
Thrombin Inhibition Prevents Endothelial Dysfunction and Reverses 20-HETE Overproduction without Affecting Blood Pressure in Angiotensin II-Induced Hypertension in Mice

 , , , , , , ,
, , , , , , ,
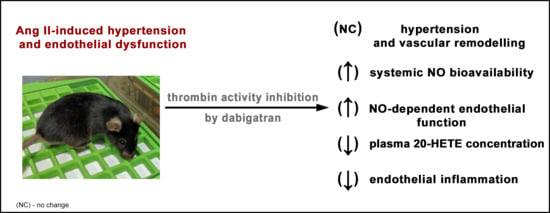
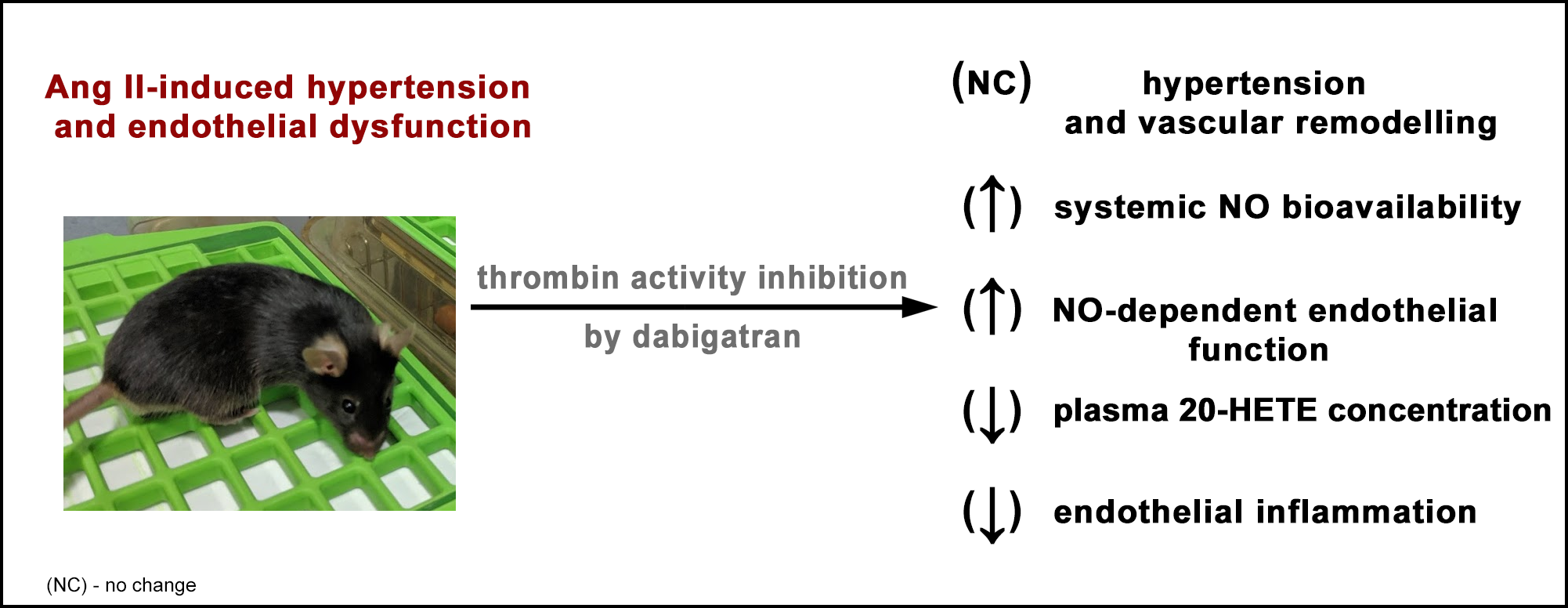
Abstract
:
1. Introduction
2. Results
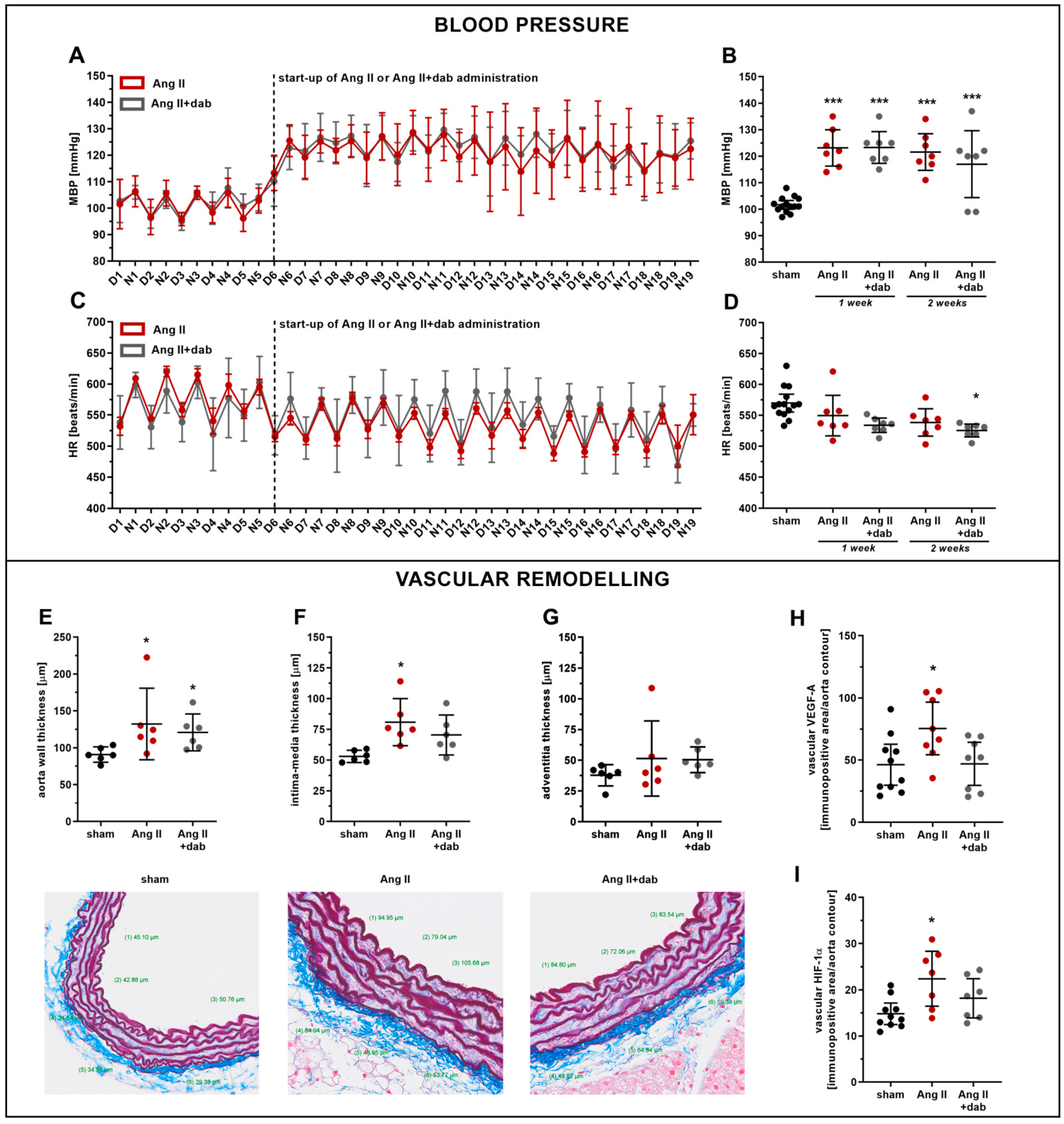
2.1. Effects of Dabigatran on Elevated Blood Pressure and Vascular Remodelling in Ang II-Induced Hypertension
2.2. Effects of Dabigatran on Endothelial Dysfunction, NO- and 20-HETE-Dependent Function in Ang II-Induced Hypertension
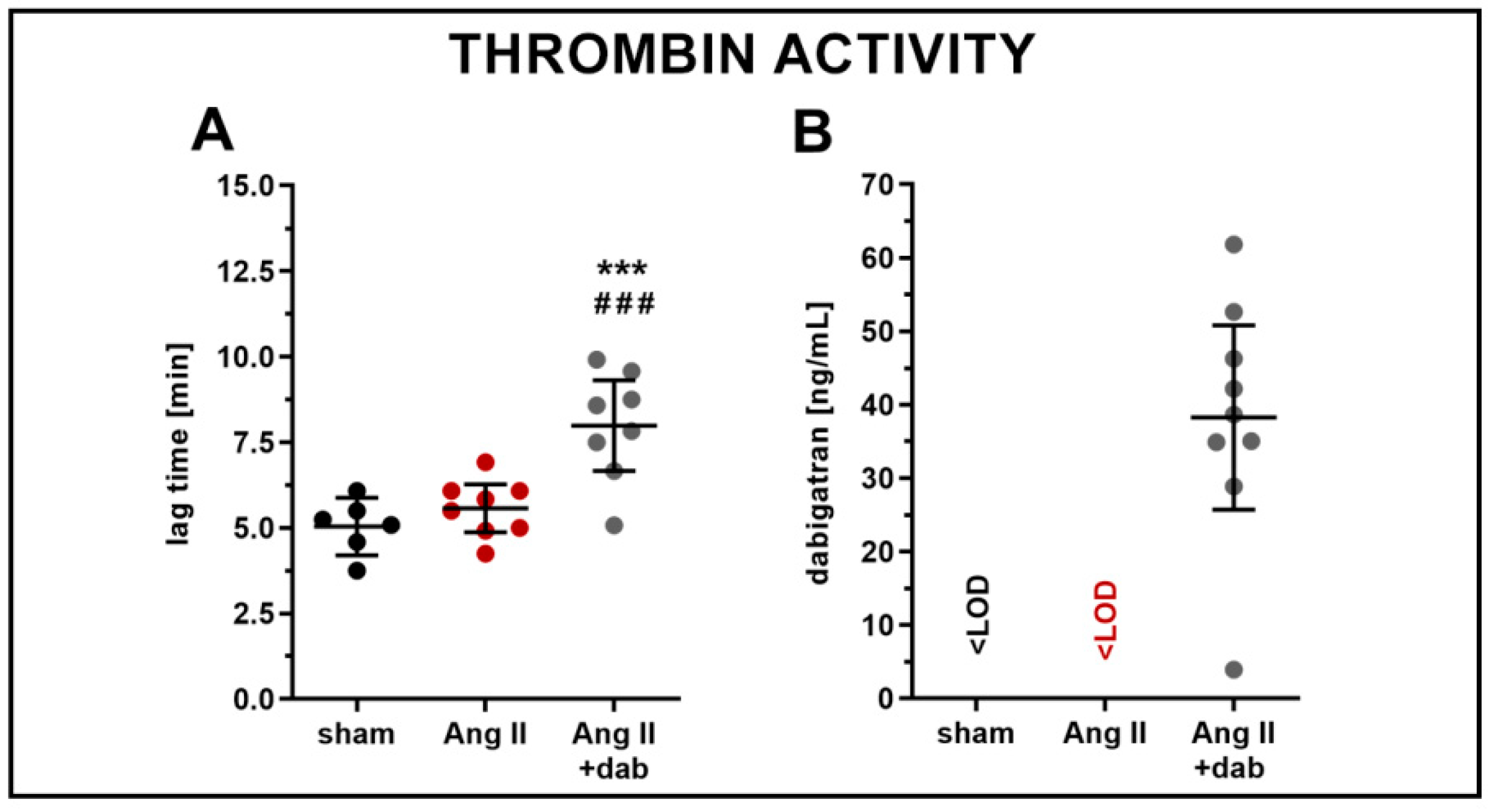
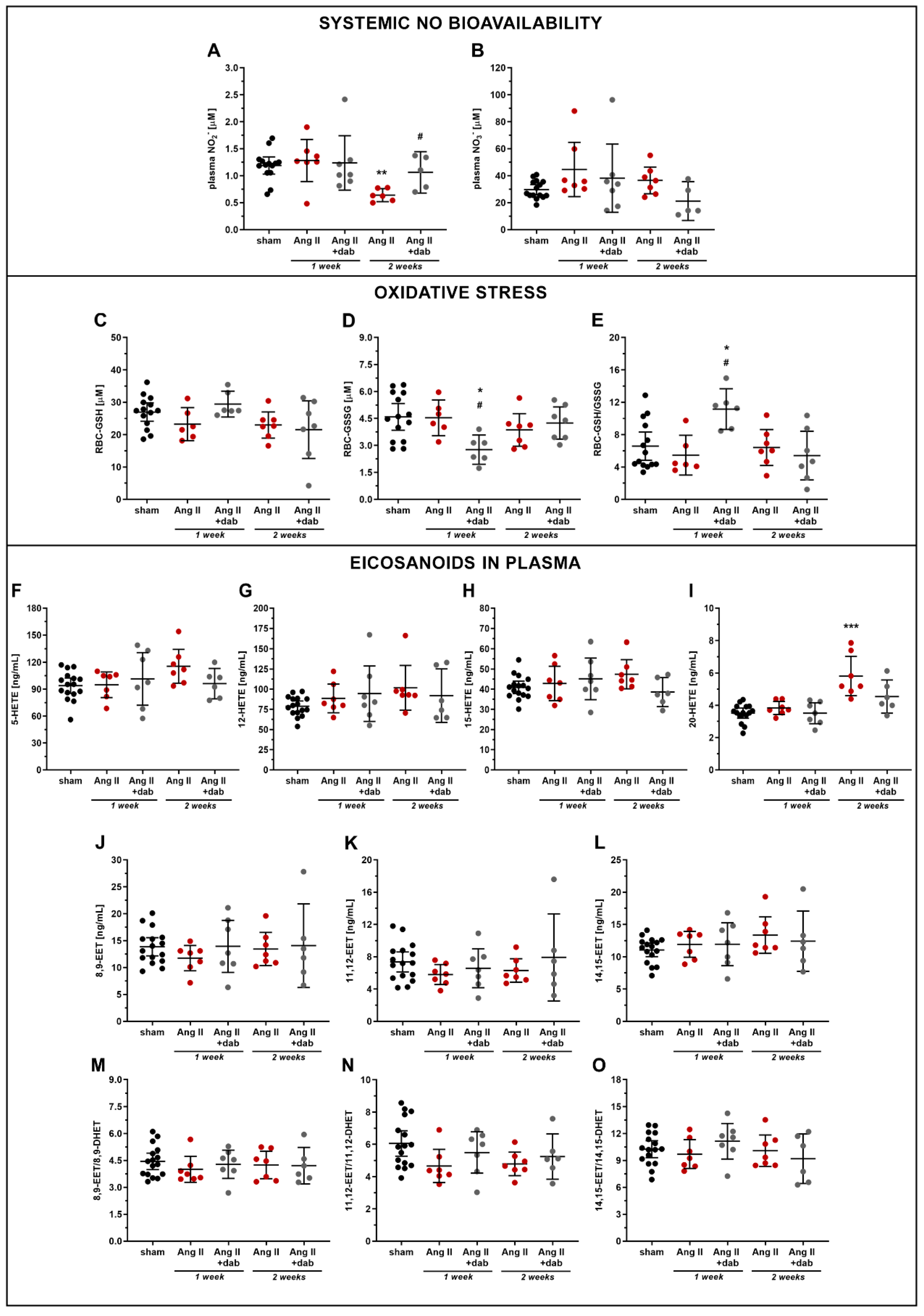
2.3. Effects of Dabigatran on Systemic NO Bioavailability and Plasma Concentration of 20-HETE in Ang II-Induced Hypertension
3. Discussion
4. Materials and Methods
4.1. Animals
4.1.1. Subcutaneous Ang II Administration via Micro-Osmotic Pumps
4.1.2. Intravenous Ang II Administration and Blood Pressure Measurements
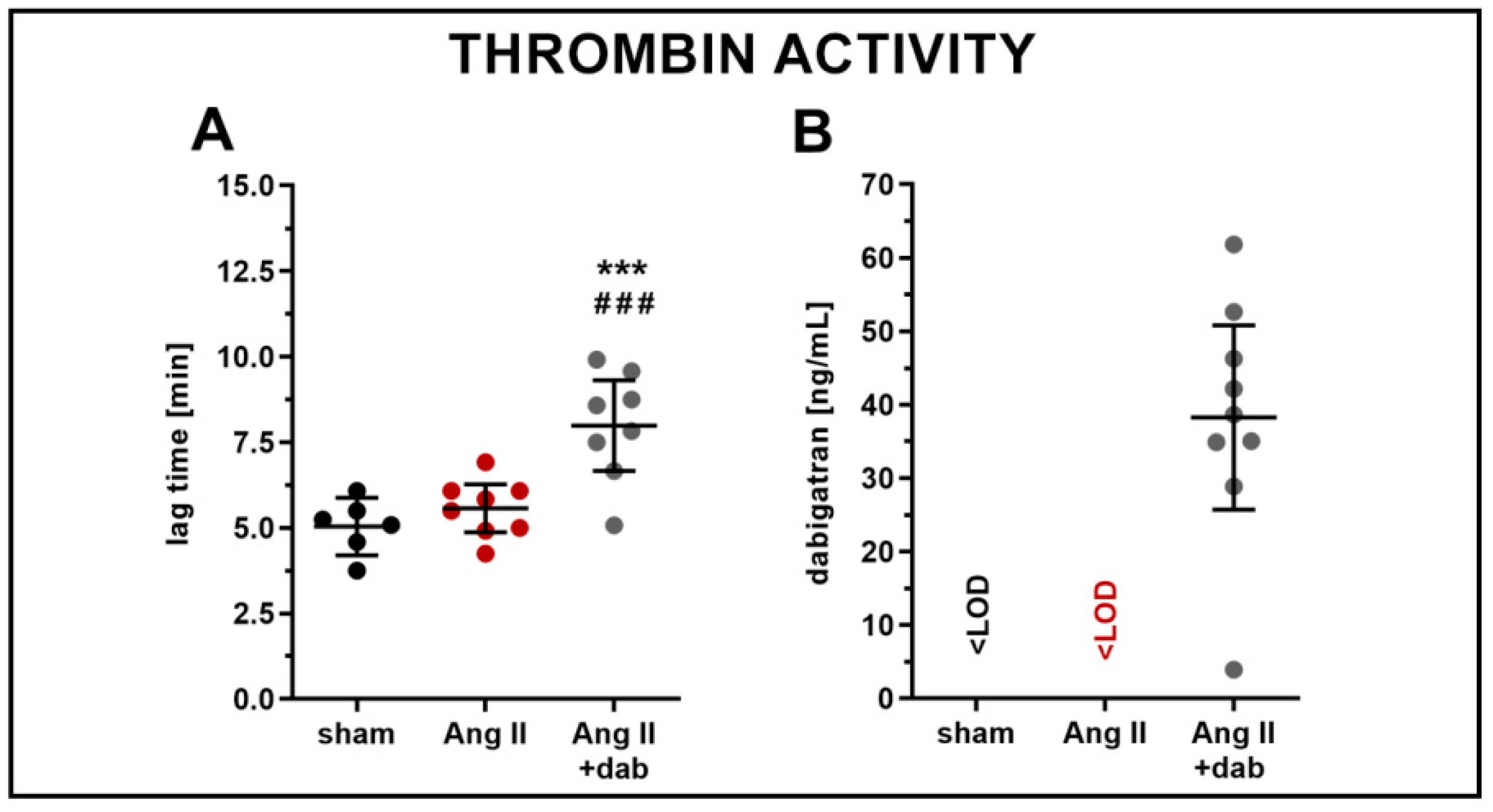
4.2. Measurements of Thrombin Activity (CAT) and Dabigatran Concentration in Plasma
4.3. Assessment of In Vivo Endothelial Function by Magnetic Resonance Imaging (MRI)
4.4. Measurements of Endothelium-Derived NO Production in Aorta by EPR
4.5. Measurements of NO Metabolites in Plasma
4.6. Analysis of Endothelial Phenotype by Immunohistochemistry (IHC)
4.7. Assessment of Aorta Vascular Wall Thickness by Histology
4.8. Measurements of Eicosanoid Production in Full Blood
4.9. Measurements of Eicosanoid Production in Aorta
4.10. UPLC-MS/MS Eicosanoid Analysis
4.11. Measurements of GSH and GSSG in Red Blood Cells
4.12. Total Protein Determination in Aorta Homogenates
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial barrier and its abnormalities in cardiovascular disease. Front. Physiol. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Reriani, M.K.; Lerman, L.O.; Lerman, A. Endothelial function as a functional expression of cardiovascular risk factors. Biomark. Med. 2010, 4, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Bernatova, I. Endothelial dysfunction in experimental models of arterial hypertension: Cause or consequence? Biomed. Res. Int. 2014, 2014, 598271. [Google Scholar] [CrossRef]
- Foëx, P.; Sear, J.W. Hypertension: Pathophysiology and treatment. Contin. Educ. Anaesthesia Crit. Care Pain 2004, 4, 71–75. [Google Scholar] [CrossRef]
- Pirani, N.; Khiavi, F. Population attributable fraction for cardiovascular diseases risk factors in selected countries: A comparative study. Mater. Socio Med. 2017, 29, 35–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubcevic, J.; Santisteban, M.M.; Perez, P.D.; Arocha, R.; Hiller, H.; Malphurs, W.L.; Colon-Perez, L.M.; Sharma, R.K.; de Kloet, A.; Krause, E.G.; et al. A single angiotensin II hypertensive stimulus is associated with prolonged neuronal and immune system activation in Wistar-Kyoto rats. Front. Physiol. 2017, 8, 592. [Google Scholar] [CrossRef]
- Fan, F.; Ge, Y.; Lv, W.; Elliot, M.; Muroya, Y.; Hirata, T.; Booz, G.; Roman, R.J. Molecular mechanisms and cell signaling of 20-hydroxyeicosatetraenoic acid in vascular pathophysiology. Front. Biosci. 2016, 21, 1427–1463. [Google Scholar]
- Sodhi, K.; Wu, C.C.; Cheng, J.; Gotlinger, K.; Inoue, K.; Goli, M.; Falck, J.R.; Abraham, N.G.; Schwartzman, M.L. CYP4A2-induced hypertension is 20-hydroxyeicosatetraenoic acid- and angiotensin II-dependent. Hypertension 2010, 56, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, I. Vascular cytochrome P450 enzymes: Physiology and pathophysiology. Trends Cardiovasc. Med. 2008, 18, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, S.; Lagrange, J.; Jäckel, S.; Jurk, K.; Ehlken, M.; Schönfelder, T.; Weihert, Y.; Knorr, M.; Brandt, M.; Xia, N.; et al. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci. Transl. Med. 2017, 9, eaah4923. [Google Scholar] [CrossRef]
- Rahadian, A.; Fukuda, D.; Salim, H.M.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Shimabukuro, M.; Sata, M. Thrombin inhibition by dabigatran attenuates endothelial dysfunction in diabetic mice. Vascul. Pharmacol. 2020, 124, 106632. [Google Scholar] [CrossRef]
- Hasan, H.; Park, S.-H.; Auger, C.; Belcastro, E.; Matsushita, K.; Marchandot, B.; Lee, H.-H.; Qureshi, A.; Kauffenstein, G.; Ohlmann, P.; et al. Thrombin induces angiotensin II-mediated senescence in atrial endothelial cells: Impact on pro-remodeling patterns. J. Clin. Med. 2019, 8, 1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrikopoulos, P.; Kieswich, J.; Harwood, S.M.; Baba, A.; Matsuda, T.; Barbeau, O.; Jones, K.; Eccles, S.A.; Yaqoob, M.M. Endothelial angiogenesis and barrier function in response to thrombin require Ca2+ influx through the Na+/Ca2+exchanger. J. Biol. Chem. 2015, 290, 18412–18428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplanski, G.; Marin, V.; Fabrigoule, M.; Boulay, V.; Benoliel, A.M.; Bongrand, P.; Kaplanski, S.; Farnarier, C. Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood 1998, 92, 1259–1267. [Google Scholar] [CrossRef]
- Gomolak, J.R.; Didion, S.P. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front. Physiol. 2014, 5, 396. [Google Scholar] [CrossRef]
- Wenzel, P.; Knorr, M.; Kossmann, S.; Stratmann, J.; Hausding, M.; Schuhmacher, S.; Karbach, S.H.; Schwenk, M.; Yogev, N.; Schulz, E.; et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 2011, 124, 1370–1381. [Google Scholar] [CrossRef] [Green Version]
- Kossmann, S.; Schwenk, M.; Hausding, M.; Karbach, S.H.; Schmidgen, M.I.; Brandt, M.; Knorr, M.; Hu, H.; Kröller-Schön, S.; Schönfelder, T.; et al. Angiotensin II-induced vascular dysfunction depends on interferon-γ- driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1313–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüler, R.; Efentakis, P.; Wild, J.; Lagrange, J.; Garlapati, V.; Molitor, M.; Kossmann, S.; Oelze, M.; Stamm, P.; Li, H.; et al. T cell-derived IL-17A induces vascular dysfunction via perivascular fibrosis formation and dysregulation of·NO/cGMP signaling. Oxid. Med. Cell. Longev. 2019, 2019, 6721531. [Google Scholar] [CrossRef] [Green Version]
- Celikel, R.; McClintock, R.A.; Roberts, J.R.; Mendolicchio, G.L.; Ware, J.; Varughese, K.I.; Ruggeri, Z.M. Modulation of α-thrombin function by distinct interactions with platelet glycoprotein Ibα. Science 2003, 301, 218–221. [Google Scholar] [CrossRef]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.F.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Elias, A.; Rock, W.; Odetalla, A.; Ron, G.; Schwartz, N.; Saliba, W.; Elias, M. Enhanced thrombin generation in patients with arterial hypertension. Thromb. Res. 2019, 174, 121–128. [Google Scholar] [CrossRef]
- Antoniak, S.; Cardenas, J.C.; Buczek, L.J.; Church, F.C.; Mackman, N.; Pawlinski, R. Protease-activated receptor 1 contributes to angiotensin II-induced cardiovascular remodeling and inflammation. Cardiology 2017, 136, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Shao, J.; Hu, R.; Chen, H.; Xie, P.; Liu, C. Angiotensin II promotes the anticoagulant effects of rivaroxaban via angiotensin type 2 receptor signaling in mice. Sci. Rep. 2017, 7, 369. [Google Scholar] [CrossRef] [Green Version]
- Leong, X.-F.; Ng, C.-Y.; Jaarin, K. Animal models in cardiovascular research: Hypertension and atherosclerosis. Biomed. Res. Int. 2015, 2015, 528757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Lee, Y.T.; Chan, Y.W.; Tse, G. Animal models for the study of primary and secondary hypertension in humans. Biomed. Reports. 2016, 5, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.M.; Andrade-Gordon, P.; Derian, C.K.; Damiano, B.P. Receptor-activating peptides distinguish thrombin receptor (PAR-1) and protease activated receptor 2 (PAR-2) mediated hemodynamic responses in vivo. Can. J. Physiol. Pharmacol. 1998, 76, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Yokono, Y.; Hanada, K.; Narita, M.; Tatara, Y.; Kawamura, Y.; Miura, N.; Kitayama, K.; Nakata, M.; Nozaka, M.; Kato, T.; et al. Blockade of PAR-1 signaling attenuates cardiac hypertrophy and fibrosis in renin-overexpressing hypertensive mice. J. Am. Heart. Assoc. 2020, 9, e015616. [Google Scholar] [CrossRef] [PubMed]
- Capers, Q., 4th; Laursen, J.B.; Fukui, T.; Rajagopalan, S.; Mori, I.; Lou, P.; Freeman, B.A.; Berrington, W.R.; Griendling, K.K.; Harrison, D.G.; et al. Vascular thrombin receptor regulation in hypertensive rats. Circ. Res. 1997, 80, 838–844. [Google Scholar] [CrossRef]
- Bar, A.; Targosz-Korecka, M.; Suraj, J.; Proniewski, B.; Jasztal, A.; Marczyk, B.; Sternak, M.; Przybyło, M.; Kurpinska, A.; Walczak, M.; et al. Degradation of glycocalyx and multiple manifestations of endothelial dysfunction coincide in the early phase of endothelial dysfunction before atherosclerotic plaque development in apolipoprotein E/low-density lipoprotein receptor-deficient mice. J. Am. Heart. Assoc. 2019, 8, e011171. [Google Scholar] [CrossRef] [Green Version]
- Bar, A.; Kieronska-Rudek, A.; Proniewski, B.; Suraj-Prazmowska, J.; Czamara, K.; Marczyk, B.; Matyjaszczyk-Gwarda, K.; Jasztal, A.; Kus, E.; Majka, Z.; et al. In vivo magnetic resonance imaging-based detection of heterogeneous endothelial response in thoracic and abdominal aorta to short-term high-fat diet ascribed to differences in perivascular adipose tissue in mice. J. Am. Heart. Assoc. 2020, 9, e016929. [Google Scholar] [CrossRef]
- Cheng, J.; Garcia, V.; Ding, Y.; Wu, C.C.; Thakar, K.; Falck, J.R.; Ramu, E.; Schwartzman, M.L. Induction of angiotensin-converting enzyme and activation of the renin-angiotensin system contribute to 20-hydroxyeicosatetraenoic acid-mediated endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1917–1924. [Google Scholar] [CrossRef] [Green Version]
- Minuz, P.; Jiang, H.; Fava, C.; Turolo, L.; Tacconelli, S.; Ricci, M.; Patrignani, P.; Morganti, A.; Lechi, A.; McGiff, J.C. Altered release of cytochrome P450 metabolites of arachidonic acid in renovascular disease. Hypertension 2008, 51, 1379–1385. [Google Scholar] [CrossRef] [Green Version]
- Rocic, P.; Schwartzman, M.L. 20-HETE in the regulation of vascular and cardiac function. Pharmacol. Ther. 2018, 192, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Schwartzman, M.L. The role of 20-HETE in androgen-mediated hypertension. Prostaglandins Other. Lipid Mediat. 2011, 96, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-X.; Xue, K.-Y.; Xin, J.-J.; Yan, X.; Li, R.L.; Wang, X.X.; Wang, X.L.; Tong, M.-M.; Gan, L.; Li, H.; et al. 5-Lipoxagenase deficiency attenuates L-NAME-induced hypertension and vascular remodeling. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 2379–2392. [Google Scholar] [CrossRef] [PubMed]
- Al-Naamani, N.; Sagliani, K.D.; Dolnikowski, G.G.; Warburton, R.R.; Toksoz, D.; Kayyali, U.; Hill, N.S.; Fanburg, B.L.; Roberts, K.E.; Preston, I.R. Plasma 12- and 15-hydroxyeicosanoids are predictors of survival in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Imig, J.D.; Zhao, X.; Capdevila, J.H.; Morisseau, C.; Hammock, B.D. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 2002, 39, 690–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, O.; Brandes, R.P.; Kim, I.; Schweda, F.; Schmidt, R.; Hammock, B.D.; Busse, R.; Fleming, I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 2005, 45, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeners, M.P.; Wesseling, S.; Ulu, A.; Sepúlveda, R.L.; Morisseau, C.; Braam, B.; Hammock, B.D.; Joles, J.A. Soluble epoxide hydrolase in the generation and maintenance of high blood pressure in spontaneously hypertensive rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E691–E698. [Google Scholar] [CrossRef] [Green Version]
- Paar, V.; Jirak, P.; Gruber, S.; Prodinger, C.; Cadamuro, J.; Wernyl, B.; Motloch, L.; Haschke-Becher, E.; Hoppe, U.; Lichtenauer, M. Influence of dabigatran on pro-inflammatory cytokines; growth factors and chemokines–Slowing the vicious circle of coagulation and inflammation. Life Sci. 2020, 262, 118474. [Google Scholar] [CrossRef]
- Lin, J.; He, S.; Sun, X.; Franck, G.; Deng, Y.; Yang, D.; Haemmig, S.; Wara, A.K.M.; Icli, B.; Li, D.; et al. MicroRNA-181b inhibits thrombin-mediated endothelial activation and arterial thrombosis by targeting caspase recruitment domain family member 10. FASEB J. 2016, 30, 3216–3226. [Google Scholar] [CrossRef] [Green Version]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation: Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D. Epoxyeicosatrienoic acids and 20-hydroxyeicosatetraenoic acid on endothelial and vascular function. Adv. Pharmacol. 2016, 77, 105–141. [Google Scholar] [PubMed] [Green Version]
- Laursen, S.B.; Finsen, S.; Marcussen, N.; Quaggin, S.E.; Hansen, B.L.; Dimke, H. Endothelial mineralocorticoid receptor ablation does not alter blood pressure; kidney function or renal vessel contractility. PLoS ONE 2018, 13, e0193032. [Google Scholar] [CrossRef] [Green Version]
- Tchaikovski, S.N.; van Vlijmen, B.J.M.; Rosing, J.; Tans, G. Development of a calibrated automated thrombography based thrombin generation test in mouse plasma. J. Thromb. Haemost. 2007, 5, 2079–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemker, H.C.; Kremers, R. Data management in thrombin generation. Thromb. Res. 2013, 131, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Smeda, M.; Przyborowski, K.; Proniewski, B.; Zakrzewska, A.; Kaczor, D.; Stojak, M.; Buczek, E.; Nieckarz, Z.; Zoladz, J.A.; Wietrzyk, J.; et al. Breast cancer pulmonary metastasis is increased in mice undertaking spontaneous physical training in the running wheel; a call for revising beneficial effects of exercise on cancer progression. Am. J. Cancer. Res. 2017, 7, 1926–1936. [Google Scholar]
- Kij, A.; Mateuszuk, L.; Sitek, B.; Przyborowski, K.; Zakrzewska, A.; Wandzel, K.; Walczak, M.; Chlopicki, S. Simultaneous quantification of PGI2 and TXA2 metabolites in plasma and urine in NO-deficient mice by a novel UHPLC/MS/MS method. J. Pharm. Biomed. Anal. 2016, 129, 148–154. [Google Scholar] [CrossRef]
- Gajda, M.; Jasztal, A.; Banasik, T.; Jasek-Gajda, E.; Chlopicki, S. Combined orcein and martius scarlet blue (OMSB) staining for qualitative and quantitative analyses of atherosclerotic plaques in brachiocephalic arteries in apoE/LDLR−/− mice. Histochem. Cell Biol. 2017, 147, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Przyborowski, K.; Kassassir, H.; Wojewoda, M.; Kmiecik, K.; Sitek, B.; Siewiera, K.; Zakrzewska, A.; Rudolf, A.M.; Kostogrys, R.; Watala, C.; et al. Effects of a single bout of strenuous exercise on platelet activation in female ApoE/LDLR−/− mice. Platelets 2017, 28, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Kij, A.; Kus, K.; Czyzynska-Cichon, I.; Chlopicki, S.; Walczak, M. Development and validation of a rapid; specific and sensitive LC-MS/MS bioanalytical method for eicosanoid quantification-assessment of arachidonic acid metabolic pathway activity in hypertensive rats. Biochimie 2020, 171–172, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hempe, J.M.; Ory-Ascani, J. Simultaneous analysis of reduced glutathione and glutathione disulfide by capillary zone electrophoresis. Electrophoresis 2014, 35, 967–971. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eicosanoid Production in Full Blood Ex Vivo n = 9–10 | Sham | Ang II | Ang II + Dab |
|---|---|---|---|
| 5-HETE (ng/mL) A | 14.67 (13.05, 16.28) | 18.22 (15.16, 21.28) | 18.19 (15.41, 20.98) |
| 12-HETE (ng/mL) B | 496.06 (372.10, 620.01) | 553.11 (254.59, 851.62) | 526.83 (228.08, 825.59) |
| 15-HETE (ng/mL) B | 13.49 (11.59, 15.38) | 16.30 (12.68, 19.92) | 16.74 (13.81, 19.67) |
| 20-HETE (ng/mL) A | 1.26 (0.99, 1.53) | 1.31 (1.16, 1.46) | 1.25 (0.95, 1.55) |
| 8,9-EET (ng/mL) A | 4.65 (4.13, 5.17) | 5.71 (4.78, 6.65) | 5.05 (4.21, 5.88) |
| 11,12-EET (ng/mL) B | 3.30 (2.85, 3.76) | 4.08 (3.57, 4.58) | 3.67 (3.00, 4.34) |
| 14,15-EET (ng/mL) B | 2.92 (2.45, 3.39) | 3.74 (3.10, 4.39) | 3.14 (2.47, 3.81) |
| 8,9-EET/8,9-DHET B | 4.09 (3.77, 4.42) | 4.17 (3.53, 4.81) | 3.76 (3.27, 4.25) |
| 11,12-EET/11,12-DHET A | 4.21 (3.89, 4.52) | 4.91 (4.23, 5.59) | 4.54 (4.07, 5.01) |
| 14,15-EET/14,15-DHET A | 4.33 (3.77, 4.89) | 5.20 (4.75, 5.65) | 4.30 (3.53, 5.08) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kij, A.; Bar, A.; Przyborowski, K.; Proniewski, B.; Mateuszuk, L.; Jasztal, A.; Kieronska-Rudek, A.; Marczyk, B.; Matyjaszczyk-Gwarda, K.; Tworzydlo, A.; et al. Thrombin Inhibition Prevents Endothelial Dysfunction and Reverses 20-HETE Overproduction without Affecting Blood Pressure in Angiotensin II-Induced Hypertension in Mice. Int. J. Mol. Sci. 2021, 22, 8664. https://doi.org/10.3390/ijms22168664
Kij A, Bar A, Przyborowski K, Proniewski B, Mateuszuk L, Jasztal A, Kieronska-Rudek A, Marczyk B, Matyjaszczyk-Gwarda K, Tworzydlo A, et al. Thrombin Inhibition Prevents Endothelial Dysfunction and Reverses 20-HETE Overproduction without Affecting Blood Pressure in Angiotensin II-Induced Hypertension in Mice. International Journal of Molecular Sciences. 2021; 22(16):8664. https://doi.org/10.3390/ijms22168664
Chicago/Turabian StyleKij, Agnieszka, Anna Bar, Kamil Przyborowski, Bartosz Proniewski, Lukasz Mateuszuk, Agnieszka Jasztal, Anna Kieronska-Rudek, Brygida Marczyk, Karolina Matyjaszczyk-Gwarda, Anna Tworzydlo, and et al. 2021. "Thrombin Inhibition Prevents Endothelial Dysfunction and Reverses 20-HETE Overproduction without Affecting Blood Pressure in Angiotensin II-Induced Hypertension in Mice" International Journal of Molecular Sciences 22, no. 16: 8664. https://doi.org/10.3390/ijms22168664
APA StyleKij, A., Bar, A., Przyborowski, K., Proniewski, B., Mateuszuk, L., Jasztal, A., Kieronska-Rudek, A., Marczyk, B., Matyjaszczyk-Gwarda, K., Tworzydlo, A., Enggaard, C., Hansen, P. B. L., Jensen, B., Walczak, M., & Chlopicki, S. (2021). Thrombin Inhibition Prevents Endothelial Dysfunction and Reverses 20-HETE Overproduction without Affecting Blood Pressure in Angiotensin II-Induced Hypertension in Mice. International Journal of Molecular Sciences, 22(16), 8664. https://doi.org/10.3390/ijms22168664