
Graph Theoretical Methods and Workflows for Searching and Annotation of RNA Tertiary Base Motifs and Substructures


Abstract
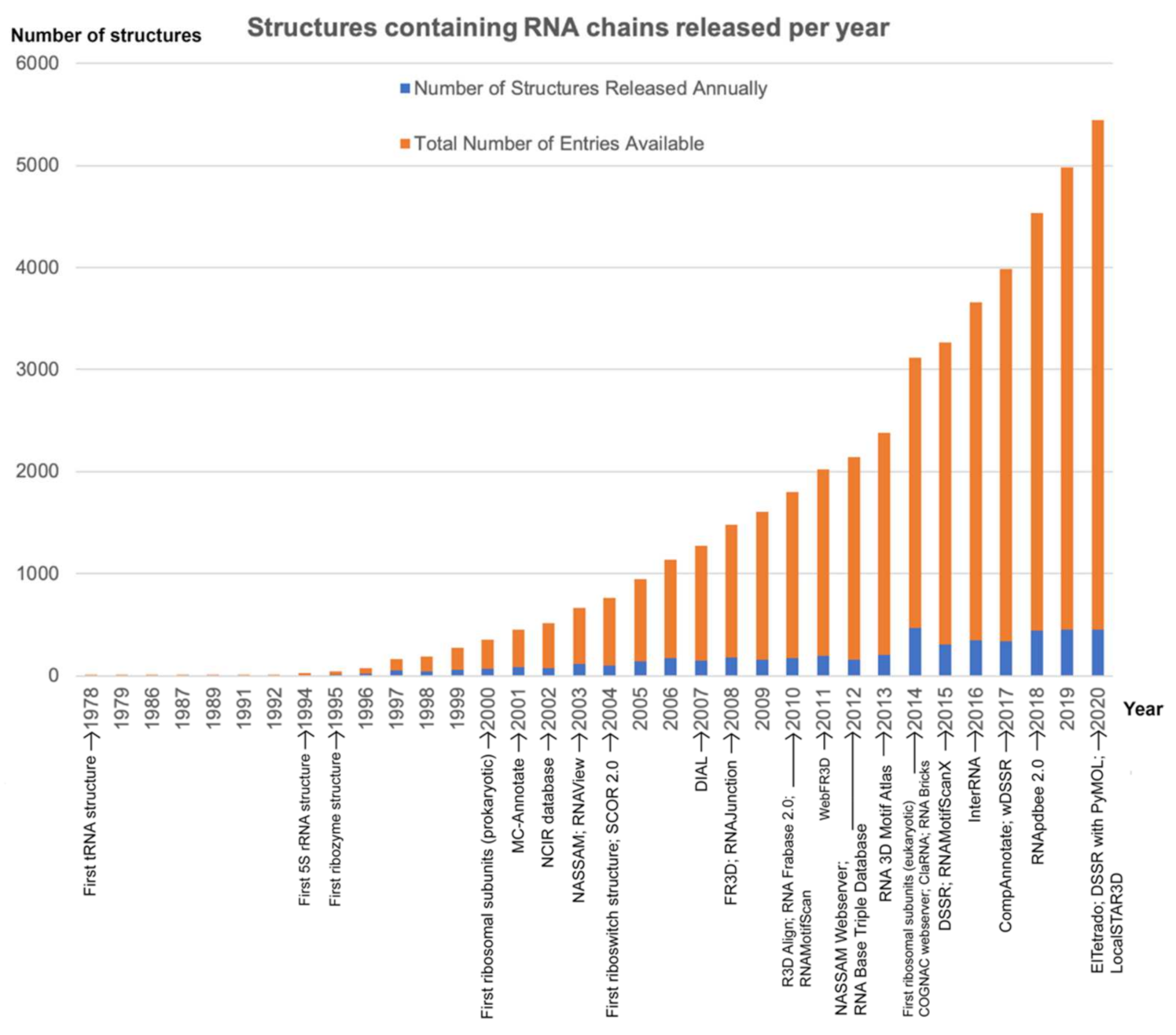
:1. Introduction
2. Algorithms for Annotating RNA 3D Base Arrangements
2.1. Comparison of Computational Approaches in Annotating RNA Base Motifs
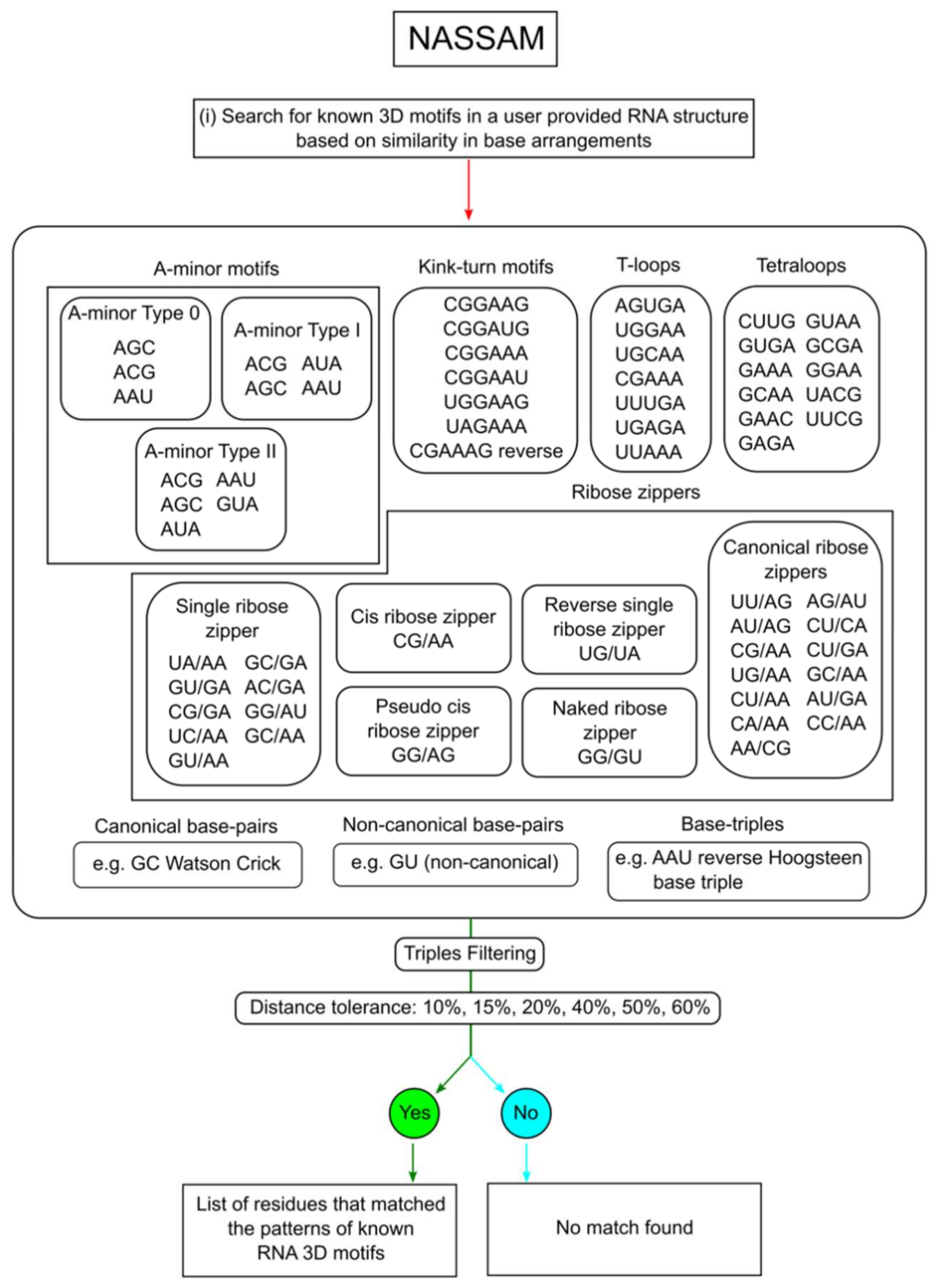
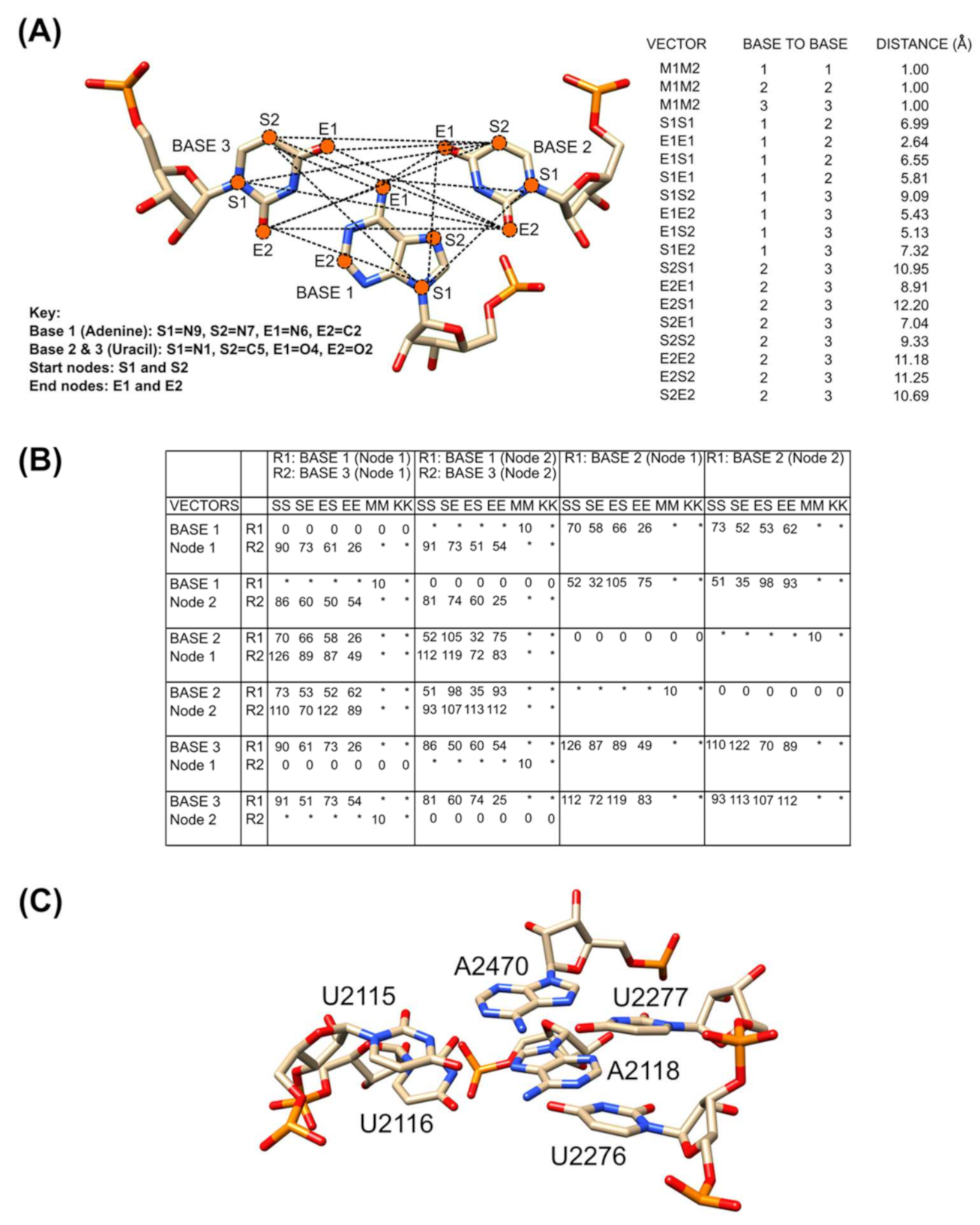
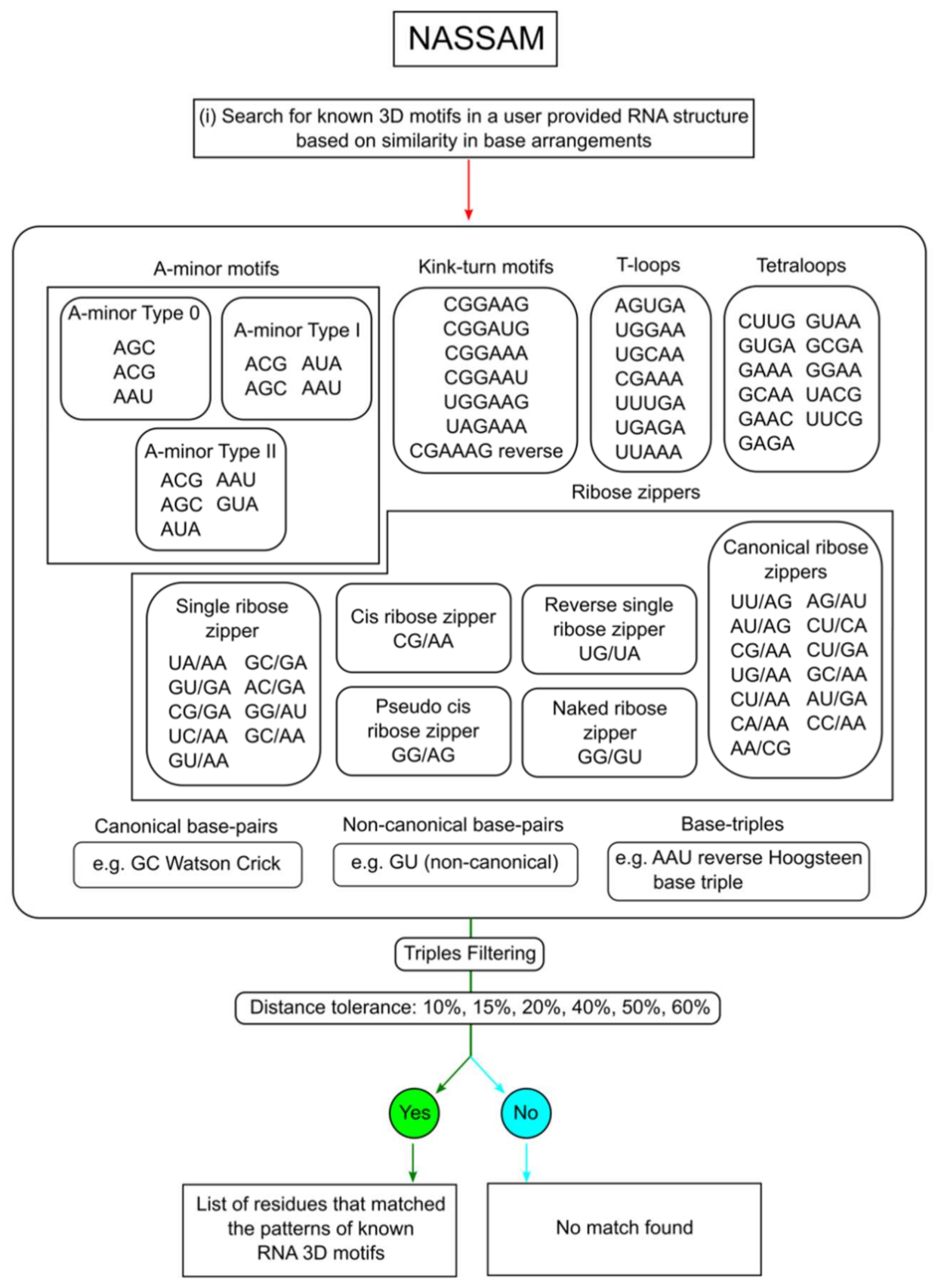
2.2. Annotation of Tertiary Base Arrangement Using the NASSAM Computer Program
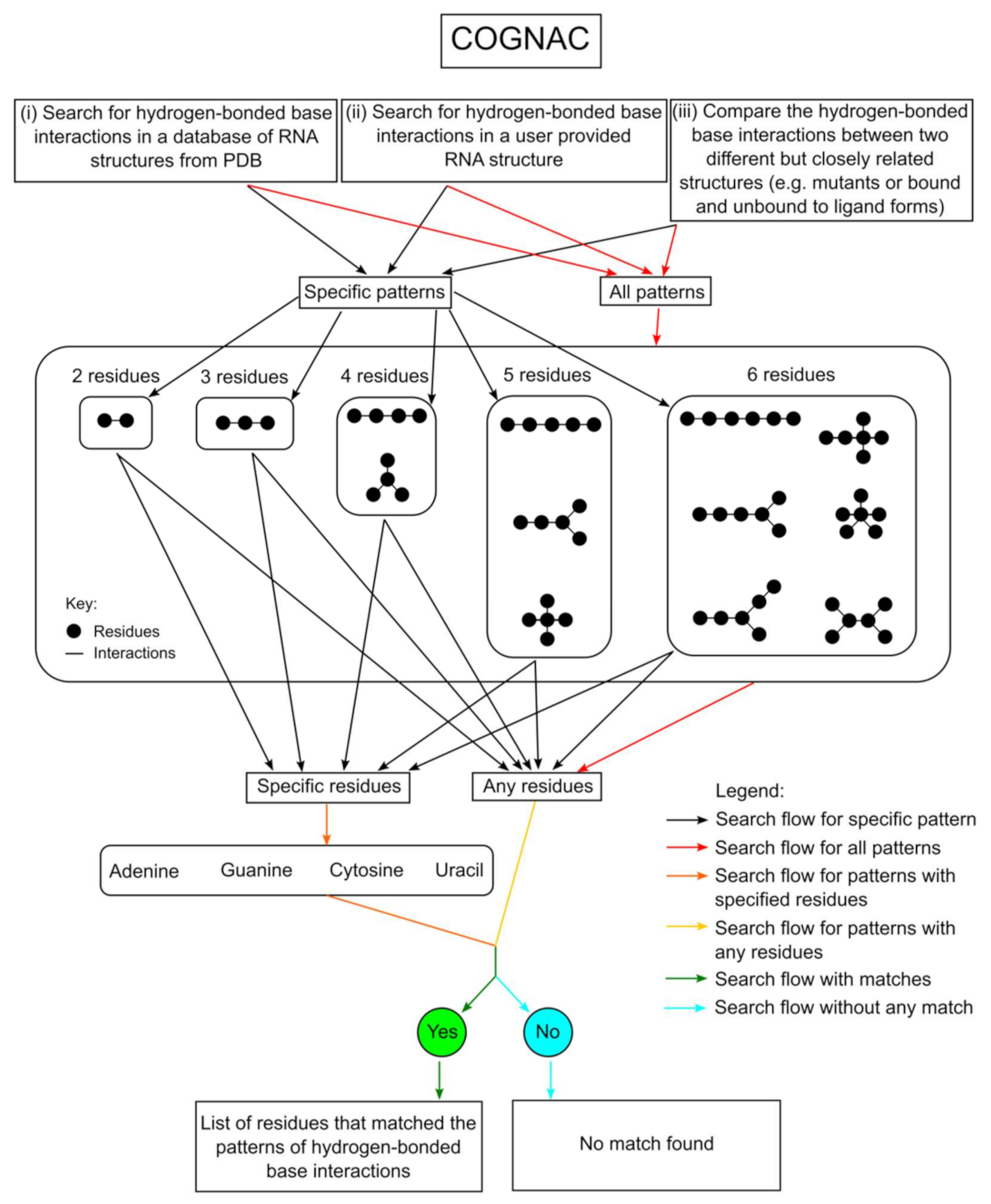
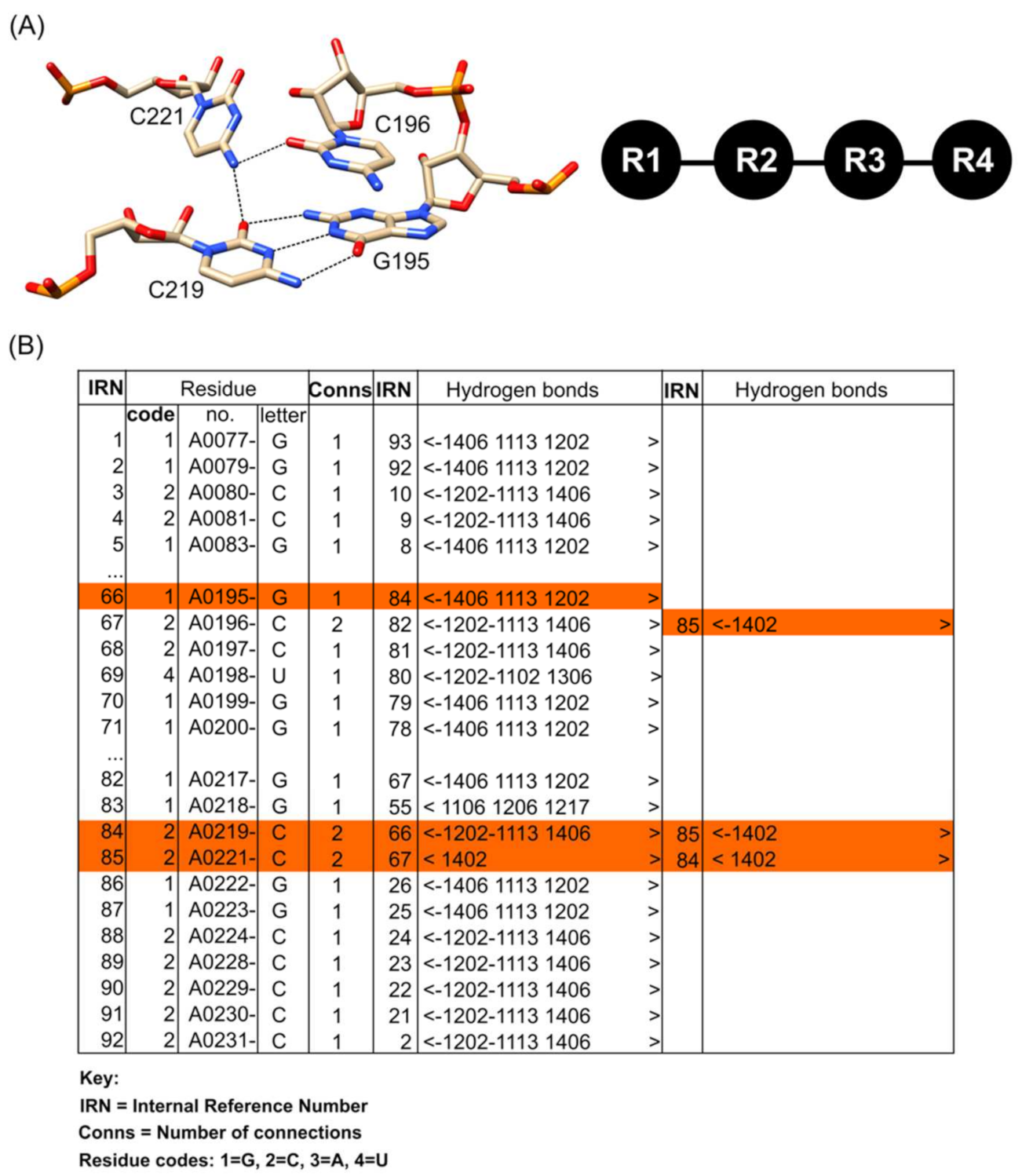
2.3. Annotation of Hydrogen-Bond-Connected Base Clusters Using the COGNAC Computer Program
3. Workflows for Annotating RNA 3D Base Arrangements
3.1. Searching for Novel RNA Base Motifs
3.2. Application of 3D Base Arrangement Comparisons to Identify Known Motifs in a Novel Context
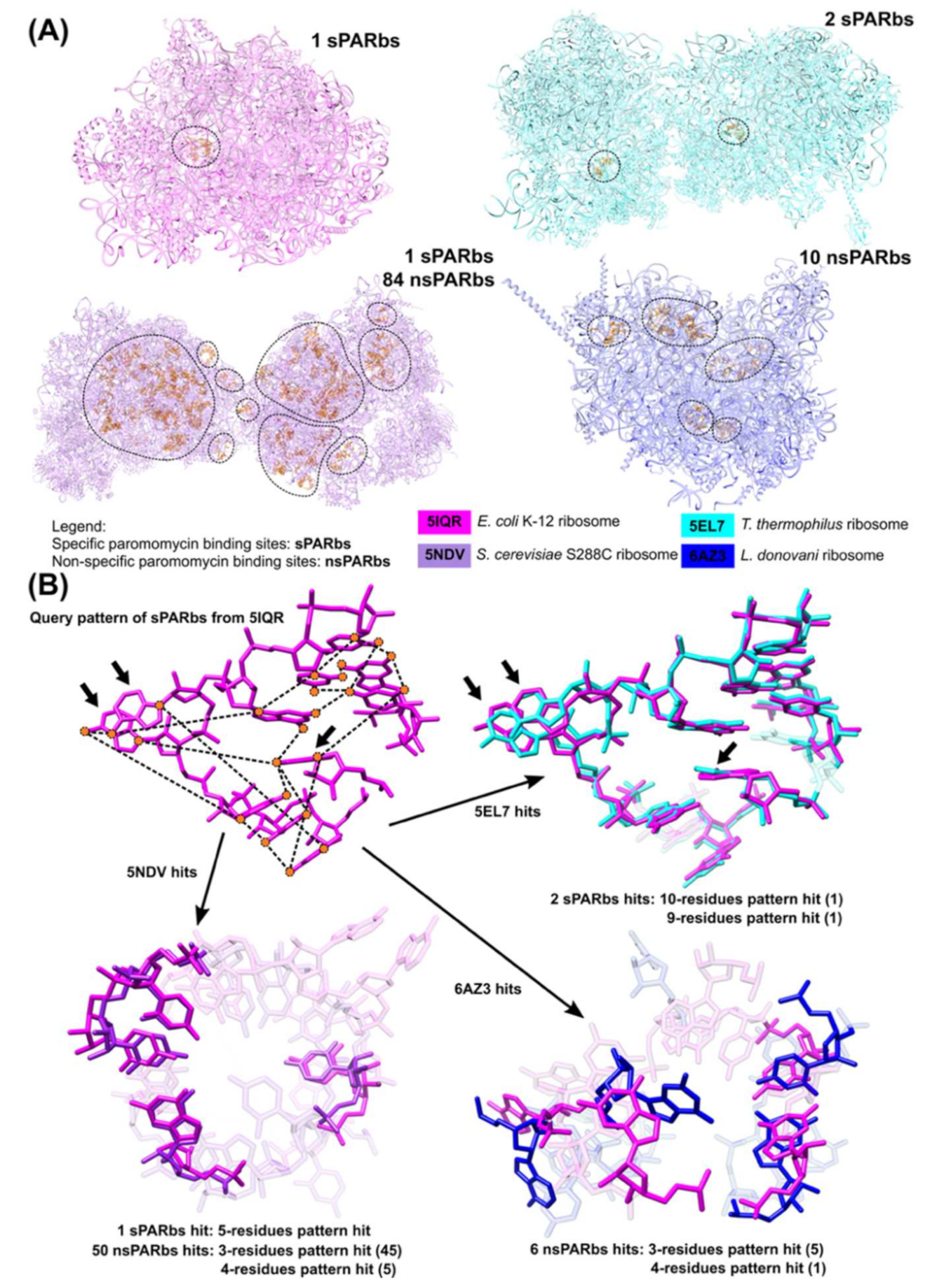
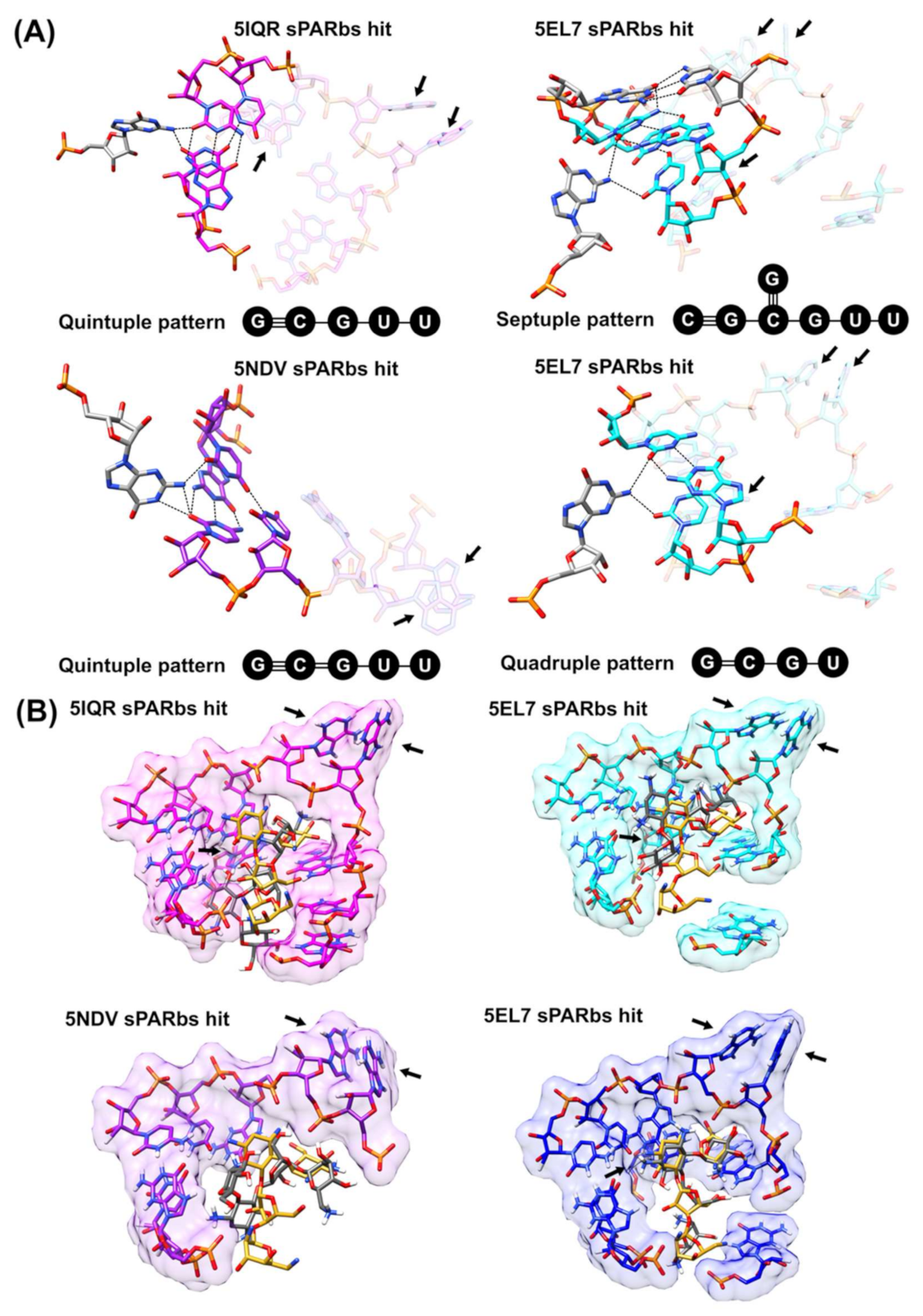
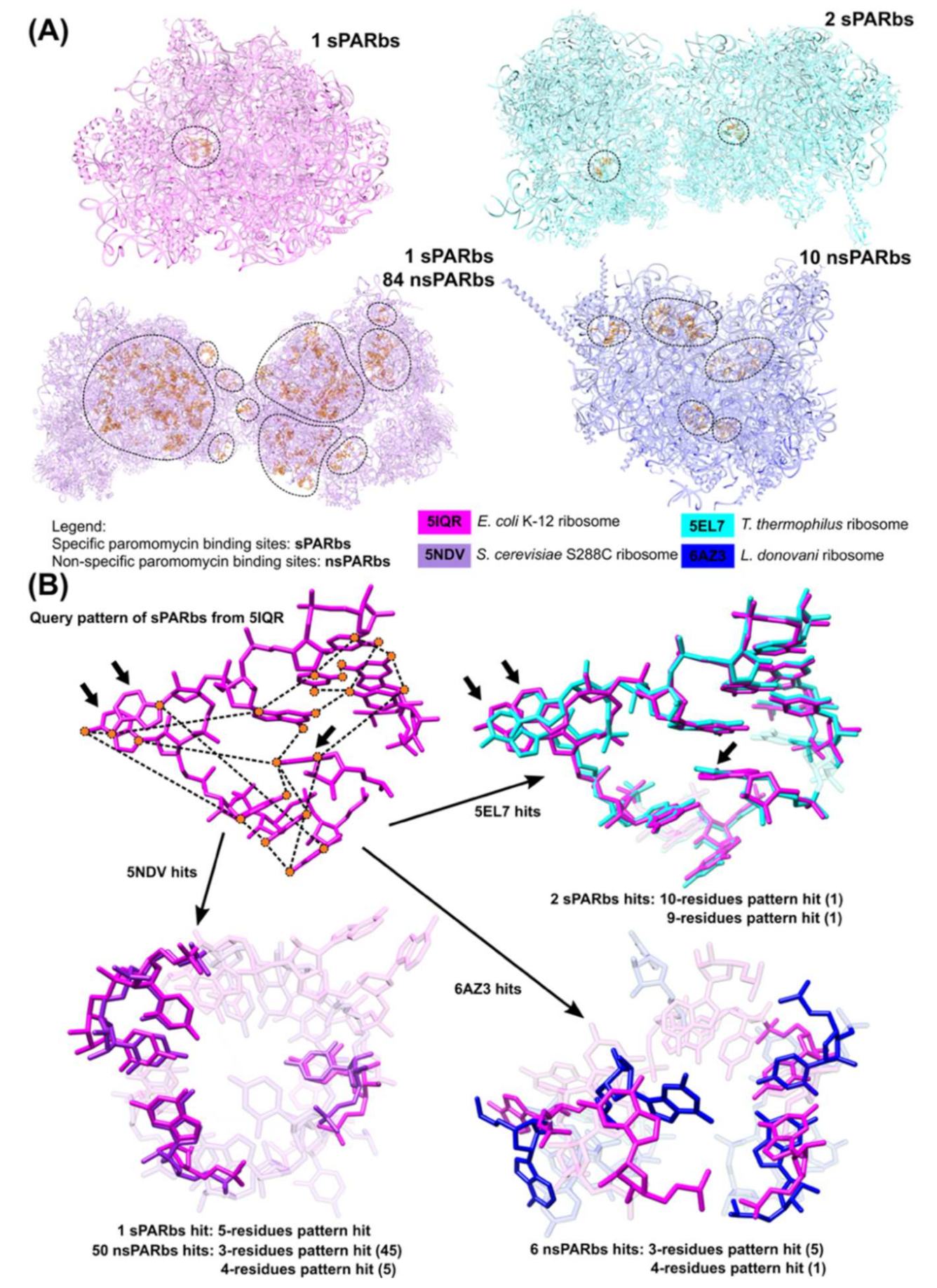
3.3. Application of 3D Base Arrangement Searching to Identify Functional Sites
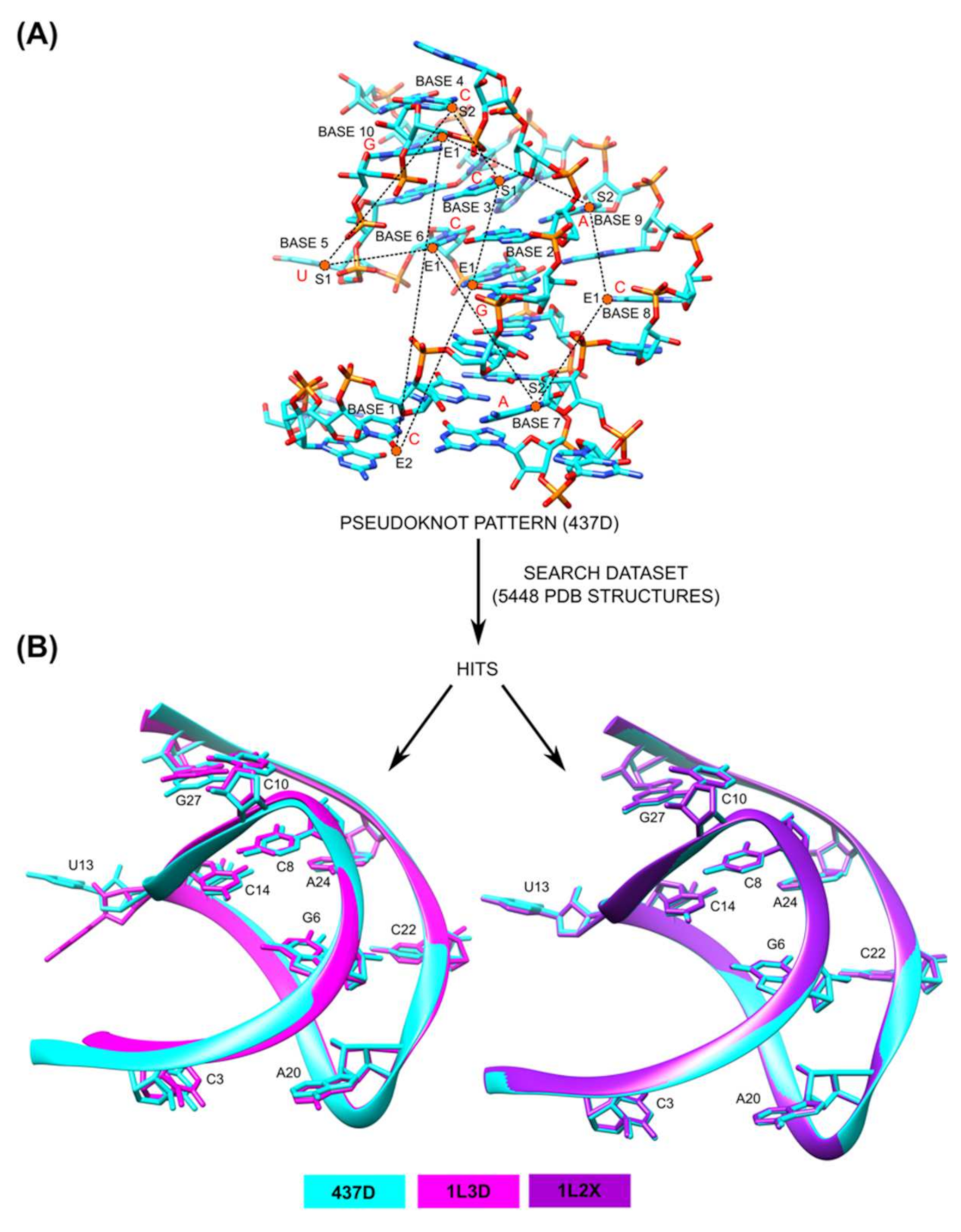
3.4. Application of 3D Base Arrangement Comparisons to Identify Pseudoknots
3.5. A Database of RNA Base Interactions
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Wilson, T.J.; Lilley, D.M.J. The potential versatility of RNA catalysis. Wiley Interdiscip. Rev. RNA 2021, e1651. [Google Scholar] [CrossRef]
- White, S.A.; Nilges, M.; Huang, A.; Brünger, A.T.; Moore, P.B. NMR analysis of helix I from the 5S RNA of Escherichia coli. Biochemistry 1992, 31, 1610–1621. [Google Scholar] [CrossRef]
- wwPDB Consortium. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [Green Version]
- Hingerty, B.; Brown, R.S.; Jack, A. Further refinement of the structure of yeast tRNAPhe. J. Mol. Biol. 1978, 124, 523–534. [Google Scholar] [CrossRef]
- Tuschl, T.; Gohlke, C.; Jovin, T.M.; Westhof, E.; Eckstein, F. A three-dimensional model for the hammerhead ribozyme based on fluorescence measurements. Science 1994, 266, 785–789. [Google Scholar] [CrossRef]
- Mueller, F.; Sommer, I.; Baranov, P.; Matadeen, R.; Stoldt, M.; Wöhnert, J.; Görlach, M.; Van Heel, M.; Brimacombe, R. The 3D arrangement of the 23 S and 5 S rRNA in the Escherichia coli 50 S ribosomal subunit based on a cryo-electron microscopic reconstruction at 7.5 Å resolution. J. Mol. Biol. 2000, 298, 35–59. [Google Scholar] [CrossRef]
- Gendron, P.; Lemieux, S.; Major, F. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001, 308, 919–936. [Google Scholar] [CrossRef] [Green Version]
- Nagaswamy, U.; Larios-Sanz, M.; Hury, J.; Collins, S.; Zhang, Z.; Zhao, Q.; Fox, G.E. NCIR: A database of non-canonical interactions in known RNA structures. Nucleic Acids Res. 2002, 30, 395–397. [Google Scholar] [CrossRef] [Green Version]
- Harrison, A.M.; South, D.R.; Willett, P.; Artymiuk, P.J. Representation, searching and discovery of patterns of bases in complex RNA structures. J. Comput. Aided. Mol. Des. 2003, 17, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jossinet, F.; Leontis, N.; Chen, L.; Westbrook, J.; Berman, H.; Westhof, E. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003, 31, 3450–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serganov, A.; Yuan, Y.-R.; Pikovskaya, O.; Polonskaia, A.; Malinina, L.; Phan, A.T.; Hobartner, C.; Micura, R.; Breaker, R.R.; Patel, D.J. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 2004, 11, 1729–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klosterman, P.S.; Tamura, M.; Holbrook, S.R.; Brenner, S.E. SCOR: A structural classification of RNA database. Nucleic Acids Res. 2002, 30, 392–394. [Google Scholar] [CrossRef] [Green Version]
- Ferrè, F.; Ponty, Y.; Lorenz, W.A.; Clote, P. DIAL: A web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Res. 2007, 35, W659–W668. [Google Scholar] [CrossRef]
- Sarver, M.; Zirbel, C.L.; Stombaugh, J.; Mokdad, A.; Leontis, N.B. FR3D: Finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol. 2008, 56, 215–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindewald, E.; Hayes, R.; Yingling, Y.G.; Kasprzak, W.; Shapiro, B.A. RNAJunction: A database of RNA junctions and kissing loops for three-dimensional structural analysis and nanodesign. Nucleic Acids Res. 2008, 36, D392–D397. [Google Scholar] [CrossRef] [Green Version]
- Rahrig, R.R.; Leontis, N.B.; Zirbel, C.L. R3D align: Global pairwise alignment of RNA 3D structures using local superpositions. Bioinformatics 2010, 26, 2689–2697. [Google Scholar] [CrossRef] [Green Version]
- Popenda, M.; Szachniuk, M.; Blazewicz, M.; Wasik, S.; Burke, E.K.; Blazewicz, J.; Adamiak, R.W. RNA FRABASE 2.0: An advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures. BMC Bioinform. 2010, 11, 231. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.; Tang, H.; Zhang, S. RNAMotifScan: Automatic identification of RNA structural motifs using secondary structural alignment. Nucleic Acids Res. 2010, 38, e176. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.I.; Zirbel, C.L.; Leontis, N.B. WebFR3D—A server for finding, aligning and analyzing recurrent RNA 3D motifs. Nucleic Acids Res. 2011, 39, W50–W55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdani, H.Y.; Appasamy, S.D.; Willett, P.; Artymiuk, P.J.; Firdaus-Raih, M. NASSAM: A server to search for and annotate tertiary interactions and motifs in three-dimensional structures of complex RNA molecules. Nucleic Acids Res. 2012, 40, W35–W41. [Google Scholar] [CrossRef]
- Abu Almakarem, A.S.; Petrov, A.I.; Stombaugh, J.; Zirbel, C.L.; Leontis, N.B. Comprehensive survey and geometric classification of base triples in RNA structures. Nucleic Acids Res. 2012, 40, 1407–1423. [Google Scholar] [CrossRef]
- Petrov, A.I.; Zirbel, C.L.; Leontis, N.B. Automated classification of RNA 3D motifs and the RNA 3D Motif Atlas. RNA 2013, 19, 1327–1340. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shem, A.; Loubresse, N.G.D.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [Green Version]
- Firdaus-Raih, M.; Hamdani, H.Y.; Nadzirin, N.; Ramlan, E.I.; Willett, P.; Artymiuk, P.J. COGNAC: A web server for searching and annotating hydrogen-bonded base interactions in RNA three-dimensional structures. Nucleic Acids Res. 2014, 12, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Walén, T.; Chojnowski, G.; Gierski, P.; Bujnicki, J.M. ClaRNA: A classifier of contacts in RNA 3D structures based on a comparative analysis of various classification schemes. Nucleic Acids Res. 2014, 42, e151. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, G.; Waleń, T.; Bujnicki, J.M. RNA Bricks—A database of RNA 3D motifs and their interactions. Nucleic Acids Res. 2014, 42, D123–D131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.J.; Bussemaker, H.J.; Olson, W.K. DSSR: An integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015, 43, e142. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.; Zhang, S. RNAMotifScanX: A graph alignment approach for RNA structural motif identification. RNA 2015, 21, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Appasamy, S.D.; Hamdani, H.Y.; Ramlan, E.I.; Firdaus-Raih, M. InterRNA: A database of base interactions in RNA structures. Nucleic Acids Res. 2016, 44, D266–D271. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Ge, P.; Zhang, S. CompAnnotate: A comparative approach to annotate base-pairing interactions in RNA 3D structures. Nucleic Acids Res. 2017, 45, e136. [Google Scholar] [CrossRef] [Green Version]
- Hanson, R.M.; Lu, X.J. DSSR-enhanced visualization of nucleic acid structures in Jmol. Nucleic Acids Res. 2017, 45, W528–W533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zok, T.; Antczak, M.; Zurkowski, M.; Popenda, M.; Blazewicz, J.; Adamiak, R.W.; Szachniuk, M. RNApdbee 2.0: Multifunctional tool for RNA structure annotation. Nucleic Acids Res. 2018, 46, W30–W35. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Khan, N.S.; Zhang, S. LocalSTAR3D: A local stack-based RNA 3D structural alignment tool. Nucleic Acids Res. 2020, 48, e77. [Google Scholar] [CrossRef]
- Zok, T.; Popenda, M.; Szachniuk, M. ElTetrado: A tool for identification and classification of tetrads and quadruplexes. BMC Bioinform. 2020, 21, 40. [Google Scholar] [CrossRef]
- Lu, X.J. DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL. Nucleic Acids Res. 2020, 48, e74. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 1–11. [Google Scholar] [CrossRef]
- Das, R. RNA structure: A renaissance begins? Nat. Methods 2021, 18, 439. [Google Scholar] [CrossRef] [PubMed]
- Jednačak, T.; Mikulandra, I.; Novak, P. Advanced methods for studying structure and interactions of macrolide antibiotics. Int. J. Mol. Sci. 2020, 21, 7799. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.; Sobczak, K. Intrinsic regulatory role of RNA structural arrangement in alternative splicing control. Int. J. Mol. Sci. 2020, 21, 5161. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Ippolito, J.A.; Ban, N.; Moore, P.B.; Steitz, T.A. RNA tertiary interactions in the large ribosomal subunit: The A-minor motif. Proc. Natl. Acad. Sci. USA 2001, 98, 4899–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef]
- Wimberly, B.T.; Brodersen, D.E.; Clemons, W.M., Jr.; Morgan-Warren, R.J.; Carter, A.P.; Vonrhein, C.; Hartsch, T.; Ramakrishnan, V. Structure of the 30S ribosomal subunit. Nature 2000, 407, 327. [Google Scholar] [CrossRef] [PubMed]
- Wadley, L.M.; Pyle, A.M. The identification of novel RNA structural motifs using COMPADRES: An automated approach to structural discovery. Nucleic Acids Res. 2004, 32, 6650–6659. [Google Scholar] [CrossRef] [Green Version]
- Leontis, N.B.; Westhof, E. Geometric nomenclature and classification of RNA base pairs. RNA 2001, 7, 499–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadzirin, N.; Gardiner, E.J.; Willett, P.; Artymiuk, P.J.; Firdaus-Raih, M. SPRITE and ASSAM: Web servers for side chain 3D-motif searching in protein structures. Nucleic Acids Res. 2012, 40, W380–W386. [Google Scholar] [CrossRef] [Green Version]
- Firdaus-Raih, M.; Harrison, A.M.; Willett, P.; Artymiuk, P.J. Novel base triples in RNA structures revealed by graph theoretical searching methods. BMC Bioinform. 2011, 12, S2. [Google Scholar] [CrossRef] [Green Version]
- Hamdani, H.Y.; Firdaus-Raih, M. Identification of structural motifs using networks of hydrogen-bonded base interactions in RNA crystallographic structures. Crystals 2019, 9, 550. [Google Scholar] [CrossRef] [Green Version]
- Rozov, A.; Demeshkina, N.; Khusainov, I.; Westhof, E.; Yusupov, M.; Yusupova, G. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 2016, 7, 10457. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Fernández, I.S.; Gordiyenko, Y.; Ramakrishnan, V. Ribosome-dependent activation of stringent control. Nature 2016, 534, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Prokhorova, I.; Altman, R.B.; Djumagulov, M.; Shrestha, J.P.; Urzhumtsev, A.; Ferguson, A.; Chang, C.W.T.; Yusupov, M.; Blanchard, S.C.; Yusupova, G. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, E10899–E10908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalev-Benami, M.; Zhang, Y.; Rozenberg, H.; Nobe, Y.; Taoka, M.; Matzov, D.; Zimmerman, E.; Bashan, A.; Isobe, T.; Jaffe, C.L.; et al. Atomic resolution snapshot of Leishmania ribosome inhibition by the aminoglycoside paromomycin. Nat. Commun. 2017, 8, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leontis, N.B.; Zirbel, C.L. Nonredundant 3D Structure Datasets for RNA Knowledge Extraction and Benchmarking. In RNA 3D Structure Analysis and Prediction; Springer: Berlin/Heidelberg, Germany, 2012; Volume 27, pp. 281–298. ISBN 978-3-642-25739-1. [Google Scholar]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forconi, M.; Sengupta, R.N.; Piccirilli, J.A.; Herschlag, D. A rearrangement of the guanosine-binding site establishes an extended network of functional interactions in the Tetrahymena group I ribozyme active site. Biochemistry 2010, 49, 2753–2762. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, A.; Zhou, K.; Ding, F.; Cate, J.H.D.; Doudna, J.A. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 2004, 429, 201–205. [Google Scholar] [CrossRef]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef]
- Theimer, C.A.; Blois, C.A.; Feigon, J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol. Cell 2005, 17, 671–682. [Google Scholar] [CrossRef]
- Egli, M.; Minasov, G.; Su, L.; Rich, A. Metal ions and flexibility in a viral RNA pseudoknot at atomic resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 4302–4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Chen, L.; Egli, M.; Berger, J.M.; Rich, A. Minor groove RNA triplex in the crystal structure of a ribosomal frameshifting viral pseudoknot. Nat. Struct. Biol. 1999, 6, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Staple, D.W.; Butcher, S.E. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005, 3, e213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antczak, M.; Popenda, M.; Zok, T.; Zurkowski, M.; Adamiak, R.W.; Szachniuk, M. New algorithms to represent complex pseudoknotted RNA structures in dot-bracket notation. Bioinformatics 2018, 34, 1304–1312. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Programs | MC-Annotate | NCIR | RNAView | SCOR | DIAL | RNAJunction | R3D Align | RNA Frabase 2.0 | WebFR3D | Nassam Webserver | RNA Base Triple Database |
| Method | Structural graph | Literature survey | Coordinate frame as reference and least-square fit | Manual curation | 3D-structure alignment | Predicts helices and determines their connectivity | Local alignment graph | Matching pattern in dot-bracket format | Geometric or symbolic matching (FR3D) | Sub-graph matching of vector arrangements | Symbolic matching (FR3D) |
| Input | 3D structure | N.A. | 3D structure | N.A. | 3D structure | Sequences or PDB structure identifiers | 3D structure | Sequence or secondary structure | Symbols (up to 15 nucleotides) or PDB structure identifiers | 3D structure | N.A. |
| Availability | Webserver | Database | Webserver | Database | Webserver | Database | Webserver | Database | Webserver | Webserver | Database |
| Motifs annotated | Base pairs, Base triples, U-turn | Base pairs, base triples, base quadruples | Base pairs | Ribose zippers, T-loops, A-minor, pseudoknots, tetraloops | No specific motifs | RNA junctions, kissing loops | No specific motifs | Base pairs, base triples, base quadruples, base quintuples, hairpin loops, internal loops, junctions | GNRA loops, T-loops, sarcin-ricin loops, kissing loops, C-loops, A-minor, kink-turn | Base pairs, base triples, A-minor, T-loop, ribose-zippers, kink-turn, tetraloops | Base triples |
| Programs | RNA 3D Motif Atlas | COGNAC Webserver | ClaRNA | RNA Bricks | RNAMotifScanX | InterRNA | CompAnnotate | RNApdbee 2.0 | Local STAR3D | DSSR with PyMOL | ElTetrado |
| Method | 3D-structure alignment (FR3D) | Sub-graph matching of connection table graph representation | Geometric matching to reference dataset of ribonucleotide doublet | Secondary structure graph and superposition of 3D motifs to query structures | Alignment of interaction graph | Sub-graph matching using NASSAM and COGNAC | 3D-structure alignment and comparative geometric assessments using high-resolution reference | Annotation of secondary structures to predict 3D interactions | Local com-patible graph alignment | DSSR geometric algorithm | Categorize quadruplexes based on secondary structure topology and component tetrads |
| Input | N.A. | 3D structure | 3D structure | 3D structure | 3D structure | N.A. | 3D structure and base-pairing annotations | 3D structure | 3D structure | 3D structure | 3D structure |
| Availability | Database | Webserver | Webserver | Database | Offline executable | Database | Offline executable | Webserver | Offline executable | Webserver, Plugin in PyMOL | Offline executable |
| Motifs annotated | Sarcin-ricin loops, GNRA loops, T-loops, kink-turn, C-loop | Base pairs, base triples, base quadruples, base quintuples, base sextuples | Base pairs,base stacking, base-phosphate, base-ribose | Loops, stems, single-stranded | Kink-turn, C-loop, sarcin-ricin loops, reverse kink-turn, E-loop | Base pair, base interaction (triples-sextuples), ribose-zippers, A/G-minor motifs, hairpin loop, internal loop | Base pairs, kink-turn, C-loop, sarcin-ricin loops | Base pairs, loops, stems, single-stranded, quadruplexes, pseudoknots | No specific motifs | Base pairs, kissing loops, junctions | quadruplexes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emrizal, R.; Hamdani, H.Y.; Firdaus-Raih, M. Graph Theoretical Methods and Workflows for Searching and Annotation of RNA Tertiary Base Motifs and Substructures. Int. J. Mol. Sci. 2021, 22, 8553. https://doi.org/10.3390/ijms22168553
Emrizal R, Hamdani HY, Firdaus-Raih M. Graph Theoretical Methods and Workflows for Searching and Annotation of RNA Tertiary Base Motifs and Substructures. International Journal of Molecular Sciences. 2021; 22(16):8553. https://doi.org/10.3390/ijms22168553
Chicago/Turabian StyleEmrizal, Reeki, Hazrina Yusof Hamdani, and Mohd Firdaus-Raih. 2021. "Graph Theoretical Methods and Workflows for Searching and Annotation of RNA Tertiary Base Motifs and Substructures" International Journal of Molecular Sciences 22, no. 16: 8553. https://doi.org/10.3390/ijms22168553
APA StyleEmrizal, R., Hamdani, H. Y., & Firdaus-Raih, M. (2021). Graph Theoretical Methods and Workflows for Searching and Annotation of RNA Tertiary Base Motifs and Substructures. International Journal of Molecular Sciences, 22(16), 8553. https://doi.org/10.3390/ijms22168553