
Immune Prophylaxis and Therapy for Human Cytomegalovirus Infection

Abstract
:1. Introduction
2. Virology
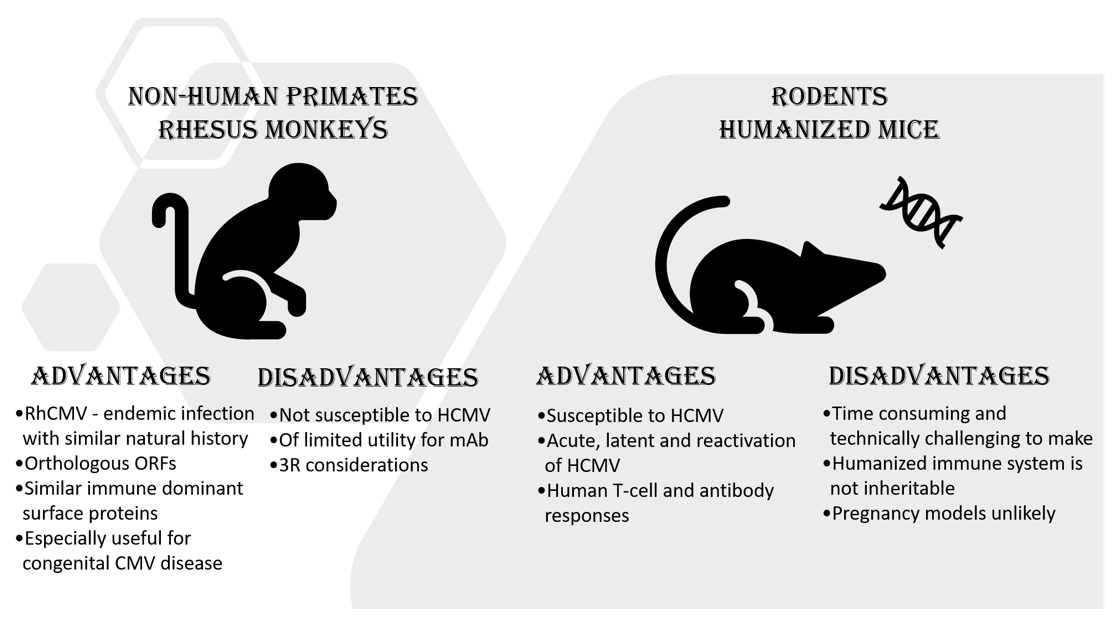
Animal Models
3. Immune Prophylaxis and Therapy
3.1. Vaccines
3.2. Polyclonal Antibody Therapy
3.3. Monoclonal Antibody Therapy
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pediatrics, A.A.O. Cytomegalovirus Infection. In Red Book: 2018 Report of the Committee on Infectious Diseases; Kimberlin, D.W., Long, S.S., Brady, M.T., Jackson, M.A., Eds.; American Academy of Pediatrics: Itasca, IL, USA, 2018; pp. 310–317. [Google Scholar]
- About Cytomegalovirus (CMV). Available online: https://www.cdc.gov/cmv/overview.html (accessed on 12 August 2021).
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Britt, W. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar] [CrossRef] [PubMed]
- Limaye, A.P.; Babu, T.M.; Boeckh, M. Progress and Challenges in the Prevention, Diagnosis, and Management of Cytomegalovirus Infection in Transplantation. Clin. Microbiol. Rev. 2020, 34, 19. [Google Scholar] [CrossRef]
- Njue, A.; Coyne, C.; Margulis, A.V.; Wang, D.; Marks, M.A.; Russell, K.; Das, R.; Sinha, A. The Role of Congenital Cytomegalovirus Infection in Adverse Birth Outcomes: A Review of the Potential Mechanisms. Viruses 2020, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kamil, J.P. Viral Regulation of Cell Tropism in Human Cytomegalovirus. J. Virol. 2016, 90, 626–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrum, F. Human Cytomegalovirus Latency: Approaching the Gordian Knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diggins, N.L.; Skalsky, R.L.; Hancock, M.H. Regulation of Latency and Reactivation by Human Cytomegalovirus miRNAs. Pathogens 2021, 10, 200. [Google Scholar] [CrossRef]
- Kari, B.; Gehrz, R. A human cytomegalovirus glycoprotein complex designated gC-II is a major heparin-binding component of the envelope. J. Virol. 1992, 66, 1761–1764. [Google Scholar] [CrossRef] [Green Version]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 2008, 455, 391–395. [Google Scholar] [CrossRef]
- Wang, X.; Huong, S.M.; Chiu, M.L.; Raab-Traub, N.; Huang, E.S. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 2003, 424, 456–461. [Google Scholar] [CrossRef]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. USA 2004, 101, 15470–15475. [Google Scholar] [CrossRef] [Green Version]
- Feire, A.L.; Roy, R.M.; Manley, K.; Compton, T. The glycoprotein B disintegrin-like domain binds beta 1 integrin to mediate cytomegalovirus entry. J. Virol. 2010, 84, 10026–10037. [Google Scholar] [CrossRef] [Green Version]
- Cooper, R.S.; Heldwein, E.E. Herpesvirus gB: A Finely Tuned Fusion Machine. Viruses 2015, 7, 2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentz, G.L.; Jarquin-Pardo, M.; Chan, G.; Smith, M.S.; Sinzger, C.; Yurochko, A.D. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J. Virol. 2006, 80, 11539–11555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldman, W.J.; Knight, D.A.; Huang, E.H.; Sedmak, D.D. Bidirectional transmission of infectious cytomegalovirus between monocytes and vascular endothelial cells: An in vitro model. J. Infect. Dis. 1995, 171, 263–272. [Google Scholar] [CrossRef]
- Compton, T. Receptors and immune sensors: The complex entry path of human cytomegalovirus. Trends Cell Biol. 2004, 14, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef]
- Wang, X.; Huang, D.Y.; Huong, S.M.; Huang, E.S. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat. Med. 2005, 11, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.A.; Nelson, J.A. Human cytomegalovirus tropism for endothelial cells: Not all endothelial cells are created equal. J. Virol. 2007, 81, 2095–2101. [Google Scholar] [CrossRef] [Green Version]
- Gerna, G.; Revello, M.G.; Baldanti, F.; Percivalle, E.; Lilleri, D. The pentameric complex of human Cytomegalovirus: Cell tropism, virus dissemination, immune response and vaccine development. J. Gen. Virol. 2017, 98, 2215–2234. [Google Scholar] [CrossRef] [PubMed]
- Fouts, A.E.; Chan, P.; Stephan, J.P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Valencia, S.M.; Pfeifer, S.P.; Jensen, J.D.; Kowalik, T.F.; Permar, S.R. Common Polymorphisms in the Glycoproteins of Human Cytomegalovirus and Associated Strain-Specific Immunity. Viruses 2021, 13, 1106. [Google Scholar] [CrossRef] [PubMed]
- Burwitz, B.J.; Malouli, D.; Bimber, B.N.; Reed, J.S.; Ventura, A.B.; Hancock, M.H.; Uebelhoer, L.S.; Bhusari, A.; Hammond, K.B.; Espinosa Trethewy, R.G.; et al. Cross-Species Rhesus Cytomegalovirus Infection of Cynomolgus Macaques. PLoS Pathog. 2016, 12, e1006014. [Google Scholar] [CrossRef]
- Yue, Y.; Barry, P.A. Rhesus cytomegalovirus a nonhuman primate model for the study of human cytomegalovirus. Adv. Virus Res. 2008, 72, 207–226. [Google Scholar] [CrossRef]
- Roark, H.K.; Jenks, J.A.; Permar, S.R.; Schleiss, M.R. Animal Models of Congenital Cytomegalovirus Transmission: Implications for Vaccine Development. J. Infect. Dis. 2020, 221, S60–S73. [Google Scholar] [CrossRef] [Green Version]
- Itell, H.L.; Kaur, A.; Deere, J.D.; Barry, P.A.; Permar, S.R. Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr. Opin. Virol. 2017, 25, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Wang, Z.; Abel, K.; Li, J.; Strelow, L.; Mandarino, A.; Eberhardt, M.K.; Schmidt, K.A.; Diamond, D.J.; Barry, P.A. Evaluation of recombinant modified vaccinia Ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med. Microbiol. Immunol. 2008, 197, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Wussow, F.; Yue, Y.; Martinez, J.; Deere, J.D.; Longmate, J.; Herrmann, A.; Barry, P.A.; Diamond, D.J. A vaccine based on the rhesus cytomegalovirus UL128 complex induces broadly neutralizing antibodies in rhesus macaques. J. Virol. 2013, 87, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.S.; Cruz, D.V.; Tran, D.; Bialas, K.M.; Stamper, L.; Wu, H.; Gilbert, M.; Blair, R.; Alvarez, X.; Itell, H.; et al. Preexisting antibodies can protect against congenital cytomegalovirus infection in monkeys. JCI Insight 2017, 2, 122. [Google Scholar] [CrossRef] [Green Version]
- Cheeran, M.C.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of congenital cytomegalovirus infection: Disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 2009, 22, 99–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleiss, M.R.; McVoy, M.A. Guinea Pig Cytomegalovirus (GPCMV): A Model for the Study of the Prevention and Treatment of Maternal-Fetal Transmission. Future Virol. 2010, 5, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.A.; Lloyd, M.L. A Review of Murine Cytomegalovirus as a Model for Human Cytomegalovirus Disease—Do Mice Lie? Int. J. Mol. Sci. 2020, 22, 214. [Google Scholar] [CrossRef]
- Koenig, J.; Theobald, S.J.; Stripecke, R. Modeling Human Cytomegalovirus in Humanized Mice for Vaccine Testing. Vaccines 2020, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Abeynaike, S.; Paust, S. Humanized Mice for the Evaluation of Novel HIV-1 Therapies. Front. Immunol. 2021, 12, 636775. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.L.; Bility, M.T.; Zhang, L.; Kovalev, G.I.; Buntzman, A.; Frelinger, J.A.; Barry, W.; Ploss, A.; Rice, C.M.; Su, L. A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology 2011, 140, 1334–1344. [Google Scholar] [CrossRef] [Green Version]
- Crawford, L.B.; Caposio, P. Development of a huBLT Mouse Model to Study HCMV Latency, Reactivation, and Immune Response. Methods Mol. Biol. 2021, 2244, 343–363. [Google Scholar] [CrossRef]
- Theobald, S.J.; Khailaie, S.; Meyer-Hermann, M.; Volk, V.; Olbrich, H.; Danisch, S.; Gerasch, L.; Schneider, A.; Sinzger, C.; Schaudien, D.; et al. Signatures of T and B Cell Development, Functional Responses and PD-1 Upregulation After HCMV Latent Infections and Reactivations in Nod.Rag.Gamma Mice Humanized With Cord Blood CD34(+) Cells. Front. Immunol. 2018, 9, 2734. [Google Scholar] [CrossRef] [Green Version]
- Mian, S.A.; Anjos-Afonso, F.; Bonnet, D. Advances in Human Immune System Mouse Models for Studying Human Hematopoiesis and Cancer Immunotherapy. Front. Immunol. 2020, 11, 619236. [Google Scholar] [CrossRef] [PubMed]
- Basso, S.; Compagno, F.; Zelini, P.; Giorgiani, G.; Boghen, S.; Bergami, E.; Bagnarino, J.; Siciliano, M.; Del Fante, C.; Luppi, M.; et al. Harnessing T Cells to Control Infections After Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2020, 11, 567531. [Google Scholar] [CrossRef]
- Gottlieb, D.J.; Clancy, L.E.; Withers, B.; McGuire, H.M.; Luciani, F.; Singh, M.; Hughes, B.; Gloss, B.; Kliman, D.; Ma, C.K.K.; et al. Prophylactic antigen-specific T-cells targeting seven viral and fungal pathogens after allogeneic haemopoietic stem cell transplant. Clin. Transl. Immunol. 2021, 10, e1249. [Google Scholar] [CrossRef]
- Lambour, J.; Naranjo-Gomez, M.; Piechaczyk, M.; Pelegrin, M. Converting monoclonal antibody-based immunotherapies from passive to active: Bringing immune complexes into play. Emerg. Microbes Infect. 2016, 5, e92. [Google Scholar] [CrossRef] [Green Version]
- Bowers, P.M.; Boyle, W.J.; Damoiseaux, R. The Use of Somatic Hypermutation for the Affinity Maturation of Therapeutic Antibodies. Methods Mol. Biol. 2018, 1827, 479–489. [Google Scholar] [CrossRef]
- Cyster, J.G.; Allen, C.D.C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Mariuzza, R.A. Insights into the Structural Basis of Antibody Affinity Maturation from Next-Generation Sequencing. Front. Immunol. 2018, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struble, E.B.; Kirschbaum, N.; Liu, J.; Marszal, E.; Shapiro, M. Characterization of Therapeutic Proteins. In Protein Therapeutics; Sauna, Z.E., Kimchi-Sarfaty, C., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 69–121. [Google Scholar] [CrossRef]
- Bootz, A.; Karbach, A.; Spindler, J.; Kropff, B.; Reuter, N.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog. 2017, 13, e1006601. [Google Scholar] [CrossRef]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.S.; Huffman, T.; Jenks, J.A.; Cisneros de la Rosa, E.; Xie, G.; Vandergrift, N.; Pass, R.F.; Pollara, J.; Permar, S.R. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc. Natl. Acad. Sci. USA 2018, 115, 6267–6272. [Google Scholar] [CrossRef] [Green Version]
- Pecetta, S.; Finco, O.; Seubert, A. Quantum leap of monoclonal antibody (mAb) discovery and development in the COVID-19 era. Semin. Immunol. 2020, 50, 101427. [Google Scholar] [CrossRef]
- Cytogam Prescribing Information. Available online: https://www.fda.gov/media/77671/download (accessed on 29 July 2021).
- Stratton, K.; Durch, J.; Lawrence, R. Vaccines for the 21st Century: A Tool for Decision Making; National Academy Press: Washington, DC, USA, 2000. [Google Scholar]
- Nelson, C.S.; Baraniak, I.; Lilleri, D.; Reeves, M.B.; Griffiths, P.D.; Permar, S.R. Immune Correlates of Protection Against Human Cytomegalovirus Acquisition, Replication, and Disease. J. Infect. Dis. 2020, 221, S45–S59. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Farquhar, J.; Horberger, E. Clinical trials of immunization with the Towne 125 strain of human cytomegalovirus. J. Infect. Dis. 1976, 134, 470–475. [Google Scholar] [CrossRef]
- Adler, S.P.; Starr, S.E.; Plotkin, S.A.; Hempfling, S.H.; Buis, J.; Manning, M.L.; Best, A.M. Immunity induced by primary human cytomegalovirus infection protects against secondary infection among women of childbearing age. J. Infect. Dis. 1995, 171, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.A.; Starr, S.E.; Friedman, H.M.; Gonczol, E.; Weibel, R.E. Protective effects of Towne cytomegalovirus vaccine against low-passage cytomegalovirus administered as a challenge. J. Infect. Dis. 1989, 159, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Higgins, R.; Kurtz, J.B.; Morris, P.J.; Campbell, D.A., Jr.; Shope, T.C.; Spector, S.A.; Dankner, W.M. Multicenter trial of Towne strain attenuated virus vaccine in seronegative renal transplant recipients. Transplantation 1994, 58, 1176–1178. [Google Scholar] [PubMed]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Munoz, F.M.; Callahan, S.T.; Rupp, R.; Wootton, S.H.; Edwards, K.M.; Turley, C.B.; Stanberry, L.R.; Patel, S.M.; McNeal, M.M.; et al. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: A randomized clinical trial. Vaccine 2016, 34, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O’Beirne, J.; et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Sabbaj, S.; Pass, R.F.; Goepfert, P.A.; Pichon, S. Glycoprotein B vaccine is capable of boosting both antibody and CD4 T-cell responses to cytomegalovirus in chronically infected women. J. Infect. Dis. 2011, 203, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Krause, P.R.; Roberts, J. Scientific and Regulatory Considerations for Efficacy Studies of Cytomegalovirus Vaccines. J. Infect. Dis. 2020, 221, S103–S108. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Freed, D.C.; He, X.; Li, F.; Tang, A.; Cox, K.S.; Dubey, S.A.; Cole, S.; Medi, M.B.; Liu, Y.; et al. A replication-defective human cytomegalovirus vaccine for prevention of congenital infection. Sci. Transl. Med. 2016, 8, 362ra145. [Google Scholar] [CrossRef]
- Ourahmane, A.; Cui, X.; He, L.; Catron, M.; Dittmer, D.P.; Al Qaffasaa, A.; Schleiss, M.R.; Hertel, L.; McVoy, M.A. Inclusion of Antibodies to Cell Culture Media Preserves the Integrity of Genes Encoding RL13 and the Pentameric Complex Components During Fibroblast Passage of Human Cytomegalovirus. Viruses 2019, 11, 221. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.; Li, F.; Troutman, M.C.; Freed, D.C.; Tang, A.; Loughney, J.W.; Wang, D.; Wang, I.M.; Vlasak, J.; Nickle, D.C.; et al. Neutralization of Diverse Human Cytomegalovirus Strains Conferred by Antibodies Targeting Viral gH/gL/pUL128-131 Pentameric Complex. J. Virol. 2017, 91, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, S.P.; Lewis, N.; Conlon, A.; Christiansen, M.P.; Al-Ibrahim, M.; Rupp, R.; Fu, T.M.; Bautista, O.; Tang, H.; Wang, D.; et al. Phase 1 Clinical Trial of a Conditionally Replication-Defective Human Cytomegalovirus (CMV) Vaccine in CMV-Seronegative Subjects. J. Infect. Dis. 2019, 220, 411–419. [Google Scholar] [CrossRef]
- Liu, Y.; Freed, D.C.; Li, L.; Tang, A.; Li, F.; Murray, E.M.; Adler, S.P.; McVoy, M.A.; Rupp, R.E.; Barrett, D.; et al. A Replication-Defective Human Cytomegalovirus Vaccine Elicits Humoral Immune Responses Analogous to Those with Natural Infection. J. Virol. 2019, 93, 19. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.S.; Zhang, L.; Freed, D.C.; Tang, A.; Zhang, S.; Zhou, Y.; Wang, I.M.; Rupp, R.E.; Adler, S.P.; Musey, L.K.; et al. Functional Evaluation and Genetic Evolution of Human T-cell Responses after Vaccination with a Conditionally Replication-Defective Cytomegalovirus Vaccine. J. Infect. Dis. 2020, 7, 631. [Google Scholar] [CrossRef]
- GSK. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01357915?cond=cytomegalovirus+gsk&draw=2&rank=1 (accessed on 29 July 2021).
- Plotkin, S.A.; Wang, D.; Oualim, A.; Diamond, D.J.; Kotton, C.N.; Mossman, S.; Carfi, A.; Anderson, D.; Dormitzer, P.R. The Status of Vaccine Development Against the Human Cytomegalovirus. J. Infect. Dis. 2020, 221, S113–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchmeier, M.; Fluckiger, A.C.; Soare, C.; Bozic, J.; Ontsouka, B.; Ahmed, T.; Diress, A.; Pereira, L.; Schodel, F.; Plotkin, S.; et al. Enveloped virus-like particle expression of human cytomegalovirus glycoprotein B antigen induces antibodies with potent and broad neutralizing activity. Clin. Vaccine Immunol. 2014, 21, 174–180. [Google Scholar] [CrossRef] [Green Version]
- VBI. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02826798?cond=cytomegalovirus+vbi&draw=2&rank=1 (accessed on 29 July 2021).
- Walsh, S.R.; Wilck, M.B.; Dominguez, D.J.; Zablowsky, E.; Bajimaya, S.; Gagne, L.S.; Verrill, K.A.; Kleinjan, J.A.; Patel, A.; Zhang, Y.; et al. Safety and immunogenicity of modified vaccinia Ankara in hematopoietic stem cell transplant recipients: A randomized, controlled trial. J. Infect. Dis. 2013, 207, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, C.; Longmate, J.; Martinez, J.; Zhou, Q.; Kaltcheva, T.I.; Tsai, W.; Drake, J.; Carroll, M.; Wussow, F.; Chiuppesi, F.; et al. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T cells in healthy adults. Blood 2017, 129, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; La Rosa, C.; Baden, L.R.; Longmate, J.; Ariza-Heredia, E.J.; Rida, W.N.; Lingaraju, C.R.; Zhou, Q.; Martinez, J.; Kaltcheva, T.; et al. Poxvirus Vectored Cytomegalovirus Vaccine to Prevent Cytomegalovirus Viremia in Transplant Recipients: A Phase 2, Randomized Clinical Trial. Ann. Intern. Med. 2020, 172, 306–316. [Google Scholar] [CrossRef]
- Flatz, L.; Hegazy, A.N.; Bergthaler, A.; Verschoor, A.; Claus, C.; Fernandez, M.; Gattinoni, L.; Johnson, S.; Kreppel, F.; Kochanek, S.; et al. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat. Med. 2010, 16, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R.; Berka, U.; Watson, E.; Aistleithner, M.; Kiefmann, B.; Mangeat, B.; Swanson, E.C.; Gillis, P.A.; Hernandez-Alvarado, N.; Fernandez-Alarcon, C.; et al. Additive Protection against Congenital Cytomegalovirus Conferred by Combined Glycoprotein B/pp65 Vaccination Using a Lymphocytic Choriomeningitis Virus Vector. Clin. Vaccine Immunol. 2017, 24. [Google Scholar] [CrossRef] [Green Version]
- Schwendinger, M.; Thiry, G.; De Vos, B.; Leroux-Roels, G.; Bruhwyler, J.; Huygens, A.; Ganeff, C.; Buchinger, H.; Orlinger, K.K.; Pinschewer, D.D.; et al. A Randomized Dose-Escalating Phase I Trial of a Replication-Deficient Lymphocytic Choriomeningitis Virus Vector-Based Vaccine Against Human Cytomegalovirus. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.; Yuzhakov, O.; Woods, A.; Deterling, J.; Hassett, K.; Shaw, C.A.; Ciaramella, G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 2018, 36, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef]
- Wen, Y.; Monroe, J.; Linton, C.; Archer, J.; Beard, C.W.; Barnett, S.W.; Palladino, G.; Mason, P.W.; Carfi, A.; Lilja, A.E. Human cytomegalovirus gH/gL/UL128/UL130/UL131A complex elicits potently neutralizing antibodies in mice. Vaccine 2014, 32, 3796–3804. [Google Scholar] [CrossRef] [PubMed]
- Cytotect Prescribing Information. Available online: https://www.sps.nhs.uk/medicines/cytomegalovirus-immunoglobulin/ (accessed on 29 July 2021).
- Germer, M.; Herbener, P.; Schuttrumpf, J. Functional Properties of Human Cytomegalovirus Hyperimmunoglobulin and Standard Immunoglobulin Preparations. Ann. Transplant. 2016, 21, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Staras, S.A.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.F.; Cannon, M.J. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin. Infect. Dis 2006, 43, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Zaia, J.A.; Levin, M.J.; Leszczynski, J.; Wright, G.G.; Grady, G.F. Cytomegalovirus immune globulin: Production from selected normal donor blood. Transplantation 1979, 27, 66–67. [Google Scholar]
- Cohn, E.J.; Strong, L.E.; Mendelovich, S.L.; Hutt, D.; Levin, M.J. Preparation and properties of serum and plasma proteins; a system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids. J. Am. Chem. Soc. 1946, 68, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Oncley, J.L.; Melin, M.; Oikawa, M.T.; Toren, A.; Strong, L.E.; Bielorai, B. The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and beta1-lipoprotein into subfractions of human plasma. J. Am. Chem Soc. 1949, 71, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Planitzer, C.B.; Saemann, M.D.; Gajek, H.; Farcet, M.R.; Kreil, T.R. Cytomegalovirus neutralization by hyperimmune and standard intravenous immunoglobulin preparations. Transplantation 2011, 92, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Rutenberg, T.F.; Mendelovich, S.L.; Hutt, D.; Oikawa, M.T.; Toren, A.; Bielorai, B. The role of immunoglobulin prophylaxis for prevention of cytomegalovirus infection in pediatric hematopoietic stem cell transplantation recipients. Pediatr. Blood Cancer 2017, 64, e26420. [Google Scholar] [CrossRef]
- Sechet, A.; Bridoux, F.; Bauwens, M.; Ayache, R.A.; Belmouaz, S.; Touchard, G. Prevention of cytomegalovirus infection and disease in high-risk renal transplant recipients with polyvalent intravenous immunoglobulins. Transplant. Proc. 2002, 34, 812–813. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Scott, D.E.; Murata, H.; Struble, E.B. Binding and neutralizing anti-cytomegalovirus activities in immune globulin products. Biologicals 2017, 50, 35–41. [Google Scholar] [CrossRef]
- Miescher, S.M.; Huber, T.M.; Kuhne, M.; Lieby, P.; Snydman, D.R.; Vensak, J.L.; Berger, M. In vitro evaluation of cytomegalovirus-specific hyperimmune globulins vs. standard intravenous immunoglobulins. Vox Sang. 2015, 109, 71–78. [Google Scholar] [CrossRef]
- Forthal, D.N.; Phan, T.; Landucci, G. Antibody inhibition of cytomegalovirus: The role of natural killer and macrophage effector cells. Transpl. Infect. Dis. 2001, 3, 31–34. [Google Scholar] [CrossRef]
- Li, F.; Freed, D.C.; Tang, A.; Rustandi, R.R.; Troutman, M.C.; Espeseth, A.S.; Zhang, N.; An, Z.; McVoy, M.; Zhu, H.; et al. Complement enhances in vitro neutralizing potency of antibodies to human cytomegalovirus glycoprotein B (gB) and immune sera induced by gB/MF59 vaccination. NPJ Vaccines 2017, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Fujita, A.; Murayama, T.; Iba, Y.; Kurosawa, Y.; Yoshikawa, T.; Asano, Y. Recombinant human monoclonal antibodies to human cytomegalovirus glycoprotein B neutralize virus in a complement-dependent manner. Microbes Infect. 2009, 11, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.M.; Shelden, S.L.; Godkin, R.R.; Posner, M.P.; Fisher, R.A. Cytomegalovirus prophylaxis with ganciclovir, acyclovir, and CMV hyperimmune globulin in liver transplant patients receiving OKT3 induction. Transplant. Proc. 1995, 27, 31–33. [Google Scholar] [PubMed]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A.; The Transplantation Society International, C.M.V.C.G. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation. Transplantation 2018, 102, 900–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, T.; Mangold, J.F.; Cantrell, S.; Permar, S.R. Impact of Maternal Immunity on Congenital Cytomegalovirus Birth Prevalence and Infant Outcomes: A Systematic Review. Vaccines 2019, 7, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revello, M.G.; Lazzarotto, T.; Guerra, B.; Spinillo, A.; Ferrazzi, E.; Kustermann, A.; Guaschino, S.; Vergani, P.; Todros, T.; Frusca, T.; et al. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N. Engl. J. Med. 2014, 370, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Kagan, K.O.; Enders, M.; Schampera, M.S.; Baeumel, E.; Hoopmann, M.; Geipel, A.; Berg, C.; Goelz, R.; De Catte, L.; Wallwiener, D.; et al. Prevention of maternal-fetal transmission of cytomegalovirus after primary maternal infection in the first trimester by biweekly hyperimmunoglobulin administration. Ultrasound Obstet. Gynecol. 2019, 53, 383–389. [Google Scholar] [CrossRef]
- Hamprecht, K.; Kagan, K.O.; Goelz, R. Hyperimmune globulin to prevent congenital CMV infection. N. Engl. J. Med. 2014, 370, 2543. [Google Scholar] [CrossRef]
- Leruez-Ville, M.; Foulon, I.; Pass, R.; Ville, Y. Cytomegalovirus infection during pregnancy: State of the science. Am. J. Obstet. Gynecol. 2020, 223, 330–349. [Google Scholar] [CrossRef] [PubMed]
- Marsico, C.; Kimberlin, D.W. Congenital Cytomegalovirus infection: Advances and challenges in diagnosis, prevention and treatment. Ital. J. Pediatr. 2017, 43, 38. [Google Scholar] [CrossRef] [PubMed]
- Nokta, M.; Tolpin, M.D.; Nadler, P.I.; Pollard, R.B. Human monoclonal anti-cytomegalovirus (CMV) antibody (MSL 109): Enhancement of in vitro foscarnet- and ganciclovir-induced inhibition of CMV replication. Antivir. Res. 1994, 24, 17–26. [Google Scholar] [CrossRef]
- Jabs, D.A.; Gilpin, A.M.; Min, Y.I.; Erice, A.; Kempen, J.H.; Quinn, T.C.; Studies of Ocular Complications of, A.R.G. HIV and cytomegalovirus viral load and clinical outcomes in AIDS and cytomegalovirus retinitis patients: Monoclonal Antibody Cytomegalovirus Retinitis Trial. AIDS 2002, 16, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Ishida, J.H.; Burgess, T.; Derby, M.A.; Brown, P.A.; Maia, M.; Deng, R.; Emu, B.; Feierbach, B.; Fouts, A.E.; Liao, X.C.; et al. Phase 1 Randomized, Double-Blind, Placebo-Controlled Study of RG7667, an Anticytomegalovirus Combination Monoclonal Antibody Therapy, in Healthy Adults. Antimicrob. Agents Chemother. 2015, 59, 4919–4929. [Google Scholar] [CrossRef] [Green Version]
- Ishida, J.H.; Patel, A.; Mehta, A.K.; Gatault, P.; McBride, J.M.; Burgess, T.; Derby, M.A.; Snydman, D.R.; Emu, B.; Feierbach, B.; et al. Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial of RG7667, a Combination Monoclonal Antibody, for Prevention of Cytomegalovirus Infection in High-Risk Kidney Transplant Recipients. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.D.; Nikitin, P.; Gesner, T.; Lin, J.J.; Barkan, D.T.; Ciferri, C.; Carfi, A.; Akbarnejad Yazdi, T.; Skewes-Cox, P.; Wiedmann, B.; et al. In Vitro Characterization of Human Cytomegalovirus-Targeting Therapeutic Monoclonal Antibodies LJP538 and LJP539. Antimicrob. Agents Chemother. 2016, 60, 4961–4971. [Google Scholar] [CrossRef] [Green Version]
- Maertens, J.; Logan, A.C.; Jang, J.; Long, G.; Tang, J.L.; Hwang, W.Y.K.; Koh, L.P.; Chemaly, R.; Gerbitz, A.; Winkler, J.; et al. Phase 2 Study of Anti-Human Cytomegalovirus Monoclonal Antibodies for Prophylaxis in Hematopoietic Cell Transplantation. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [Green Version]
- FDA Approves First Treatment for Ebola Virus. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-ebola-virus (accessed on 29 July 2021).

{kind=link}
| Licensed for Marketing (USA) | Registered in clinicaltrials.gov (accessed on 15 June 2021) | ||
|---|---|---|---|
| Total Trials (Completed) | Unique Molecular Entities | ||
| Vaccines | None | 21 (9) | 14 |
| Monoclonal antibodies | None | 1 (0) | 1 |
| Polyclonal antibodies | Cytogam | 2 (1) | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Struble, E.B.; Murata, H.; Komatsu, T.; Scott, D. Immune Prophylaxis and Therapy for Human Cytomegalovirus Infection. Int. J. Mol. Sci. 2021, 22, 8728. https://doi.org/10.3390/ijms22168728
Struble EB, Murata H, Komatsu T, Scott D. Immune Prophylaxis and Therapy for Human Cytomegalovirus Infection. International Journal of Molecular Sciences. 2021; 22(16):8728. https://doi.org/10.3390/ijms22168728
Chicago/Turabian StyleStruble, Evi B., Haruhiko Murata, Takashi Komatsu, and Dorothy Scott. 2021. "Immune Prophylaxis and Therapy for Human Cytomegalovirus Infection" International Journal of Molecular Sciences 22, no. 16: 8728. https://doi.org/10.3390/ijms22168728
APA StyleStruble, E. B., Murata, H., Komatsu, T., & Scott, D. (2021). Immune Prophylaxis and Therapy for Human Cytomegalovirus Infection. International Journal of Molecular Sciences, 22(16), 8728. https://doi.org/10.3390/ijms22168728