; Cook, A.G.; Wallace, E.W.J. Repeated Evolution of Inactive Pseudonucleases in a Fungal Branch of the Dis3/RNase II Family of Nucleases. Mol. Biol. Evol. 2021, 38, 1837–1846. [Google Scholar] [CrossRef]
- Chou, H.-J.; Donnard, E.; Gustafsson, H.T.; Garber, M.; Rando, O.J. Transcriptome-wide Analysis of Roles for tRNA Modifications in Translational Regulation. Mol. Cell 2017, 68, 978–992.e4. [Google Scholar] [CrossRef]
- Rezgui, V.A.N.; Tyagi, K.; Ranjan, N.; Konevega, A.L.; Mittelstaet, J.; Rodnina, M.V.; Peter, M.; Pedrioli, P.G.A. tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc. Natl. Acad. Sci. USA 2013, 110, 12289–12294. [Google Scholar] [CrossRef]
- Zinshteyn, B.; Gilbert, W.V. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet. 2013, 9, e1003675. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Francisco, S.; Varanda, A.S.; Santos, M.; Santos, M.A.S.; Soares, A.R. Impact of tRNA Modifications and tRNA-Modifying Enzymes on Proteostasis and Human Disease. Int. J. Mol. Sci. 2018, 19, 3738. [Google Scholar] [CrossRef] [PubMed]
- Tavares, J.F.; Davis, N.K.; Poim, A.; Reis, A.; Kellner, S.; Sousa, I.; Soares, A.R.; Moura, G.M.R.; Dedon, P.C.; Santos, M. tRNA-modifying enzyme mutations induce codon-specific mistranslation and protein aggregation in yeast. RNA Biol. 2021, 18, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.S.; Fiedler, D.; Cashikar, A.G. Ssd1 is required for thermotolerance and Hsp104-mediated protein disaggregation in Saccharomyces cerevisiae. Mol. Cell. Biol. 2009, 29, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Byström, A.S.; Johansson, M.J.O. SSD1 modifies phenotypes of Elongator mutants. Curr. Genet. 2020, 66, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, V.; Jüdes, A.; Bär, C.; Klassen, R.; Schaffrath, R. Loss of wobble uridine modification in tRNA anticodons interferes with TOR pathway signaling. Microb. Cell 2014, 1, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Reinke, A.; Anderson, S.; McCaffery, J.M.; Yates, J.; Aronova, S.; Chu, S.; Fairclough, S.; Iverson, C.; Wedaman, K.P.; Powers, T. TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 14752–14762. [Google Scholar] [CrossRef]
- Torres, J.; Di Como, C.J.; Herrero, E.; De La Torre-Ruiz, M.A. Regulation of the cell integrity pathway by rapamycin-sensitive TOR function in budding yeast. J. Biol. Chem. 2002, 277, 43495–43504. [Google Scholar] [CrossRef]
- Cao, L.; Tang, Y.; Quan, Z.; Zhang, Z.; Oliver, S.G.; Zhang, N. Chronological Lifespan in Yeast Is Dependent on the Accumulation of Storage Carbohydrates Mediated by Yak1, Mck1 and Rim15 Kinases. PLoS Genet. 2016, 12, e1006458. [Google Scholar] [CrossRef]
- Peters, T.W.; Rardin, M.J.; Czerwieniec, G.; Evani, U.S.; Reis-Rodrigues, P.; Lithgow, G.J.; Mooney, S.D.; Gibson, B.W.; Hughes, R.E. Tor1 regulates protein solubility in Saccharomyces cerevisiae. Mol. Biol. Cell 2012, 23, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar] [CrossRef] [PubMed]
- Gueldener, U.; Heinisch, J.; Koehler, G.J.; Voss, D.; Hegemann, J.H. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002, 30, e23. [Google Scholar] [CrossRef]
- Furukawa, K.; Mizushima, N.; Noda, T.; Ohsumi, Y. A protein conjugation system in yeast with homology to biosynthetic enzyme reaction of prokaryotes. J. Biol. Chem. 2000, 275, 7462–7465. [Google Scholar] [CrossRef]
- Nakayashiki, T.; Kurtzman, C.P.; Edskes, H.K.; Wickner, R.B. Yeast prions URE3 and PSI+ are diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 10575–10580. [Google Scholar] [CrossRef]
- Zachariae, W.; Shin, T.H.; Galova, M.; Obermaier, B.; Nasmyth, K. Identification of subunits of the anaphase-promoting complex of Saccharomyces cerevisiae. Science 1996, 274, 1201–1204. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Koplin, A.; Preissler, S.; Ilina, Y.; Koch, M.; Scior, A.; Erhardt, M.; Deuerling, E. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J. Cell Biol. 2010, 189, 57–68. [Google Scholar] [CrossRef]
- Longo, V.D.; Fabrizio, P. Chronological aging in Saccharomyces cerevisiae. Subcell. Biochem. 2012, 57, 101–121. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khonsari, B.; Klassen, R.; Schaffrath, R. Role of SSD1 in Phenotypic Variation of Saccharomyces cerevisiae Strains Lacking DEG1-Dependent Pseudouridylation. Int. J. Mol. Sci. 2021, 22, 8753. https://doi.org/10.3390/ijms22168753
Khonsari B, Klassen R, Schaffrath R. Role of SSD1 in Phenotypic Variation of Saccharomyces cerevisiae Strains Lacking DEG1-Dependent Pseudouridylation. International Journal of Molecular Sciences. 2021; 22(16):8753. https://doi.org/10.3390/ijms22168753
Chicago/Turabian StyleKhonsari, Bahar, Roland Klassen, and Raffael Schaffrath. 2021. "Role of SSD1 in Phenotypic Variation of Saccharomyces cerevisiae Strains Lacking DEG1-Dependent Pseudouridylation" International Journal of Molecular Sciences 22, no. 16: 8753. https://doi.org/10.3390/ijms22168753
APA StyleKhonsari, B., Klassen, R., & Schaffrath, R. (2021). Role of SSD1 in Phenotypic Variation of Saccharomyces cerevisiae Strains Lacking DEG1-Dependent Pseudouridylation. International Journal of Molecular Sciences, 22(16), 8753. https://doi.org/10.3390/ijms22168753