
The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Naturally Occurring Epialleles
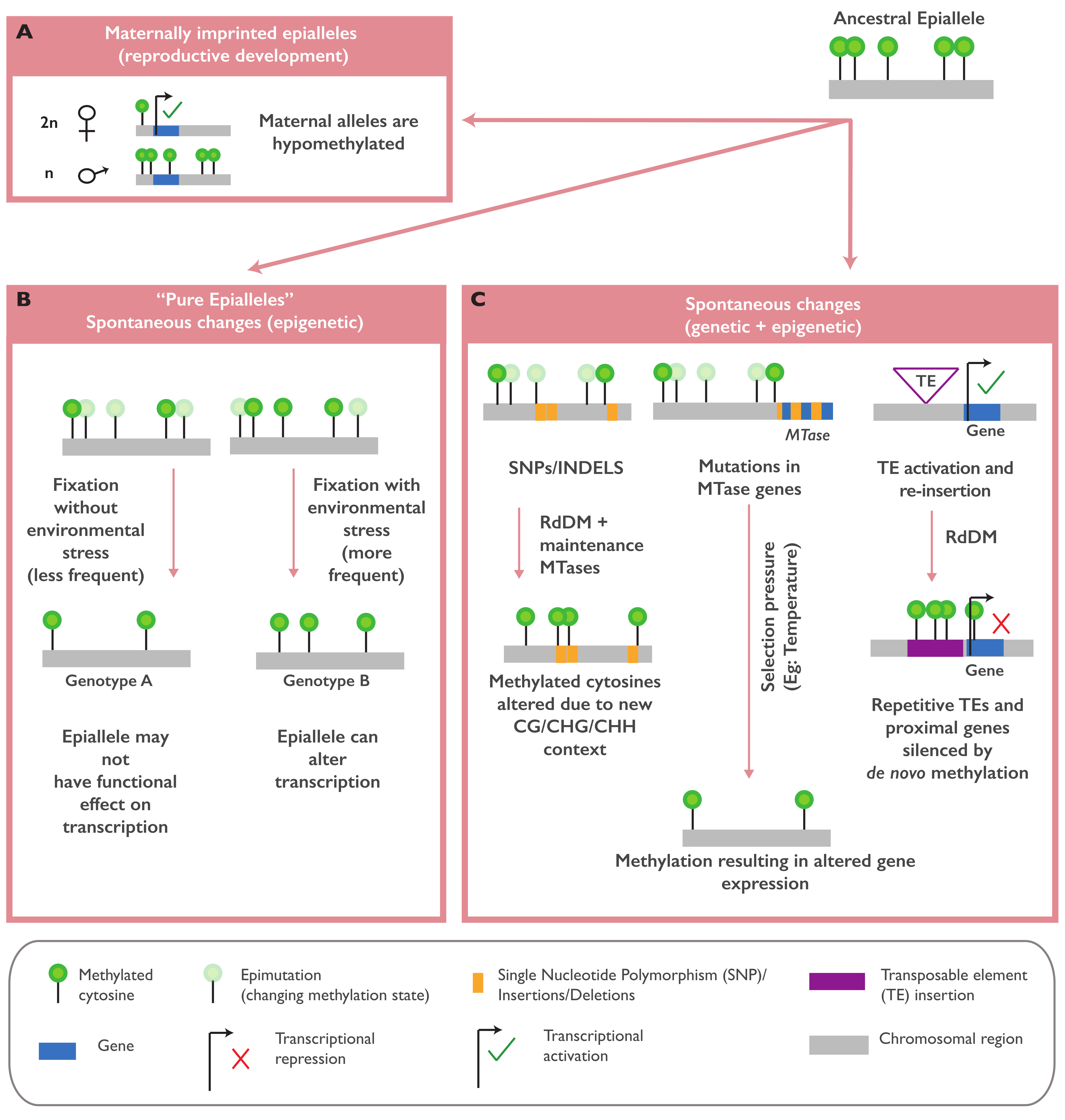
2.1. Naturally Occurring Epialleles and Their Biological Functions
2.2. The Establishment of Naturally Occurring Epialleles
3. The Origins and Functional Roles of Induced Epialleles
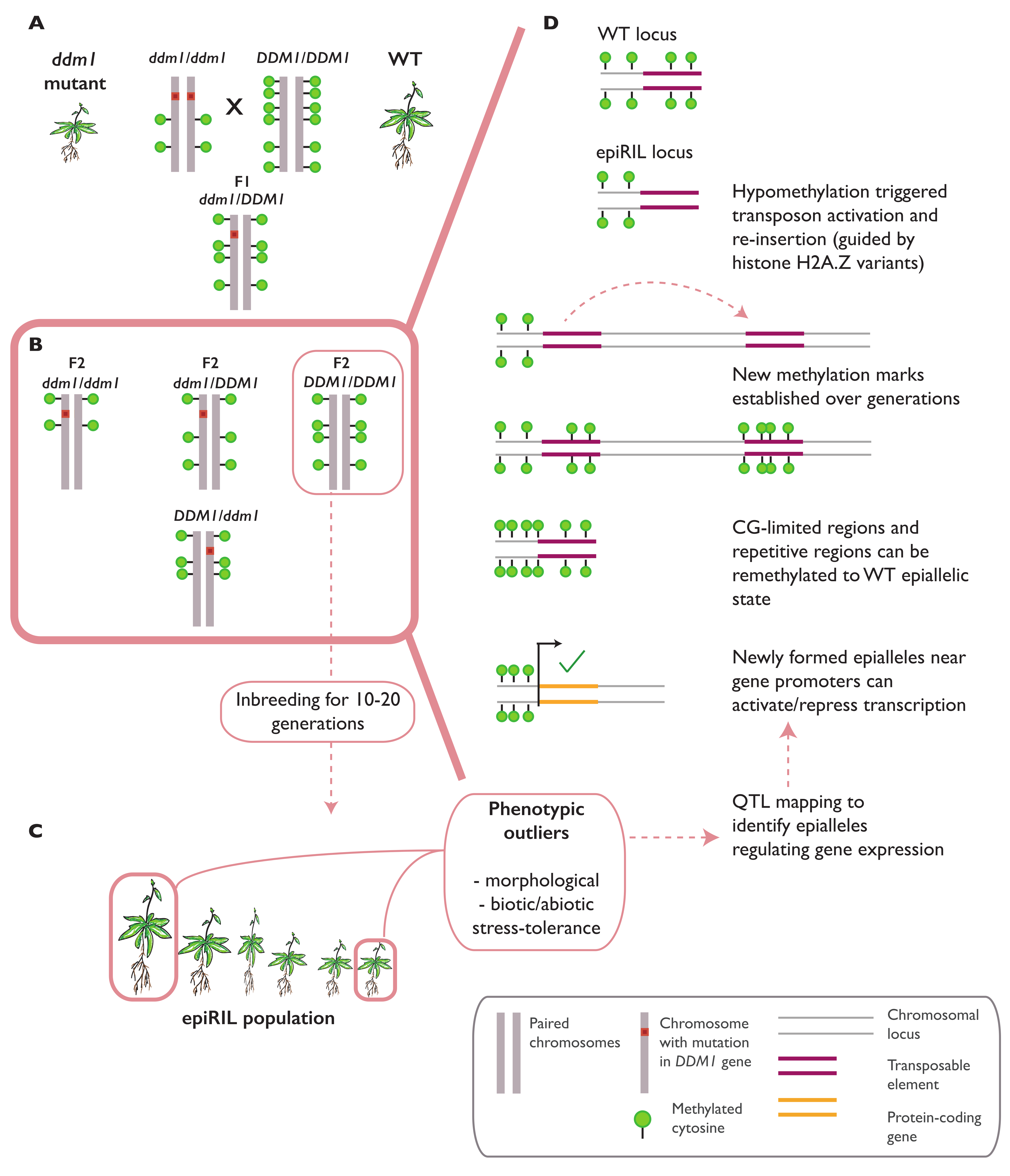
3.1. Epialleles Formed in Epigenetic Recombinant Inbred Lines (epiRILs)
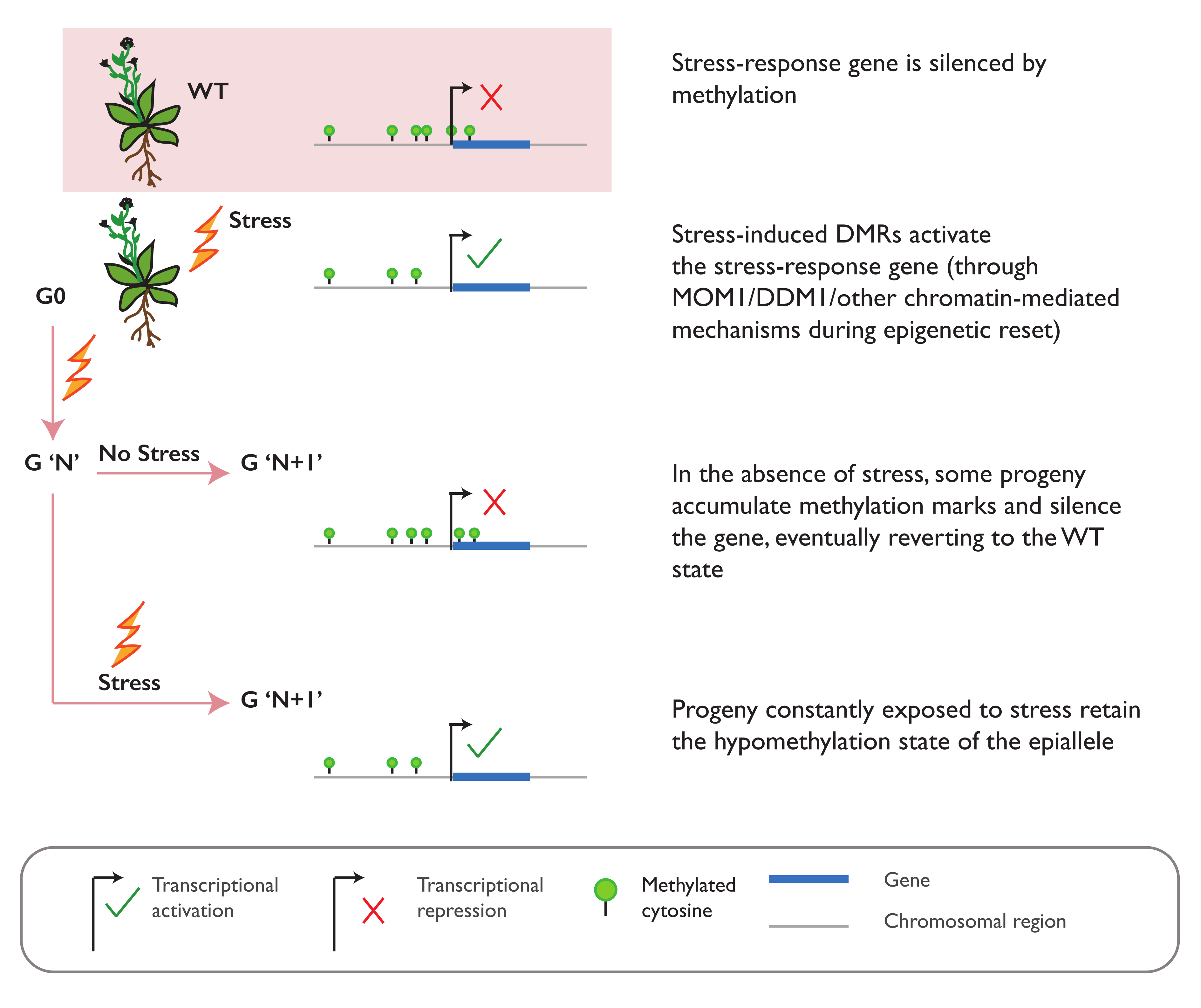
3.2. Environmental Stress Can Induce Epiallele Formation
3.3. Epiallele Formation upon Clonal Propagation
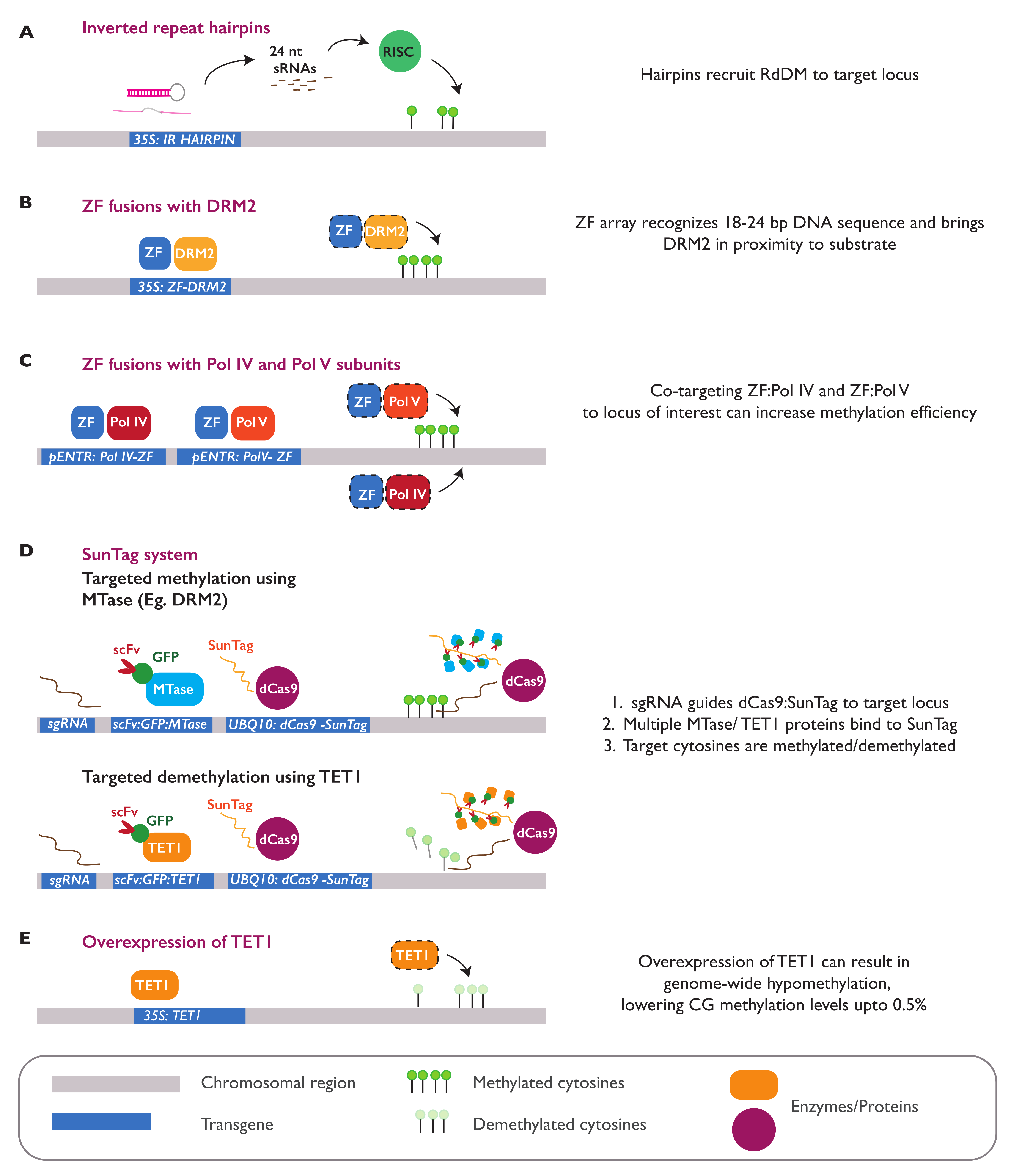
3.4. Epialleles Introduced by Epimutagenesis
3.5. Establishment and Maintenance of Induced Epialleles
4. Regulation of Gene Expressions by Epialleles
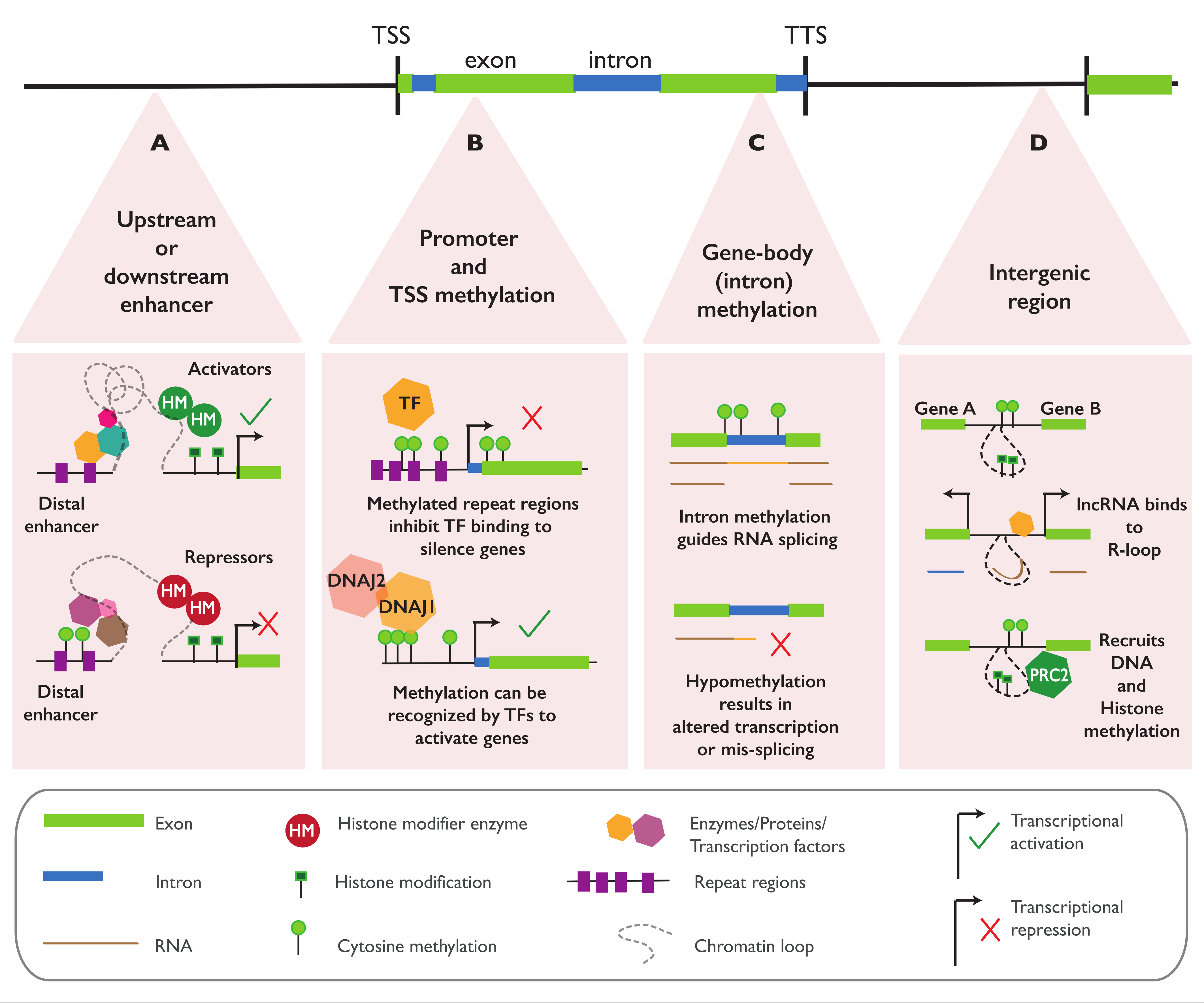
4.1. Regulatory Function of Epialleles in Proximity to Genes
4.2. Regulatory Function of Epialleles in the Gene Body
4.3. Regulatory Function of Epialleles at Distal Elements
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
| Epigenetic marks | Covalent modifications to DNA or chromatin-associated proteins that have the potential to alter gene regulation without altering the primary DNA sequence. |
| DNA methylation | An epigenetic mark involving the addition of methyl groups into nucleotides. The most common form of DNA methylation in higher eukaryotic organisms is cytosine methylation. |
| Epialleles | Heritable epigenetic variations that occur in certain loci that can potentially alter the transcription of associated genes or activity of the associated transposable elements. |
| Naturally occurring epialleles | Epialleles that have spontaneously occurred and are evolutionarily fixed in natural populations. |
| Artificially induced epialleles | Epialleles that are artificially induced by subjecting plants to various treatments and stimuli. |
| Transgenerational epigenetic inheritance | The transmission of epialleles and their associated transcriptional and phenotypic changes across generations. |
References
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]
- Eichten, S.R.; Schmitz, R.J.; Springer, N.M. Epigenetics: Beyond Chromatin Modifications and Complex Genetic Regulation. Plant Physiol. 2014, 165, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Kalisz, S.; Purugganan, M.D. Epialleles via DNA Methylation: Consequences for Plant Evolution. Trends Ecol. Evol. 2004, 19, 309–314. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and Function of DNA Methylation in Plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Hagmann, J.; Muller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous Epigenetic Variation in the Arabidopsis Thaliana Methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colomé-Tatché, M.; Johannes, F. Rate, Spectrum, and Evolutionary Dynamics of Spontaneous Epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational Epigenetic Instability Is a Source of Novel Methylation Variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef]
- Hofmeister, B.T.; Lee, K.; Rohr, N.A.; Hall, D.W.; Schmitz, R.J. Stable Inheritance of DNA Methylation Allows Creation of Epigenotype Maps and the Study of Epiallele Inheritance Patterns in the Absence of Genetic Variation. Genome Biol. 2017, 18, 155. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Huang, S.-S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic Diversity in a Global Collection of Arabidopsis Thaliana Accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread Natural Variation of DNA Methylation within Angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef]
- He, L.; Wu, W.; Zinta, G.; Yang, L.; Wang, D.; Liu, R.; Zhang, H.; Zheng, Z.; Huang, H.; Zhang, Q.; et al. A Naturally Occurring Epiallele Associates with Leaf Senescence and Local Climate Adaptation in Arabidopsis Accessions. Nat. Commun. 2018, 9, 460. [Google Scholar] [CrossRef]
- Silveira, A.B.; Trontin, C.; Cortijo, S.; Barau, J.; Del Bem, L.E.V.; Loudet, O.; Colot, V.; Vincentz, M. Extensive Natural Epigenetic Variation at a de Novo Originated Gene. PLoS Genet. 2013, 9, e1003437. [Google Scholar] [CrossRef] [PubMed]
- Luff, B.; Pawlowski, L.; Bender, J. An Inverted Repeat Triggers Cytosine Methylation of Identical Sequences in Arabidopsis. Mol. Cell 1999, 3, 505–511. [Google Scholar] [CrossRef]
- Durand, S.; Bouché, N.; Perez Strand, E.; Loudet, O.; Camilleri, C. Rapid Establishment of Genetic Incompatibility through Natural Epigenetic Variation. Curr. Biol. 2012, 22, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Liégard, B.; Baillet, V.; Etcheverry, M.; Joseph, E.; Lariagon, C.; Lemoine, J.; Evrard, A.; Colot, V.; Gravot, A.; Manzanares-Dauleux, M.J.; et al. Quantitative Resistance to Clubroot Infection Mediated by Transgenerational Epigenetic Variation in Arabidopsis. New Phytol. 2019, 222, 468–479. [Google Scholar] [CrossRef]
- Pignatta, D.; Erdmann, R.M.; Scheer, E.; Picard, C.L.; Bell, G.W.; Gehring, M. Natural Epigenetic Polymorphisms Lead to Intraspecific Variation in Arabidopsis Gene Imprinting. eLife 2014, 3, e03198. [Google Scholar] [CrossRef]
- Pignatta, D.; Novitzky, K.; Satyaki, P.R.V.; Gehring, M. A Variably Imprinted Epiallele Impacts Seed Development. PLoS Genet. 2018, 14, e1007469. [Google Scholar] [CrossRef]
- Bondada, R.; Somasundaram, S.; Marimuthu, M.P.; Badarudeen, M.A.; Puthiyaveedu, V.K.; Maruthachalam, R. Natural Epialleles of Arabidopsis SUPERMAN Display Superwoman Phenotypes. Commun. Biol. 2020, 3, 772. [Google Scholar] [CrossRef]
- Quadrana, L.; Almeida, J.; Asís, R.; Duffy, T.; Dominguez, P.G.; Bermúdez, L.; Conti, G.; Corrêa da Silva, J.V.; Peralta, I.E.; Colot, V.; et al. Natural Occurring Epialleles Determine Vitamin E Accumulation in Tomato Fruits. Nat. Commun. 2014, 5, 3027. [Google Scholar] [CrossRef]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A Naturally Occurring Epigenetic Mutation in a Gene Encoding an SBP-Box Transcription Factor Inhibits Tomato Fruit Ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Pitrat, M.; Dogimont, C.; Bendahmane, A. A Transposon-Induced Epigenetic Change Leads to Sex Determination in Melon. Nature 2009, 461, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Song, X.; Wei, L.; Tang, S.; Sun, J.; Hu, P.; Cao, X. An Epiallele of Rice AK1 Affects Photosynthetic Capacity. J. Integr. Plant Biol. 2017, 59, 158–163. [Google Scholar] [CrossRef]
- Miura, K.; Agetsuma, M.; Kitano, H.; Yoshimura, A.; Matsuoka, M.; Jacobsen, S.E.; Ashikari, M. A Metastable DWARF1 Epigenetic Mutant Affecting Plant Stature in Rice. Proc. Natl. Acad. Sci. USA 2009, 106, 11218–11223. [Google Scholar] [CrossRef]
- Cubas, P.; Vincent, C.; Coen, E. An Epigenetic Mutation Responsible for Natural Variation in Floral Symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef]
- Sekhon, R.S.; Peterson, T.; Chopra, S. Epigenetic Modifications of Distinct Sequences of the p1 Regulatory Gene Specify Tissue-Specific Expression Patterns in Maize. Genetics 2007, 175, 1059–1070. [Google Scholar] [CrossRef][Green Version]
- Xu, J.; Chen, G.; Hermanson, P.J.; Xu, Q.; Sun, C.; Chen, W.; Kan, Q.; Li, M.; Crisp, P.A.; Yan, J.; et al. Population-Level Analysis Reveals the Widespread Occurrence and Phenotypic Consequence of DNA Methylation Variation Not Tagged by Genetic Variation in Maize. Genome Biol. 2019, 20, 243. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Clarke, C.R.; Larose, H.; Tran, H.T.; Haak, D.C.; Zhang, L.; Askew, S.; Barney, J.; Westwood, J.H. Herbicide Injury Induces DNA Methylome Alterations in Arabidopsis. Peer J. 2017, 5, e3560. [Google Scholar] [CrossRef]
- Lu, Y.C.; Feng, S.J.; Zhang, J.J.; Luo, F.; Zhang, S.; Yang, H. Genome-Wide Identification of DNA Methylation Provides Insights into the Association of Gene Expression in Rice Exposed to Pesticide Atrazine. Sci. Rep. 2016, 6, 18985. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, P.T.; Orłowska, R. Plant Tissue Culture Environment as a Switch-Key of (epi) Genetic Changes. Plant Cell Tissue Organ Cult. 2020, 140, 245–257. [Google Scholar] [CrossRef]
- Reinders, J.; Wulff, B.B.H.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised Stability of DNA Methylation and Transposon Immobilization in Mosaic Arabidopsis Epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the Impact of Transgenerational Epigenetic Variation on Complex Traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Antunez-Sanchez, J.; Naish, M.; Ramirez-Prado, J.S.; Ohno, S.; Huang, Y.; Dawson, A.; Opassathian, K.; Manza-Mianza, D.; Ariel, F.; Raynaud, C.; et al. A New Role for Histone Demethylases in the Maintenance of Plant Genome Integrity. eLife 2020, 9, e58533. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis Thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.; Schmitz, R.J. Spontaneous Epimutations in Plants. New Phytol. 2019, 221, 1253–1259. [Google Scholar] [CrossRef]
- Schmid, M.W.; Heichinger, C.; Coman Schmid, D.; Guthörl, D.; Gagliardini, V.; Bruggmann, R.; Aluri, S.; Aquino, C.; Schmid, B.; Turnbull, L.A.; et al. Contribution of Epigenetic Variation to Adaptation in Arabidopsis. Nat. Commun. 2018, 9, 4446. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-Wide Inheritance of Cytosine Methylation Variants in a Recombinant Inbred Population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, J.; Liu, Y.; Liu, S.; Liu, Z.; Duan, Z.; Wang, Z.; Zhu, B.; Guo, Y.-L.; Tian, Z. DNA Methylation Footprints during Soybean Domestication and Improvement. Genome Biol. 2018, 19, 128. [Google Scholar] [CrossRef]
- Eichten, S.R.; Swanson-Wagner, R.A.; Schnable, J.C.; Waters, A.J.; Hermanson, P.J.; Liu, S.; Yeh, C.-T.; Jia, Y.; Gendler, K.; Freeling, M.; et al. Heritable Epigenetic Variation among Maize Inbreds. PLoS Genet. 2011, 7, e1002372. [Google Scholar] [CrossRef]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P.; et al. Epigenetic and Genetic Influences on DNA Methylation Variation in Maize Populations. Plant Cell 2013, 25, 2783–2797. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M. Inheritance Patterns and Stability of DNA Methylation Variation in Maize near-Isogenic Lines. Genetics 2014, 196, 667–676. [Google Scholar] [CrossRef]
- Eichten, S.R.; Stuart, T.; Srivastava, A.; Lister, R.; Borevitz, J.O. DNA Methylation Profiles of Diverse Brachypodium Distachyon Align with Underlying Genetic Diversity. Genome Res. 2016, 26, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Paszkowski, J. Epigenetic Memory in Plants. EMBO J. 2014, 33, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Wyler, M.; Stritt, C.; Walser, J.-C.; Baroux, C.; Roulin, A.C. Impact of Transposable Elements on Methylation and Gene Expression across Natural Accessions of Brachypodium Distachyon. bioRxiv 2020, arXiv:2020.06.16.154047. [Google Scholar]
- Shen, X.; De Jonge, J.; Forsberg, S.K.G.; Pettersson, M.E.; Sheng, Z.; Hennig, L.; Carlborg, Ö. Natural CMT2 Variation Is Associated with Genome-Wide Methylation Changes and Temperature Seasonality. PLoS Genet. 2014, 10, e1004842. [Google Scholar] [CrossRef]
- Sasaki, E.; Kawakatsu, T.; Ecker, J.R.; Nordborg, M. Common Alleles of CMT2 and NRPE1 Are Major Determinants of CHH Methylation Variation in Arabidopsis Thaliana. PLoS Genet. 2019, 15, e1008492. [Google Scholar] [CrossRef]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.-S.; Osborne, E.J.; Paolo Casale, F.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjálmsson, B.; et al. DNA Methylation in Arabidopsis Has a Genetic Basis and Shows Evidence of Local Adaptation. eLife 2015, 4, e05255. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Eichten, S.R.; Cahn, J.; Karpievitch, Y.V.; Borevitz, J.O.; Lister, R. Population Scale Mapping of Transposable Element Diversity Reveals Links to Gene Regulation and Epigenomic Variation. eLife 2016, 5, e20777. [Google Scholar] [CrossRef]
- Quadrana, L.; Bortolini Silveira, A.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis Thaliana Mobilome and Its Impact at the Species Level. eLife 2016, 5, e15716. [Google Scholar] [CrossRef]
- Baduel, P.; Leduque, B.; Ignace, A.; Gy, I.; Gil, J., Jr.; Loudet, O.; Colot, V.; Quadrana, L. Genetic and Environmental Modulation of Transposition Shapes the Evolutionary Potential of Arabidopsis Thaliana. Genome Biol. 2021, 22, 138. [Google Scholar] [CrossRef]
- Noshay, J.M.; Anderson, S.N.; Zhou, P.; Ji, L.; Ricci, W.; Lu, Z.; Stitzer, M.C.; Crisp, P.A.; Hirsch, C.N.; Zhang, X.; et al. Monitoring the Interplay between Transposable Element Families and DNA Methylation in Maize. PLoS Genet. 2019, 15, e1008291. [Google Scholar] [CrossRef]
- Choi, J.Y.; Purugganan, M.D. Evolutionary Epigenomics of Retrotransposon-Mediated Methylation Spreading in Rice. Mol. Biol. Evol. 2018, 35, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wendte, J.M.; Ji, L.; Schmitz, R.J. Natural Variation in DNA Methylation Homeostasis and the Emergence of Epialleles. Proc. Natl. Acad. Sci. USA 2020, 117, 4874–4884. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Schmitz, R.J. Covering Your Bases: Inheritance of DNA Methylation in Plant Genomes. Mol. Plant 2014, 7, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Saze, H.; Mittelsten Scheid, O.; Paszkowski, J. Maintenance of CpG Methylation Is Essential for Epigenetic Inheritance during Plant Gametogenesis. Nat. Genet. 2003, 34, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Colomé-Tatché, M.; Cortijo, S.; Wardenaar, R.; Morgado, L.; Lahouze, B.; Sarazin, A.; Etcheverry, M.; Martin, A.; Feng, S.; Duvernois-Berthet, E.; et al. Features of the Arabidopsis Recombination Landscape Resulting from the Combined Loss of Sequence Variation and DNA Methylation. Proc. Natl. Acad. Sci. USA 2012, 109, 16240–16245. [Google Scholar] [CrossRef]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the Origin and Evolutionary Consequences of Gene Body DNA Methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef]
- Catoni, M.; Griffiths, J.; Becker, C.; Zabet, N.R.; Bayon, C.; Dapp, M.; Lieberman-Lazarovich, M.; Weigel, D.; Paszkowski, J. DNA Sequence Properties That Predict Susceptibility to Epiallelic Switching. EMBO J. 2017, 36, 617–628. [Google Scholar] [CrossRef]
- Cortijo, S.; Wardenaar, R.; Colomé-Tatché, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.-M.; Wincker, P.; et al. Mapping the Epigenetic Basis of Complex Traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Kooke, R.; Johannes, F.; Wardenaar, R.; Becker, F.; Etcheverry, M.; Colot, V.; Vreugdenhil, D.; Keurentjes, J.J.B. Epigenetic Basis of Morphological Variation and Phenotypic Plasticity in Arabidopsis Thaliana. Plant Cell 2015, 27, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Furci, L.; Jain, R.; Stassen, J.; Berkowitz, O.; Whelan, J.; Roquis, D.; Baillet, V.; Colot, V.; Johannes, F.; Ton, J. Identification and Characterisation of Hypomethylated DNA Loci Controlling Quantitative Resistance in Arabidopsis. eLife 2019, 8, e40655. [Google Scholar] [CrossRef]
- Teixeira, F.K.; Heredia, F.; Sarazin, A.; Roudier, F.; Boccara, M.; Ciaudo, C.; Cruaud, C.; Poulain, J.; Berdasco, M.; Fraga, M.F.; et al. A Role for RNAi in the Selective Correction of DNA Methylation Defects. Science 2009, 323, 1600–1604. [Google Scholar] [CrossRef]
- Rigal, M.; Becker, C.; Pélissier, T.; Pogorelcnik, R.; Devos, J.; Ikeda, Y.; Weigel, D.; Mathieu, O. Epigenome Confrontation Triggers Immediate Reprogramming of DNA Methylation and Transposon Silencing in Arabidopsis Thaliana F1 Epihybrids. Proc. Natl. Acad. Sci. USA 2016, 113, E2083–E2092. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread Dynamic DNA Methylation in Response to Biotic Stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Stassen, J.H.M.; López, A.; Jain, R.; Pascual-Pardo, D.; Luna, E.; Smith, L.M.; Ton, J. The Relationship between Transgenerational Acquired Resistance and Global DNA Methylation in Arabidopsis. Sci. Rep. 2018, 8, 14761. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Bruce, T.J.A.; Roberts, M.R.; Flors, V.; Ton, J. Next-Generation Systemic Acquired Resistance. Plant Physiol. 2012, 158, 844–853. [Google Scholar] [CrossRef]
- Rambani, A.; Rice, J.H.; Liu, J.; Lane, T.; Ranjan, P.; Mazarei, M.; Pantalone, V.; Stewart, C.N., Jr.; Staton, M.; Hewezi, T. The Methylome of Soybean Roots during the Compatible Interaction with the Soybean Cyst Nematode. Plant Physiol. 2015, 168, 1364–1377. [Google Scholar] [CrossRef]
- Hewezi, T.; Lane, T.; Piya, S.; Rambani, A.; Rice, J.H.; Staton, M. Cyst Nematode Parasitism Induces Dynamic Changes in the Root Epigenome. Plant Physiol. 2017, 174, 405–420. [Google Scholar] [CrossRef]
- Rambani, A.; Pantalone, V.; Yang, S.; Rice, J.H.; Song, Q.; Mazarei, M.; Arelli, P.R.; Meksem, K.; Stewart, C.N.; Hewezi, T. Identification of Introduced and Stably Inherited DNA Methylation Variants in Soybean Associated with Soybean Cyst Nematode Parasitism. New Phytol. 2020, 227, 168–184. [Google Scholar] [CrossRef]
- Satgé, C.; Moreau, S.; Sallet, E.; Lefort, G.; Auriac, M.-C.; Remblière, C.; Cottret, L.; Gallardo, K.; Noirot, C.; Jardinaud, M.-F.; et al. Reprogramming of DNA Methylation Is Critical for Nodule Development in Medicago Truncatula. Nat Plants 2016, 2, 16166. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, R.T.; Schlüter, P.M.; Schiestl, F.P. Herbivore-Induced DNA Demethylation Changes Floral Signalling and Attractiveness to Pollinators in Brassica Rapa. PLoS ONE 2016, 11, e0166646. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.M.; Bazaga, P. Epigenetic Differentiation and Relationship to Adaptive Genetic Divergence in Discrete Populations of the Violet Viola Cazorlensis. New Phytol. 2010, 187, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Mithani, A.; Belfield, E.J.; Mott, R.; Hurst, L.D.; Harberd, N.P. Environmentally Responsive Genome-Wide Accumulation of de Novo Arabidopsis Thaliana Mutations and Epimutations. Genome Res. 2014, 24, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, A.; Becker, C.; Marconi, G.; Durr, J.; Price, J.; Hagmann, J.; Papareddy, R.; Putra, H.; Kageyama, J.; Becker, J.; et al. Hyperosmotic Stress Memory in Arabidopsis Is Mediated by Distinct Epigenetically Labile Sites in the Genome and Is Restricted in the Male Germline by DNA Glycosylase Activity. eLife 2016, 5, e13546. [Google Scholar] [CrossRef]
- Sani, E.; Herzyk, P.; Perrella, G.; Colot, V.; Amtmann, A. Hyperosmotic Priming of Arabidopsis Seedlings Establishes a Long-Term Somatic Memory Accompanied by Specific Changes of the Epigenome. Genome Biol. 2013, 14, R59. [Google Scholar] [CrossRef]
- Wang, W.; Qin, Q.; Sun, F.; Wang, Y.; Xu, D.; Li, Z.; Fu, B. Genome-Wide Differences in DNA Methylation Changes in Two Contrasting Rice Genotypes in Response to Drought Conditions. Front. Plant Sci. 2016, 7, 1675. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA Methylome Is Stable under Transgenerational Drought Stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef]
- Li, R.; Hu, F.; Li, B.; Zhang, Y.; Chen, M.; Fan, T.; Wang, T. Whole Genome Bisulfite Sequencing Methylome Analysis of Mulberry (Morus Alba) Reveals Epigenome Modifications in Response to Drought Stress. Sci. Rep. 2020, 10, 8013. [Google Scholar] [CrossRef]
- Van Dooren, T.J.M.; Silveira, A.B.; Gilbault, E.; Jiménez-Gómez, J.M.; Martin, A.; Bach, L.; Tisné, S.; Quadrana, L.; Loudet, O.; Colot, V. Mild Drought in the Vegetative Stage Induces Phenotypic, Gene Expression, and DNA Methylation Plasticity in Arabidopsis but No Transgenerational Effects. J. Exp. Bot. 2020, 71, 3588–3602. [Google Scholar] [CrossRef]
- Lu, X.; Wang, X.; Chen, X.; Shu, N.; Wang, J.; Wang, D.; Wang, S.; Fan, W.; Guo, L.; Guo, X.; et al. Single-Base Resolution Methylomes of Upland Cotton (Gossypium hirsutum L.) Reveal Epigenome Modifications in Response to Drought Stress. BMC Genom. 2017, 18, 297. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational Epimutations Induced by Multi-Generation Drought Imposition Mediate Rice Plant’s Adaptation to Drought Condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, Y.; Duan, W.; Huang, F.; Hou, X. Cold Acclimation Alters DNA Methylation Patterns and Confers Tolerance to Heat and Increases Growth Rate in Brassica Rapa. J. Exp. Bot. 2017, 68, 1213–1224. [Google Scholar] [CrossRef]
- Tang, X.; Wang, Q.; Yuan, H.; Huang, X. Chilling-Induced DNA Demethylation Is Associated with the Cold Tolerance of Hevea Brasiliensis. BMC Plant Biol. 2018, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Eichten, S.R.; Springer, N.M. Minimal Evidence for Consistent Changes in Maize DNA Methylation Patterns Following Environmental Stress. Front. Plant Sci. 2015, 6, 308. [Google Scholar] [CrossRef]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress Induced Gene Expression Drives Transient DNA Methylation Changes at Adjacent Repetitive Elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef]
- Yong-Villalobos, L.; González-Morales, S.I.; Wrobel, K.; Gutiérrez-Alanis, D.; Cervantes-Peréz, S.A.; Hayano-Kanashiro, C.; Oropeza-Aburto, A.; Cruz-Ramírez, A.; Martínez, O.; Herrera-Estrella, L. Methylome Analysis Reveals an Important Role for Epigenetic Changes in the Regulation of the Arabidopsis Response to Phosphate Starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E7293–E7302. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xia, Y.; Liu, T.; Dai, S.; Hou, X. The DNA Methylome and Association of Differentially Methylated Regions with Differential Gene Expression during Heat Stress in Brassica Rapa. Int. J. Mol. Sci. 2018, 19, 1414. [Google Scholar] [CrossRef] [PubMed]
- Narsai, R.; Secco, D.; Schultz, M.D.; Ecker, J.R.; Lister, R.; Whelan, J. Dynamic and Rapid Changes in the Transcriptome and Epigenome during Germination and in Developing Rice (Oryza sativa) Coleoptiles under Anoxia and Re-Oxygenation. Plant J. 2017, 89, 805–824. [Google Scholar] [CrossRef]
- Pandey, N.; Pandey-Rai, S. Deciphering UV-B-Induced Variation in DNA Methylation Pattern and Its Influence on Regulation of DBR2 Expression in Artemisia annua L. Planta 2015, 242, 869–879. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Stone, B.A.B.; Bowerman, A.F.; Eichten, S.R.; Pogson, B.J. Excess Light Priming in Arabidopsis Thaliana Genotypes with Altered DNA Methylomes. G3 2019, 9, 3611–3621. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. Maintenance of Pre-Existing DNA Methylation States through Recurring Excess-Light Stress. Plant Cell Environ. 2018, 41, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, M.; Bartucca, M.L.; Ciancaleoni, S.; Mimmo, T.; Cesco, S.; Pii, Y.; Albertini, E.; Del Buono, D. Iron Deficiency in Barley Plants: Phytosiderophore Release, Iron Translocation, and DNA Methylation. Front. Plant Sci. 2015, 6, 514. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Lepère, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.-L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; et al. Dynamics and Biological Relevance of DNA Demethylation in Arabidopsis Antibacterial Defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Wu, C.; Huang, J.; Yan, K.; Yang, G.; Zheng, C. Salt-Induced Transcription Factor MYB74 Is Regulated by the RNA-Directed DNA Methylation Pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.H.; Paszkowski, J. Heat-Induced Release of Epigenetic Silencing Reveals the Concealed Role of an Imprinted Plant Gene. PLoS Genet. 2014, 10, e1004806. [Google Scholar] [CrossRef]
- Jiang, C.; Mithani, A.; Gan, X.; Belfield, E.J.; Klingler, J.P.; Zhu, J.K.; Ragoussis, J.; Mott, R.; Harberd, N.P. Regenerant Arabidopsis Lineages Display a Distinct Genome-Wide Spectrum of Mutations Conferring Variant Phenotypes. Curr. Biol. 2011, 21, 1385–1390. [Google Scholar] [CrossRef]
- Miyao, A.; Nakagome, M.; Ohnuma, T.; Yamagata, H.; Kanamori, H.; Katayose, Y.; Takahashi, A.; Matsumoto, T.; Hirochika, H. Molecular Spectrum of Somaclonal Variation in Regenerated Rice Revealed by Whole-Genome Sequencing. Plant Cell Physiol. 2012, 53, 256–264. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Z.; Wang, N.; Gao, Y.; Liu, Y.; Wu, Y.; Bai, Y.; Zhang, Z.; Lin, X.; Dong, Y.; et al. Tissue Culture-Induced Heritable Genomic Variation in Rice, and Their Phenotypic Implications. PLoS ONE 2014, 9, e96879. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Ogawa, Y.; Iwase, A.; Sugimoto, K. Plant Regeneration: Cellular Origins and Molecular Mechanisms. Development 2016, 143, 1442–1451. [Google Scholar] [CrossRef]
- Wibowo, A.; Becker, C.; Durr, J.; Price, J.; Spaepen, S.; Hilton, S.; Putra, H.; Papareddy, R.; Saintain, Q.; Harvey, S.; et al. Partial Maintenance of Organ-Specific Epigenetic Marks during Plant Asexual Reproduction Leads to Heritable Phenotypic Variation. Proc. Natl. Acad. Sci. USA 2018, 115, E9145–E9152. [Google Scholar] [CrossRef]
- Coronel, C.J.; González, A.I.; Ruiz, M.L.; Polanco, C. Analysis of Somaclonal Variation in Transgenic and Regenerated Plants of Arabidopsis Thaliana Using Methylation Related metAFLP and TMD Markers. Plant Cell Rep. 2018, 37, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Ding, B.; Simon, S.A.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E. Plants Regenerated from Tissue Culture Contain Stable Epigenome Changes in Rice. eLife 2013, 2, e00354. [Google Scholar] [CrossRef] [PubMed]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.E.; Kok, S.Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma Transposon Methylation Underlies the Mantled Somaclonal Variant of Oil Palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef]
- Lin, W.; Xiao, X. ’ou; Zhang, H.; Li, Y.; Liu, S.; Sun, W.; Zhang, X.; Wu, Q. Whole-Genome Bisulfite Sequencing Reveals a Role for DNA Methylation in Variants from Callus Culture of Pineapple (Ananas comosus L.). Genes 2019, 10, 877. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Crisp, P.A.; Stelpflug, S.; Kaeppler, S.M.; Li, Q.; Springer, N.M. Heritable Epigenomic Changes to the Maize Methylome Resulting from Tissue Culture. Genetics 2018, 209, 983–995. [Google Scholar] [CrossRef]
- Stelpflug, S.C.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M.; Kaeppler, S.M. Consistent and Heritable Alterations of DNA Methylation Are Induced by Tissue Culture in Maize. Genetics 2014, 198, 209–218. [Google Scholar] [CrossRef]
- Rhee, Y.; Sekhon, R.S.; Chopra, S.; Kaeppler, S. Tissue Culture-Induced Novel Epialleles of a Myb Transcription Factor Encoded by Pericarp color1 in Maize. Genetics 2010, 186, 843–855. [Google Scholar] [CrossRef]
- Pecinka, A.; Liu, C.-H. Drugs for Plant Chromosome and Chromatin Research. Cytogenet. Genome Res. 2014, 143, 51–59. [Google Scholar]
- Madlung, A.; Masuelli, R.W.; Watson, B.; Reynolds, S.H.; Davison, J.; Comai, L. Remodeling of DNA Methylation and Phenotypic and Transcriptional Changes in Synthetic Arabidopsis Allotetraploids. Plant Physiol. 2002, 129, 733–746. [Google Scholar] [CrossRef]
- Janoušek, B.; Široký, J.; Vyskot, B. Epigenetic Control of Sexual Phenotype in a Dioecious plant, Melandrium Album. Mol. Gen. Genet. 1996, 250, 483–490. [Google Scholar] [CrossRef]
- Griffin, P.T.; Niederhuth, C.E.; Schmitz, R.J. A Comparative Analysis of 5-Azacytidine- and Zebularine-Induced DNA Demethylation. G3 2016, 6, 2773–2780. [Google Scholar] [CrossRef]
- Ji, L.; Jordan, W.T.; Shi, X.; Hu, L.; He, C.; Schmitz, R.J. TET-Mediated Epimutagenesis of the Arabidopsis Thaliana Methylome. Nat. Commun. 2018, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Bartolomé, J. DNA Methylation in Plants: Mechanisms and Tools for Targeted Manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef]
- Mette, M.F.; Aufsatz, W.; Van der Winden, J.; Matzke, M.A.; Matzke, A.J.M. Transcriptional Silencing and Promoter Methylation Triggered by Double-Stranded RNA. EMBO J. 2000, 19, 5194–5201. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, Q.; Smith, N.A.; Liang, G.; Wang, M.-B. RNA Silencing in Plants: Mechanisms, Technologies and Applications in Horticultural Crops. Curr. Genom. 2016, 17, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Srikant, T.; Wibowo, A.; Schwab, R.; Weigel, D. Position-Dependent Effects of Cytosine Methylation on FWA Expression in Arabidopsis Thaliana. bioRxiv 2019, arXiv:774281. [Google Scholar]
- Williams, B.P.; Pignatta, D.; Henikoff, S.; Gehring, M. Methylation-Sensitive Expression of a DNA Demethylase Gene Serves as an Epigenetic Rheostat. PLoS Genet. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Zicola, J.; Liu, L.; Tänzler, P.; Turck, F. Targeted DNA Methylation Represses Two Enhancers of FLOWERING LOCUS T in Arabidopsis Thaliana. Nat. Plants 2019, 5, 300–307. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Liu, W.; Kuo, P.H.; Feng, S.; Ghoshal, B.; Gardiner, J.; Zhao, J.M.-C.; Park, S.Y.; Chory, J.; Jacobsen, S.E. Co-Targeting RNA Polymerases IV and V Promotes Efficient De Novo DNA Methylation in Arabidopsis. Cell 2019, 176, 1068–1082.e19. [Google Scholar] [CrossRef]
- Xue, Y.; Zhong, Z.; Harris, C.J.; Gallego-Bartolomé, J.; Wang, M.; Picard, C.; Cao, X.; Hua, S.; Kwok, I.; Feng, S.; et al. Arabidopsis MORC Proteins Function in the Efficient Establishment of RNA Directed DNA Methylation. Nat. Commun. 2021, 12, 4292. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA Demethylation of the Arabidopsis Genome Using the Human TET1 Catalytic Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA Epigenome Editing Using CRISPR-Cas SunTag-Directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, B.; Picard, C.L.; Vong, B.; Feng, S.; Jacobsen, S.E. CRISPR-Based Targeting of DNA Methylation in Arabidopsis Thaliana by a Bacterial CG-Specific DNA Methyltransferase. Proc. Natl. Acad. Sci. USA 2021, 118, e2125016118. [Google Scholar] [CrossRef]
- Quadrana, L.; Colot, V. Plant Transgenerational Epigenetics. Annu. Rev. Genet. 2016, 50, 467–491. [Google Scholar] [CrossRef]
- Iwasaki, M.; Paszkowski, J. Identification of Genes Preventing Transgenerational Transmission of Stress-Induced Epigenetic States. Proc. Natl. Acad. Sci. USA 2014, 111, 8547–8552. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.-L.; Huang, H.; Zhang, G.; He, L.; Pang, J.; Lozano-Durán, R.; Lang, Z.; Zhu, J.-K. Epigenetic Memory Marks Determine Epiallele Stability at Loci Targeted by de Novo DNA Methylation. Nat. Plants 2020, 6, 661–674. [Google Scholar] [CrossRef]
- Dubin, M.J.; Mittelsten Scheid, O.; Becker, C. Transposons: A Blessing Curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Blevins, T.; Wang, J.; Pflieger, D.; Pontvianne, F.; Pikaard, C.S. Hybrid Incompatibility Caused by an Epiallele. Proc. Natl. Acad. Sci. USA 2017, 114, 3702–3707. [Google Scholar] [CrossRef] [PubMed]
- Agorio, A.; Durand, S.; Fiume, E.; Brousse, C.; Gy, I.; Simon, M.; Anava, S.; Rechavi, O.; Loudet, O.; Camilleri, C.; et al. An Arabidopsis Natural Epiallele Maintained by a Feed-Forward Silencing Loop between Histone and DNA. PLoS Genet. 2017, 13, e1006551. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Saze, H.; Kinoshita, T.; Miura, A. Control of FWA Gene Silencing in Arabidopsis Thaliana by SINE-related Direct Repeats. Plant 2007, 49, 38–45. [Google Scholar] [CrossRef]
- O’Malley, R.C.; Huang, S.-S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 166, 1598. [Google Scholar] [CrossRef]
- Habu, Y.; Mathieu, O.; Tariq, M.; Probst, A.V.; Smathajitt, C.; Zhu, T.; Paszkowski, J. Epigenetic Regulation of Transcription in Intermediate Heterochromatin. EMBO Rep. 2006, 7, 1279–1284. [Google Scholar] [CrossRef]
- Soppe, W.J.J.; Jasencakova, Z.; Houben, A.; Kakutani, T.; Meister, A.; Huang, M.S.; Jacobsen, S.E.; Schubert, I.; Fransz, P.F. DNA Methylation Controls Histone H3 Lysine 9 Methylation and Heterochromatin Assembly in Arabidopsis. EMBO J. 2002, 21, 6549–6559. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Saze, H.; Probst, A.V.; Lichota, J.; Habu, Y.; Paszkowski, J. Erasure of CpG Methylation in Arabidopsis Alters Patterns of Histone H3 Methylation in Heterochromatin. Proc. Natl. Acad. Sci. USA 2003, 100, 8823–8827. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the Relationship between DNA Methylation and Histone Lysine Methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef]
- Du, J.; Zhong, X.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A.; et al. Dual Binding of Chromomethylase Domains to H3K9me2-Containing Nucleosomes Directs DNA Methylation in Plants. Cell 2012, 151, 167–180. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Groth, M.; Feng, S.; Hale, C.J.; Li, S.; Vashisht, A.A.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Mechanism of DNA Methylation-Directed Histone Methylation by KRYPTONITE. Mol. Cell 2014, 55, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Zimmerli, L. The Histone Demethylase IBM1 Positively Regulates Arabidopsis Immunity by Control of Defense Gene Expression. Front. Plant Sci. 2019, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- West, P.T.; Li, Q.; Ji, L.; Eichten, S.R.; Song, J.; Vaughn, M.W.; Schmitz, R.J.; Springer, N.M. Genomic Distribution of H3K9me2 and DNA Methylation in a Maize Genome. PLoS ONE 2014, 9, e105267. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Mei, H.; Deng, X.; He, K.; Wu, B.; Yao, Q.; Zhang, J.; Lu, F.; Ma, J.; Cao, X. DNA Methylation Repels Targeting of Arabidopsis REF6. Nat. Commun. 2019, 10, 2063. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; He, K.; Ma, Y.; Su, N.; He, H.; Stolc, V.; Tongprasit, W.; Jin, W.; Jiang, J.; et al. High-Resolution Mapping of Epigenetic Modifications of the Rice Genome Uncovers Interplay between DNA Methylation, Histone Methylation, and Gene Expression. Plant Cell 2008, 20, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Nie, W.-F.; Lei, M.; Zhang, M.; Tang, K.; Huang, H.; Zhang, C.; Miki, D.; Liu, P.; Yang, Y.; Wang, X.; et al. Histone Acetylation Recruits the SWR1 Complex to Regulate Active DNA Demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2019, 116, 16641–16650. [Google Scholar] [CrossRef]
- Chen, X.; Liu, X.; Zhao, Y.; Zhou, D.-X. Histone H3K4me3 and H3K27me3 Regulatory Genes Control Stable Transmission of an Epimutation in Rice. Sci. Rep. 2015, 5, 13251. [Google Scholar] [CrossRef]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.-K. Regulatory Link between DNA Methylation and Active Demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef]
- Shibuya, K.; Fukushima, S.; Takatsuji, H. RNA-Directed DNA Methylation Induces Transcriptional Activation in Plants. Proc. Natl. Acad. Sci. USA 2009, 106, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical Roles of DNA Demethylation in the Activation of Ripening-Induced Genes and Inhibition of Ripening-Repressed Genes in Tomato Fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.-Q.; Lin, R.-N.; Li, L.; Chen, S.; He, X.-J. A Methylated-DNA-Binding Complex Required for Plant Development Mediates Transcriptional Activation of Promoter Methylated Genes. J. Integr. Plant Biol. 2019, 61, 120–139. [Google Scholar] [CrossRef]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.; Cornett, E.M.; Vaughan, R.M.; Li, X.; Chen, W.; Xue, Y.; Zhong, Z.; et al. A DNA Methylation Reader Complex That Enhances Gene Transcription. Science 2018, 362, 1182–1186. [Google Scholar] [CrossRef]
- Jacobsen, S.E.; Meyerowitz, E.M. Hypermethylated SUPERMAN Epigenetic Alleles in Arabidopsis. Science 1997, 277, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Breuil-Broyer, S.; Trehin, C.; Morel, P.; Boltz, V.; Sun, B.; Chambrier, P.; Ito, T.; Negrutiu, I. Analysis of the Arabidopsis Superman Allelic Series and the Interactions with Other Genes Demonstrate Developmental Robustness and Joint Specification of Male-Female Boundary, Flower Meristem Termination and Carpel Compartmentalization. Ann. Bot. 2016, 117, 905–923. [Google Scholar] [CrossRef]
- Shahzad, Z.; Moore, J.D.; Zilberman, D. Gene Body Methylation Mediates Epigenetic Inheritance of Plant Traits. bioRxiv 2021, arXiv:435374. [Google Scholar]
- Zilberman, D. An Evolutionary Case for Functional Gene Body Methylation in Plants and Animals. Genome Biol. 2017, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Laenen, B.; Takuno, S.; Slotte, T. Single-Cell Expression Noise and Gene-Body Methylation in Arabidopsis Thaliana. Heredity 2019, 123, 81–91. [Google Scholar] [CrossRef]
- Wang, X.; Hu, L.; Wang, X.; Li, N.; Xu, C.; Gong, L.; Liu, B. DNA Methylation Affects Gene Alternative Splicing in Plants: An Example from Rice. Mol. Plant 2016, 9, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Feng, S.; Duttke, S.H.; Potok, M.E.; Zhang, Y.; Gallego-Bartolomé, J.; Liu, W.; Jacobsen, S.E. DNA Methylation-Linked Chromatin Accessibility Affects Genomic Architecture in Arabidopsis. Proc. Natl. Acad. Sci. USA 2021, 118, e2023347118. [Google Scholar] [CrossRef]
- Haring, M.; Bader, R.; Louwers, M.; Schwabe, A.; van Driel, R.; Stam, M. The Role of DNA Methylation, Nucleosome Occupancy and Histone Modifications in Paramutation. Plant J. 2010, 63, 366–378. [Google Scholar] [CrossRef]
- Louwers, M.; Bader, R.; Haring, M.; van Driel, R.; de Laat, W.; Stam, M. Tissue- and Expression Level-Specific Chromatin Looping at Maize b1 Epialleles. Plant Cell 2009, 21, 832–842. [Google Scholar] [CrossRef]
- Ariel, F.; Jegu, T.; Latrasse, D.; Romero-Barrios, N.; Christ, A.; Benhamed, M.; Crespi, M. Noncoding Transcription by Alternative RNA Polymerases Dynamically Regulates an Auxin-Driven Chromatin Loop. Mol. Cell 2014, 55, 383–396. [Google Scholar] [CrossRef]
- Ariel, F.; Lucero, L.; Christ, A.; Mammarella, M.F.; Jegu, T.; Veluchamy, A.; Mariappan, K.; Latrasse, D.; Blein, T.; Liu, C.; et al. R-Loop Mediated Trans Action of the APOLO Long Noncoding RNA. Mol. Cell 2020, 77, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Hu, F.; Zhou, Y.; Wu, F.; Gaut, B.S. Maize Transposable Elements Contribute to Long Non-Coding RNAs That Are Regulatory Hubs for Abiotic Stress Response. BMC Genom. 2019, 20, 864. [Google Scholar] [CrossRef]
- Williams, B.P.; Gehring, M. Stable Transgenerational Epigenetic Inheritance Requires a DNA Methylation-Sensing Circuit. Nat. Commun. 2017, 8, 2124. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Custard, K.D.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DNA Methylation Is Critical for Arabidopsis Embryogenesis and Seed Viability. Plant Cell 2006, 18, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Butel, N.; Santos-González, J.; Simon, L.; Wärdig, C.; Köhler, C. Transgenerational Effect of Mutants in the RNA-Directed DNA Methylation Pathway on the Triploid Block in Arabidopsis. Genome Biol. 2021, 22, 141. [Google Scholar] [CrossRef]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P. Mutation Bias Shapes Gene Evolution in Arabidopsis Thaliana. bioRxiv 2020, arXiv:156752. [Google Scholar]
- Srikant, T.; Drost, H.-G. How Stress Facilitates Phenotypic Innovation through Epigenetic Diversity. Front. Plant Sci. 2020, 11, 606800. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srikant, T.; Tri Wibowo, A. The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles. Int. J. Mol. Sci. 2021, 22, 8618. https://doi.org/10.3390/ijms22168618
Srikant T, Tri Wibowo A. The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles. International Journal of Molecular Sciences. 2021; 22(16):8618. https://doi.org/10.3390/ijms22168618
Chicago/Turabian StyleSrikant, Thanvi, and Anjar Tri Wibowo. 2021. "The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles" International Journal of Molecular Sciences 22, no. 16: 8618. https://doi.org/10.3390/ijms22168618
APA StyleSrikant, T., & Tri Wibowo, A. (2021). The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles. International Journal of Molecular Sciences, 22(16), 8618. https://doi.org/10.3390/ijms22168618