
Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes

 , , and
, , and 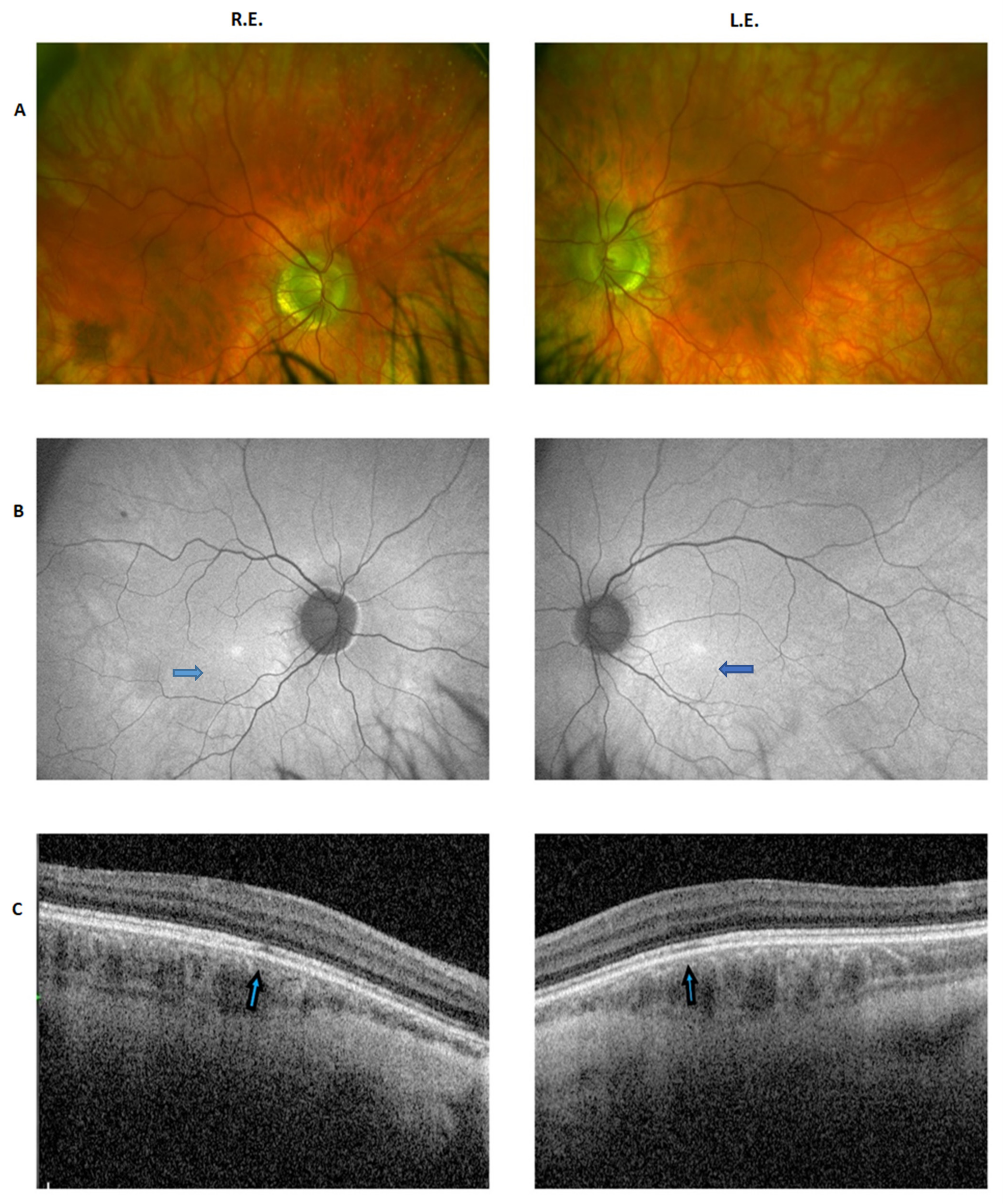{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
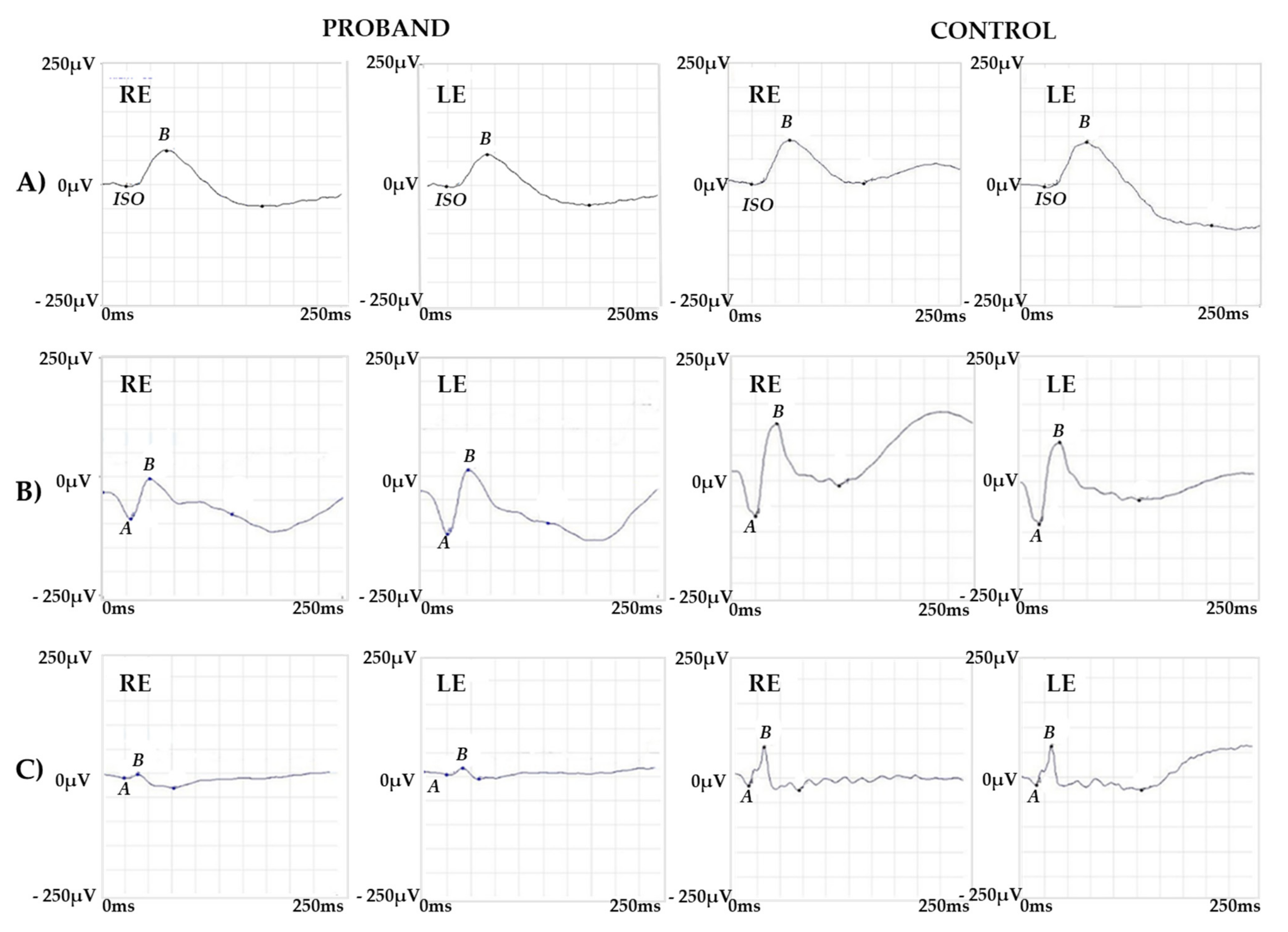
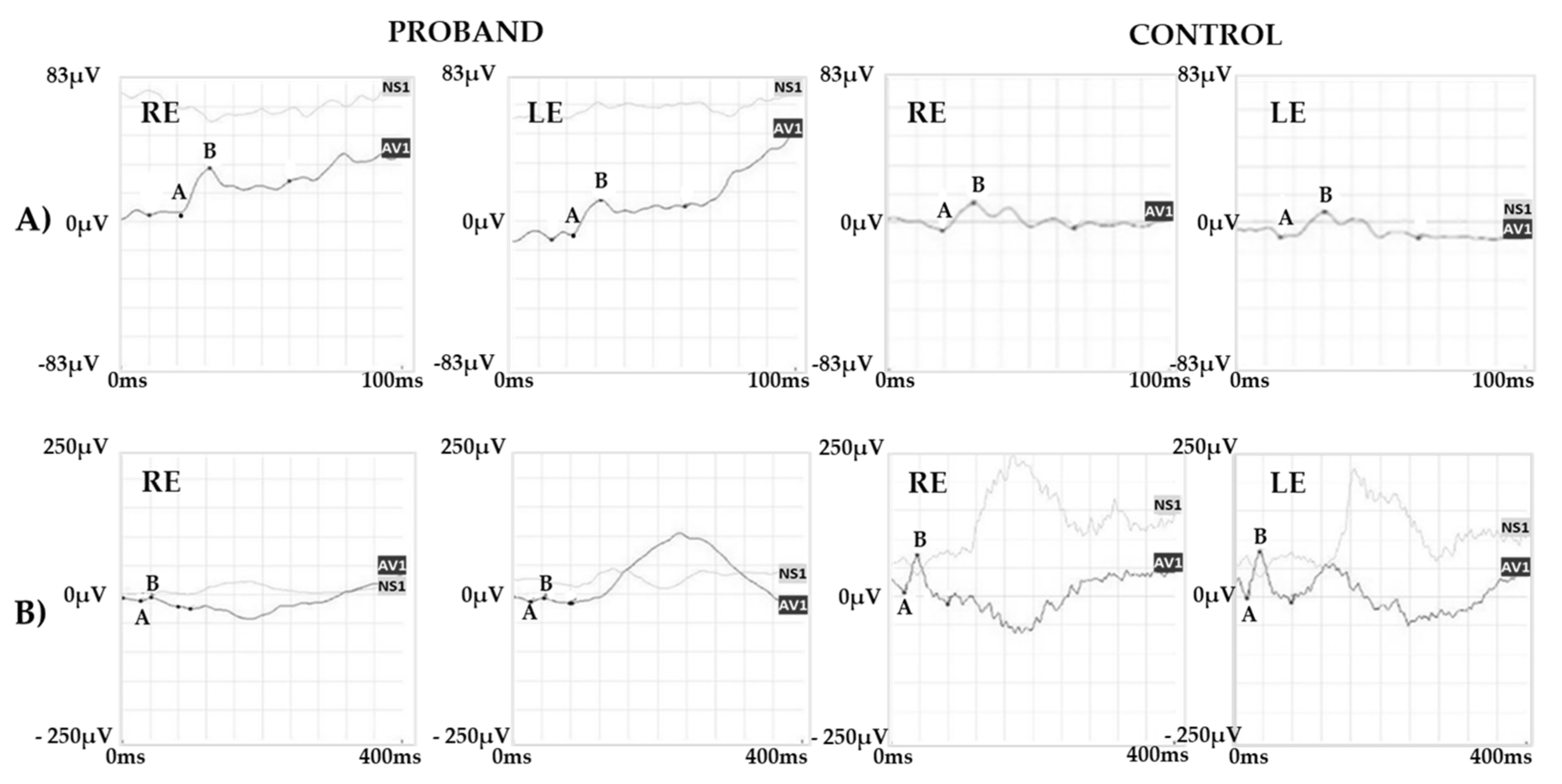
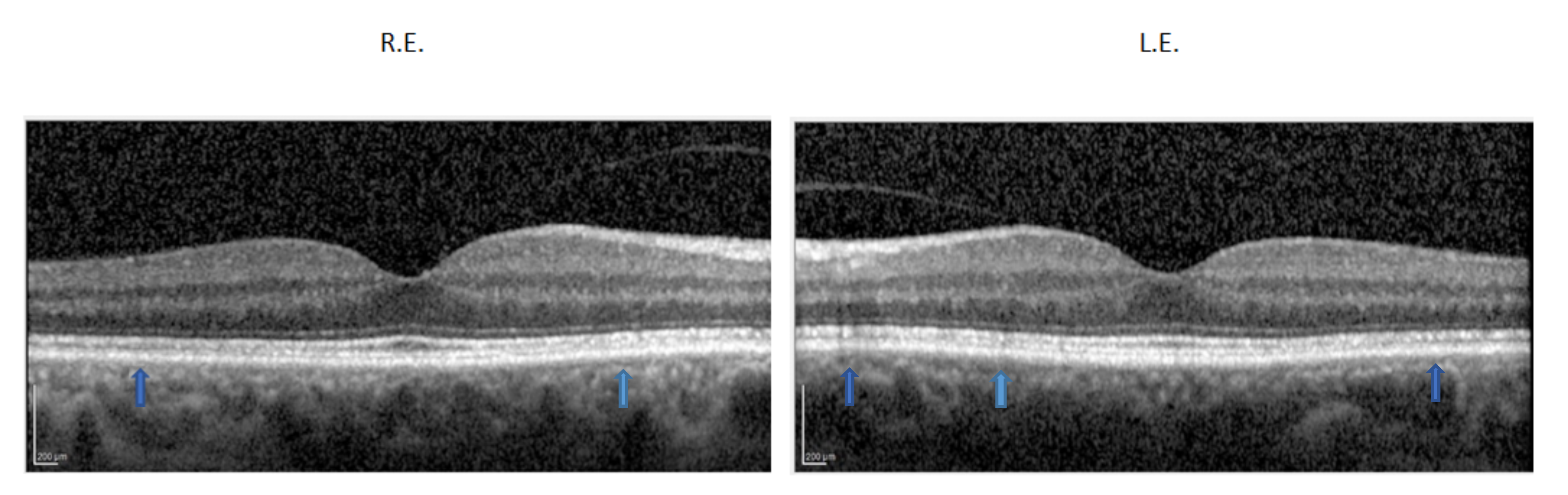
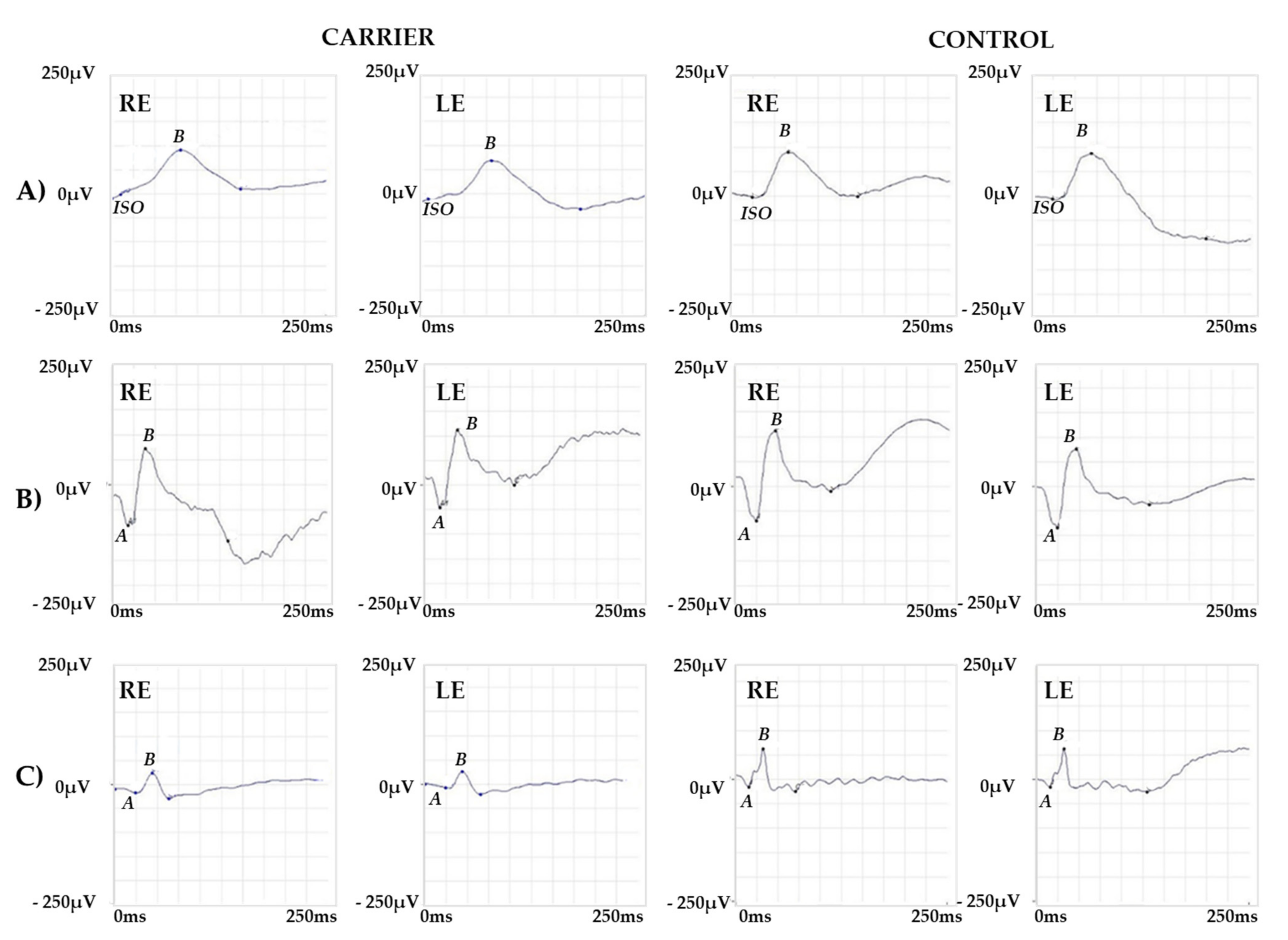
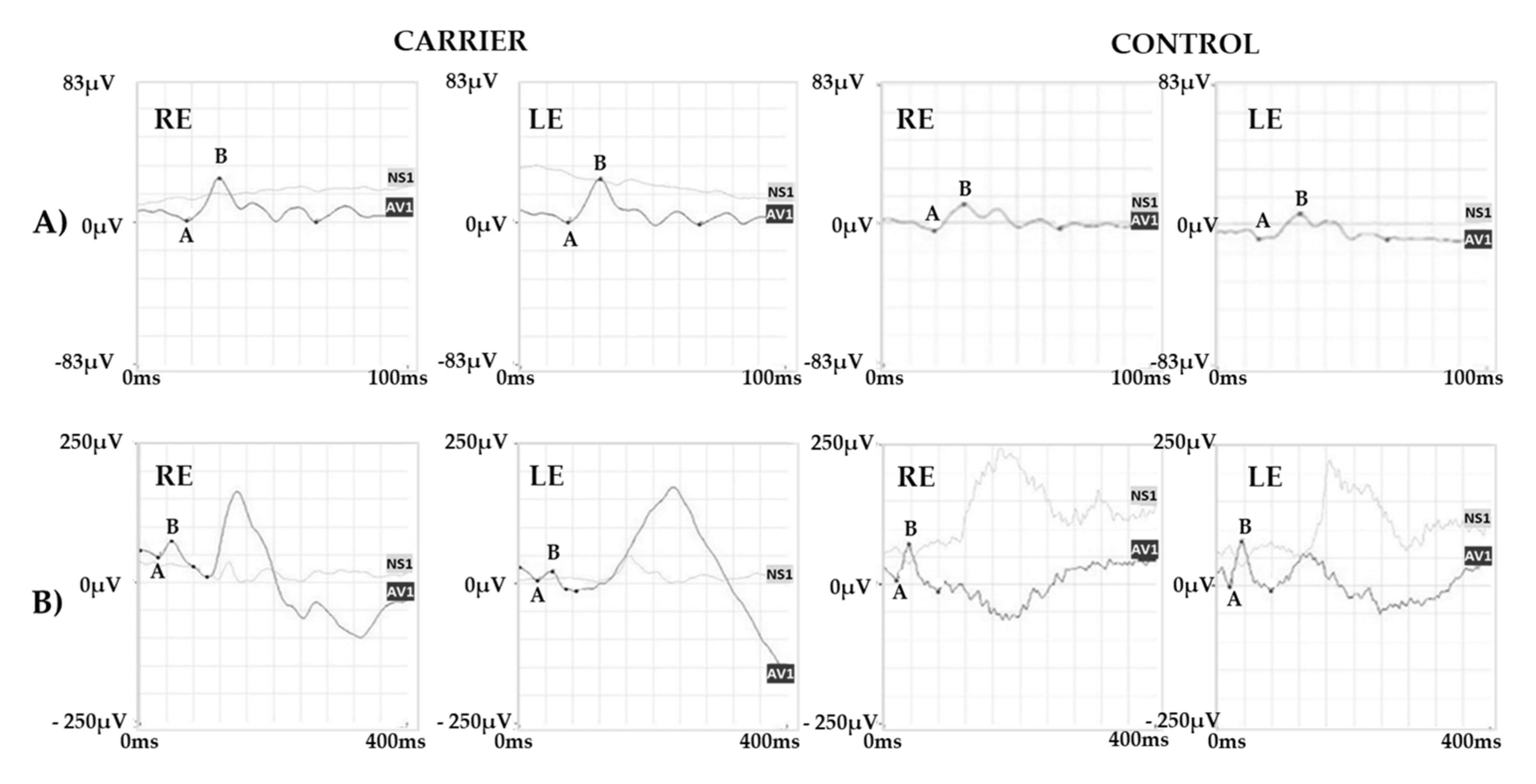
2.1. Clinical Report of the Proband and the Carrier
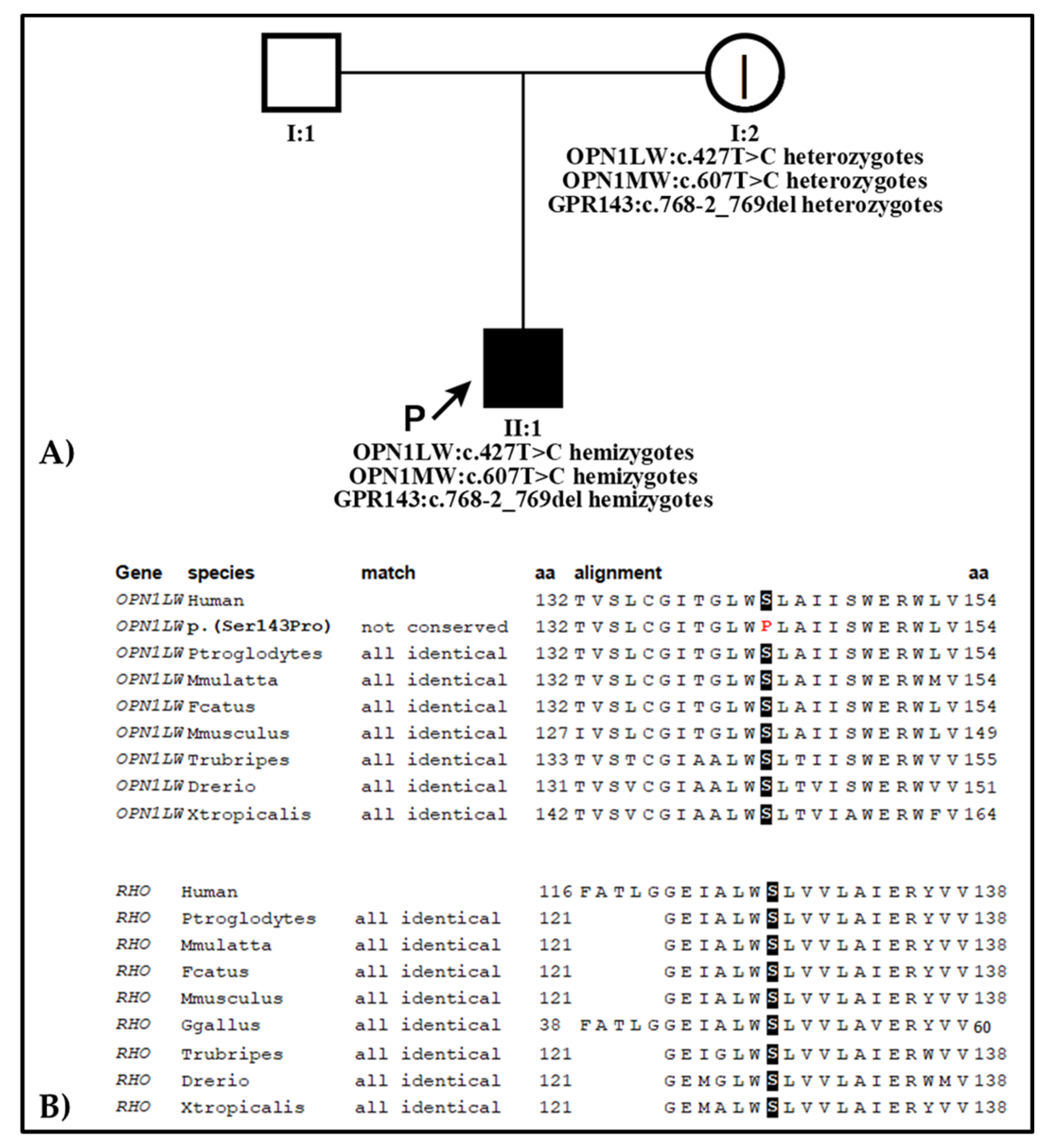
2.2. Genetic Analysis
3. Discussion
4. Materials and Methods
4.1. Patient Studies and Clinical and Ophthalmological Examinations
4.2. Genetic Testing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathans, J.; Thomas, D.; Hogness, D. Molecular genetics of human color vision: The genes encoding blue, green, and red pigments. Science 1986, 232, 193–202. [Google Scholar] [CrossRef]
- Nathans, J.; Davenport, C.; Maumenee, I.; Lewis, R.; Hejtmancik, J.; Litt, M.; Lovrien, E.; Weleber, R.; Bachynski, B.; Zwas, F.; et al. Molecular genetics of human blue cone monochromacy. Science 1989, 245, 831–838. [Google Scholar] [CrossRef]
- Neitz, J.; Neitz, M. The genetics of normal and defective color vision. Vis. Res. 2011, 51, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Hufnagel, R.B.; Carroll, J.; Sumaroka, A.; Luo, X.; Schwartz, S.B.; Dubra, A.; Land, M.; Michaelides, M.; Gardner, J.C.; et al. Human Cone Visual Pigment Deletions Spare Sufficient Photoreceptors to Warrant Gene Therapy. Hum. Gene Ther. 2013, 24, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.C.; Michaelides, M.; Holder, G.E.; Kanuga, N.; Webb, T.; Mollon, J.; Moore, A.T.; Hardcastle, A.J. Blue cone monochromacy: Causative mutations and associated phenotypes. Mol. Vis. 2009, 15, 876–884. [Google Scholar] [PubMed]
- Young, R.S.; Price, J. Wavelength discrimination deteriorates with illumination in blue cone monochromats. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1543–1549. [Google Scholar]
- Michaelides, M.; Johnson, S.; Simunovic, M.; Bradshaw, K.; Holder, G.; Mollon, J.; Moore, A.T.; Hunt, D.M. Blue cone monochromatism: A phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye 2004, 19, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.W.; Wissinger, B.; Kohl, S.; Sieving, P.A. CNGB3Achromatopsia with Progressive Loss of Residual Cone Function and Impaired Rod-Mediated Function. Investig. Opthalmol. Vis. Sci. 2007, 48, 3864–3871. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.H.J.; Somervuo, V.; Born, L.I.V.D.; Roosing, S.; van Schooneveld, M.J.; Kuijpers, R.W.A.M.; van Moll-Ramirez, N.; Cremers, F.P.M.; Hoyng, C.B.; Klaver, C.C.W. Progressive Loss of Cones in Achromatopsia: An Imaging Study Using Spectral-Domain Optical Coherence Tomography. Investig. Opthalmol. Vis. Sci. 2010, 51, 5952–5957. [Google Scholar] [CrossRef]
- Thomas, M.; McLean, R.J.; Kohl, S.; Sheth, V.; Gottlob, I. Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. Br. J. Ophthalmol. 2012, 96, 1232–1236. [Google Scholar] [CrossRef]
- Fahim, A.; Khan, N.W.; Zahid, S.; Schachar, I.H.; Branham, K.; Kohl, S.; Wissinger, B.; Elner, V.M.; Heckenlively, J.R.; Jayasundera, T. Diagnostic Fundus Autofluorescence Patterns in Achromatopsia. Am. J. Ophthalmol. 2013, 156, 1211–1219.e2. [Google Scholar] [CrossRef] [PubMed]
- Aboshiha, J.; Dubis, A.M.; Cowing, J.A.; Fahy, R.T.A.; Sundaram, V.; Bainbridge, J.; Ali, R.; Dubra, A.; Nardini, M.; Webster, A.R.; et al. A Prospective Longitudinal Study of Retinal Structure and Function in Achromatopsia. Investig. Opthalmol. Vis. Sci. 2014, 55, 5733–5743. [Google Scholar] [CrossRef]
- Brunetti-Pierri, R.; Karali, M.; Melillo, P.; Di Iorio, V.; De Benedictis, A.; Iaccarino, G.; Testa, F.; Banfi, S.; Simonelli, F. Clinical and Molecular Characterization of Achromatopsia Patients: A Longitudinal Study. Int. J. Mol. Sci. 2021, 22, 1681. [Google Scholar] [CrossRef]
- Carroll, J.; Dubra, A.; Gardner, J.C.; Mizrahi-Meissonnier, L.; Cooper, R.F.; Dubis, A.M.; Nordgren, R.; Genead, M.; Connor, T.B.; Stepien, K.E.; et al. The Effect of Cone Opsin Mutations on Retinal Structure and the Integrity of the Photoreceptor Mosaic. Investig. Opthalmol. Vis. Sci. 2012, 53, 8006–8015. [Google Scholar] [CrossRef]
- Mancuso, K.; Hauswirth, W.; Li, Q.; Connor, T.B.; Kuchenbecker, J.A.; Mauck, M.C.; Neitz, J.; Neitz, M. Gene therapy for red–green colour blindness in adult primates. Nature 2009, 461, 784–787. [Google Scholar] [CrossRef]
- Zhang, Z.; Pang, J.; Xia, F.; Guo, Q.; Li, L.; An, J.; Zhang, L.; Hauswirth, W.W.; Yang, S.; Li, Z. AAV-mediated gene therapy restores cone function in a rat with an M-cone Opsin deficiency, a model for blue cone monochromacy. Investig. Opthalmol. Vis. Sci. 2011, 52, 1403. [Google Scholar]
- Weiss, A.H.; Biersdorf, W.R. Blue cone monochromatism. J. Pediatr. Ophthalmol. Strabismus 1989, 26, 218–223. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Katagiri, S.; Iwasa, M.; Hayashi, T.; Hosono, K.; Yamashita, T.; Kuniyoshi, K.; Ueno, S.; Kondo, M.; Ueyama, H.; Ogita, H.; et al. Genotype determination of the OPN1LW/OPN1MW genes: Novel disease-causing mechanisms in Japanese patients with blue cone monochromacy. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Preising, M.N.; Forster, H.; Gonser, M.; Lorenz, B. Screening of TYR, OCA2, GPR143, and MC1R in patients with congenital nystagmus, macular hypoplasia, and fundus hypopigmentation indicating albinism. Mol. Vis. 2011, 17, 939–948. [Google Scholar]
- Hess, R.F.; Mullen, K.T.; Sharpe, L.T.; Zrenner, E. The photoreceptors in atypical achromatopsia. J. Physiol. 1989, 417, 123–149. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, H.; Blackwell, O. Rod and cone receptor mechanisms in typical and atypical congenital achromatopsia. Vis. Res. 1961, 1, 62–107. [Google Scholar] [CrossRef]
- Berson, E.L.; Sandberg, M.A.; Rosner, B.; Sullivan, P.L. Color Plates to Help Identify Patients with Blue Cone Monochromatism. Am. J. Ophthalmol. 1983, 95, 741–747. [Google Scholar] [CrossRef]
- Mizrahi-Meissonnier, L.; Merin, S.; Banin, E.; Sharon, O. Variable Retinal Phenotypes Caused by Mutations in the X-Linked Photopigment Gene Array. Investig. Opthalmol. Vis. Sci. 2010, 51, 3884–3892. [Google Scholar] [CrossRef]
- Nathans, J.; Maumenee, I.H.; Zrenner, E.; Sadowski, B.; Sharpe, L.T.; Lewis, R.A.; Hansen, E.; Rosenberg, T.; Schwartz, M.; Heckenlively, J.R.; et al. Genetic heterogeneity among blue-cone monochromats. Am. J. Hum. Genet. 1993, 53, 987–1000. [Google Scholar]
- Sumaroka, A.; Garafalo, A.V.; Cideciyan, A.V.; Charng, J.; Roman, A.J.; Choi, W.; Saxena, S.; Aksianiuk, V.; Kohl, S.; Wissinger, B.; et al. Blue Cone Monochromacy Caused by the C203R Missense Mutation or Large Deletion Mutations. Investig. Opthalmol. Vis. Sci. 2018, 59, 5762–5772. [Google Scholar] [CrossRef] [PubMed]
- Garafalo, A.V.; Cidecyan, A.V.; Hèon, E.; Sheplock, R.; Pearson, A.; Yu, C.W.; Sumaroka, A.; Aguirre, G.D.; Jacobson, S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020, 77, 100827. [Google Scholar] [CrossRef]
- Thomas, M.G.; Papageorgiou, E.; Kuht, H.J.; Gottlob, I. Normal and abnormal foveal development. Br. J. Ophthalmol. 2020, 4, 13. [Google Scholar] [CrossRef]
- McKay, B.S. Pigmentation and Vision: Is GPR143 in Control? J. Neurosci. Res. 2019, 97, 77–87. [Google Scholar] [CrossRef]
- Rufai, S.R.; Thomas, M.G.; Purohit, R.; Bunce, C.; Lee, H.; Proudlock, F.A.; Gottlob, I. Can Structural Grading of Foveal Hypoplasia Predict Future Vision in Infantile Nystagmus: A Longitudinal Study. Ophthalmology 2020, 127, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Mastey, R.R.; Georgiou, M.; Langlo, C.S.; Kalitzeos, A.; Patterson, E.J.; Kane, T.; Singh, N.; Vincent, A.; Moore, A.T.; Tsang, S.H.; et al. Characterization of Retinal Structure in ATF6-Associated Achromatopsia. Investig. Opthalmol. Vis. Sci. 2019, 60, 2631–2640. [Google Scholar] [CrossRef]
- Berson, E.L.; Sandberg, M.A.; Maguire, A.; Bromley, W.C.; Roderick, T.H. Electroretinograms in Carriers of Blue Cone Monochromatism. Am. J. Ophthalmol. 1986, 102, 254–261. [Google Scholar] [CrossRef]
- Gottlob, I. Eye movement abnormalities in carriers of blue-cone monochromatism. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3556–3560. [Google Scholar]
- Sustar, M.; Hawlina, M.; Brecelj, J. Electroretinographic evaluation of the retinal S-cone system. Doc. Ophthalmol. 2011, 123, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2018, 35, 1978–1980. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iarossi, G.; Coppè, A.M.; Passarelli, C.; Maltese, P.E.; Sinibaldi, L.; Cappelli, A.; Cetola, S.; Novelli, A.; Buzzonetti, L. Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes. Int. J. Mol. Sci. 2021, 22, 8617. https://doi.org/10.3390/ijms22168617
Iarossi G, Coppè AM, Passarelli C, Maltese PE, Sinibaldi L, Cappelli A, Cetola S, Novelli A, Buzzonetti L. Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes. International Journal of Molecular Sciences. 2021; 22(16):8617. https://doi.org/10.3390/ijms22168617
Chicago/Turabian StyleIarossi, Giancarlo, Andrea Maria Coppè, Chiara Passarelli, Paolo Enrico Maltese, Lorenzo Sinibaldi, Alessandro Cappelli, Sarah Cetola, Antonio Novelli, and Luca Buzzonetti. 2021. "Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes" International Journal of Molecular Sciences 22, no. 16: 8617. https://doi.org/10.3390/ijms22168617
APA StyleIarossi, G., Coppè, A. M., Passarelli, C., Maltese, P. E., Sinibaldi, L., Cappelli, A., Cetola, S., Novelli, A., & Buzzonetti, L. (2021). Blue Cone Monochromatism with Foveal Hypoplasia Caused by the Concomitant Effect of Variants in OPN1LW/OPN1MW and GPR143 Genes. International Journal of Molecular Sciences, 22(16), 8617. https://doi.org/10.3390/ijms22168617