
Modeling Neuroimmune Interactions in Human Subjects and Animal Models to Predict Subtype-Specific Multidrug Treatments for Gulf War Illness

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. The GWI Cohort
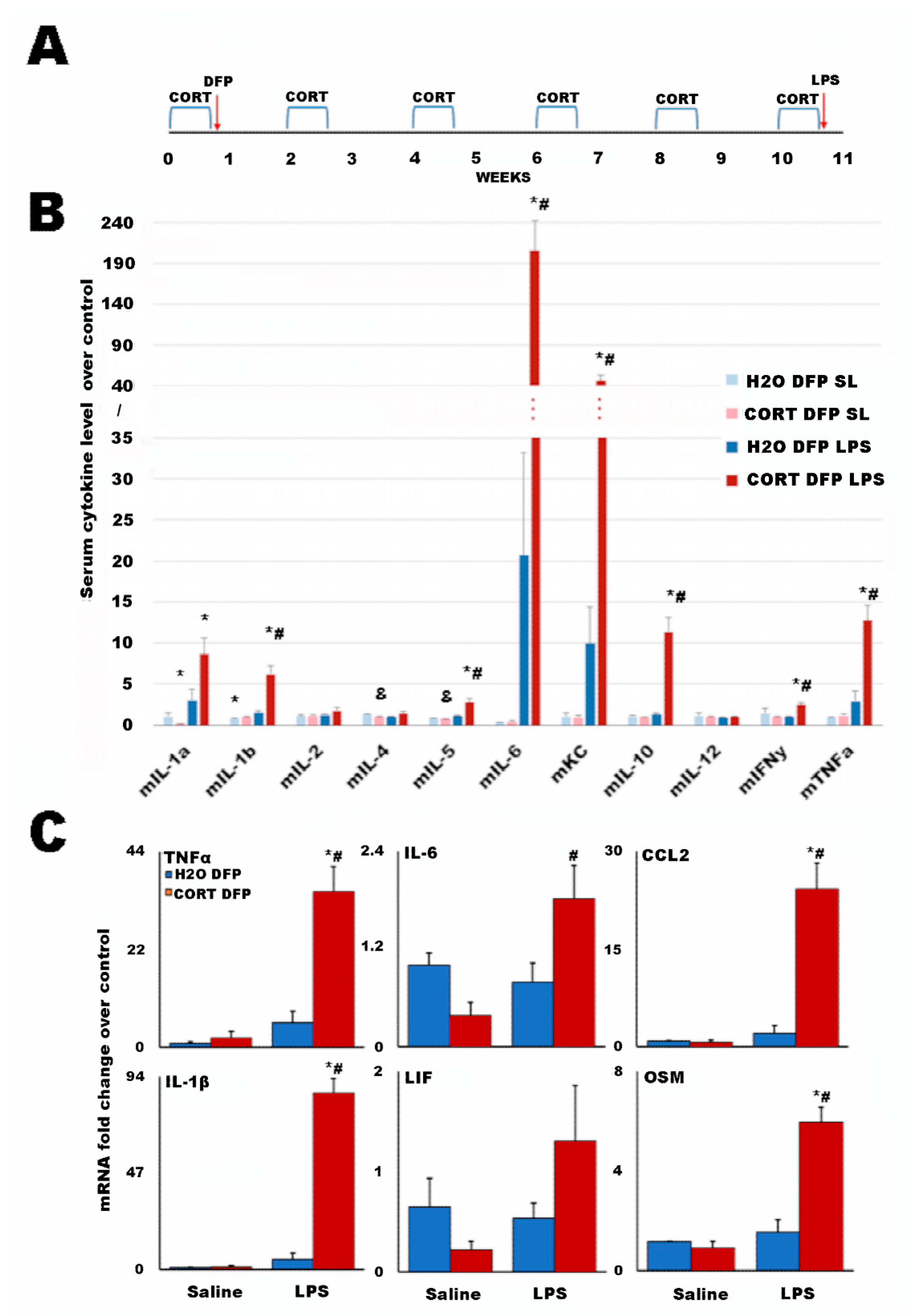
2.2. A Long-Term Mouse Model of Gulf War Illness
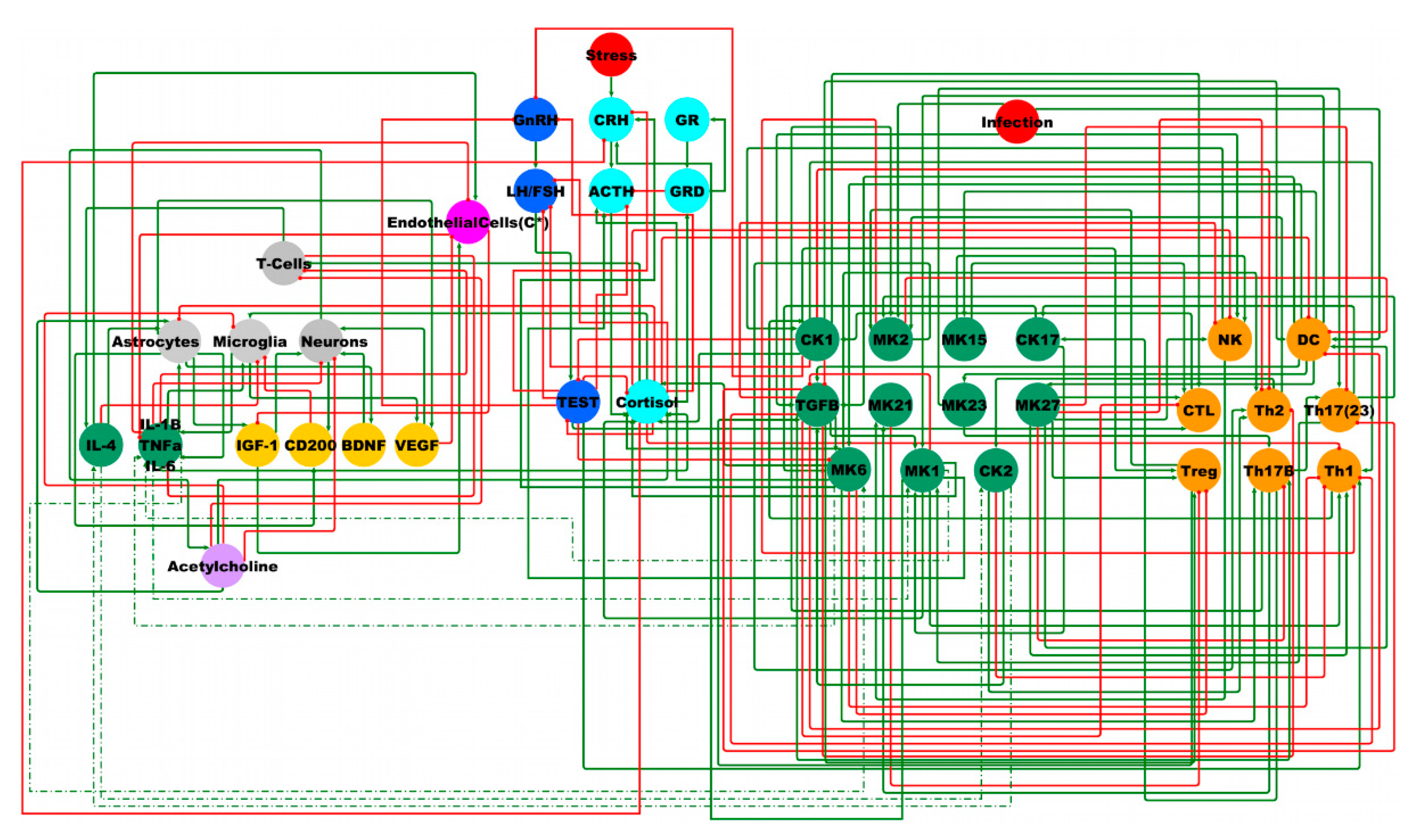
2.3. Male BBB Neuro-Endrocrine-Immune Model
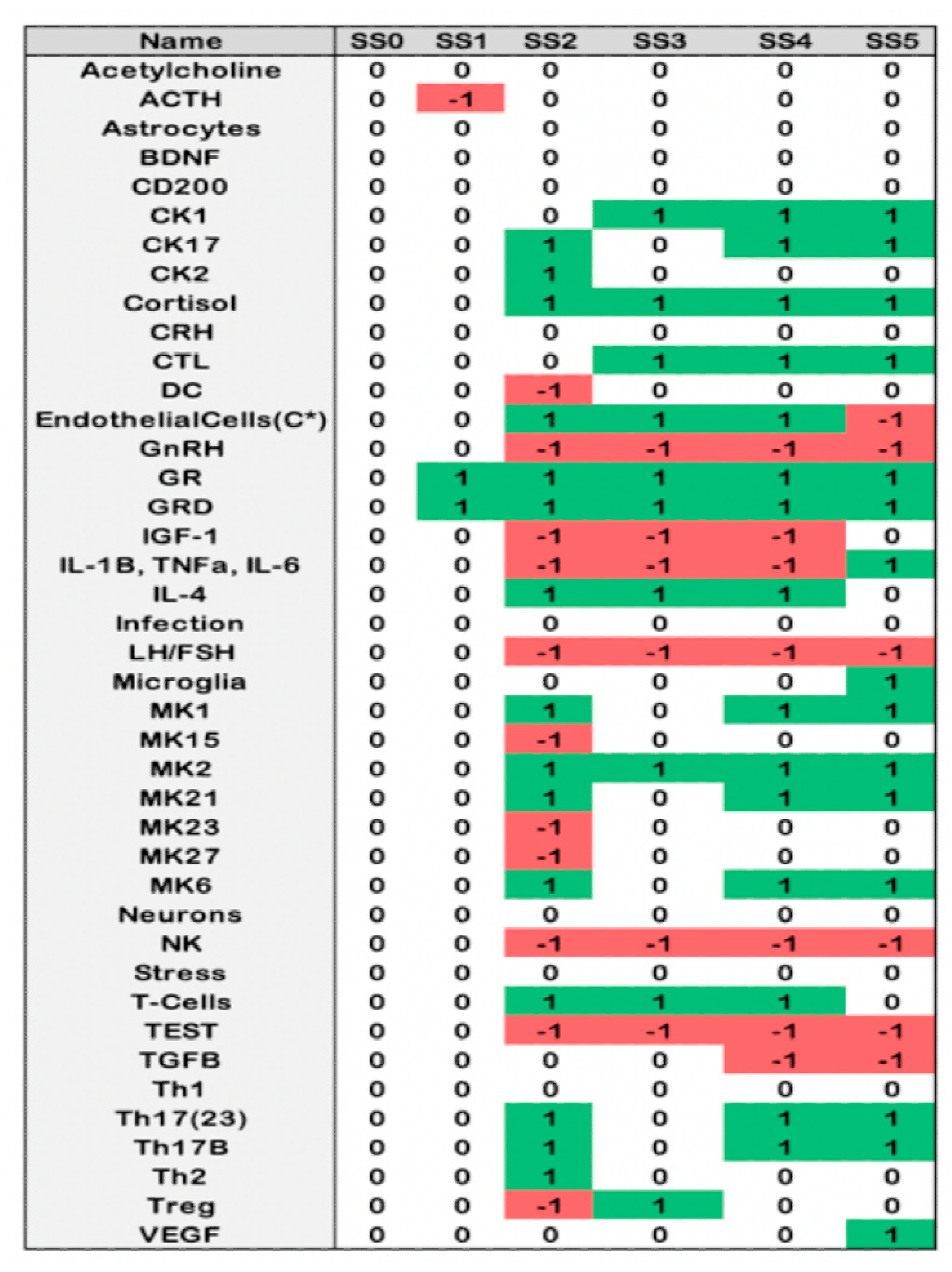
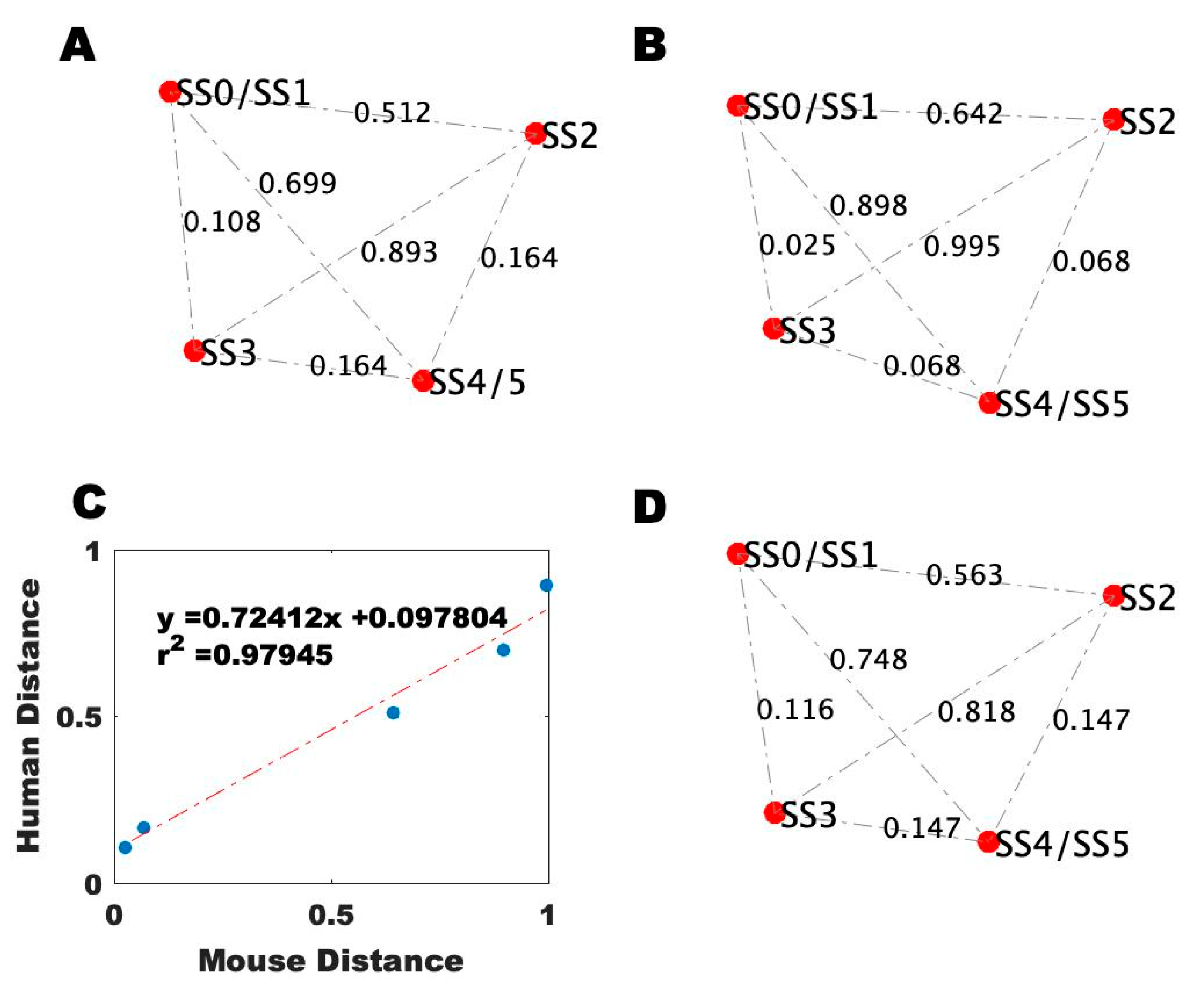
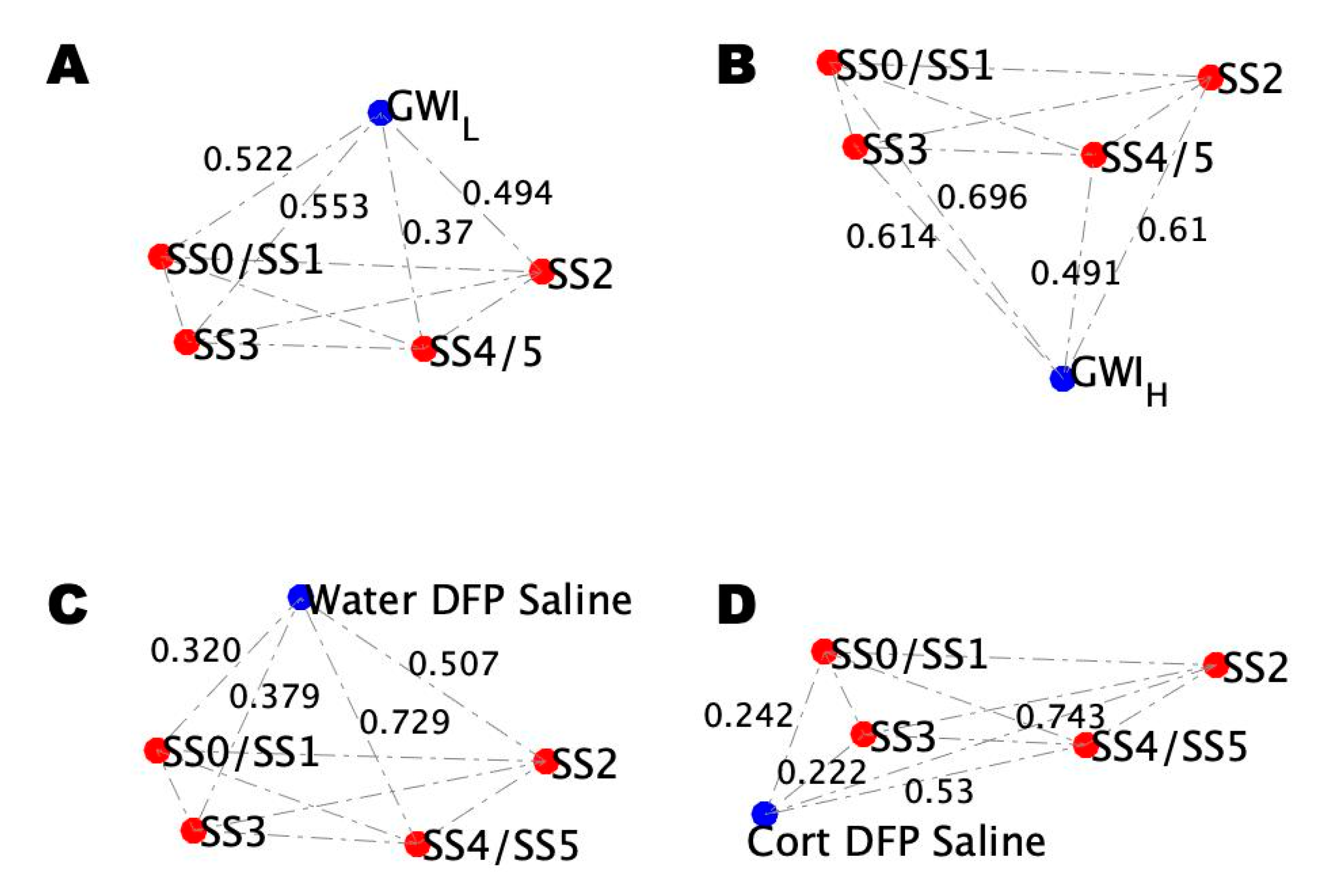
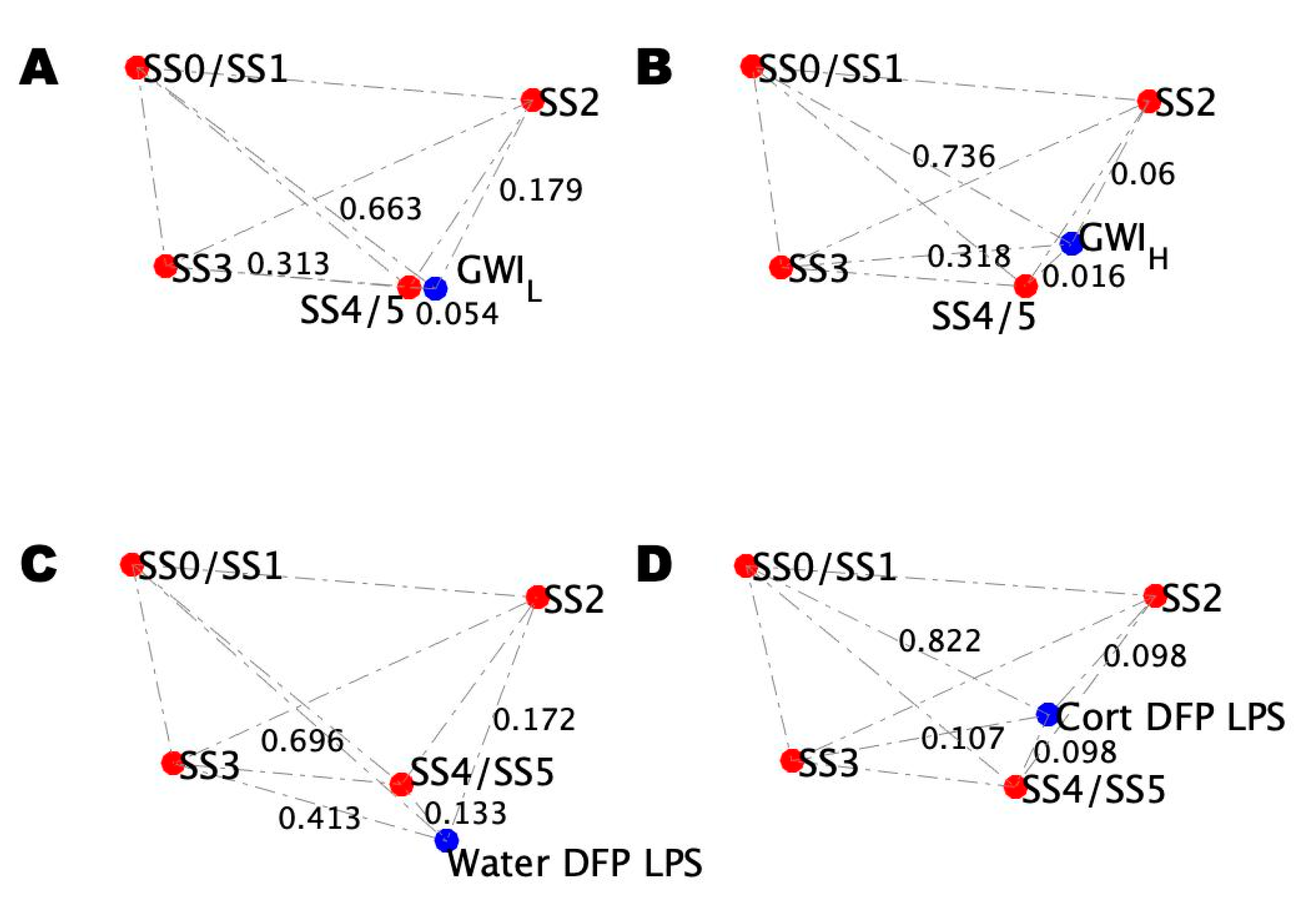
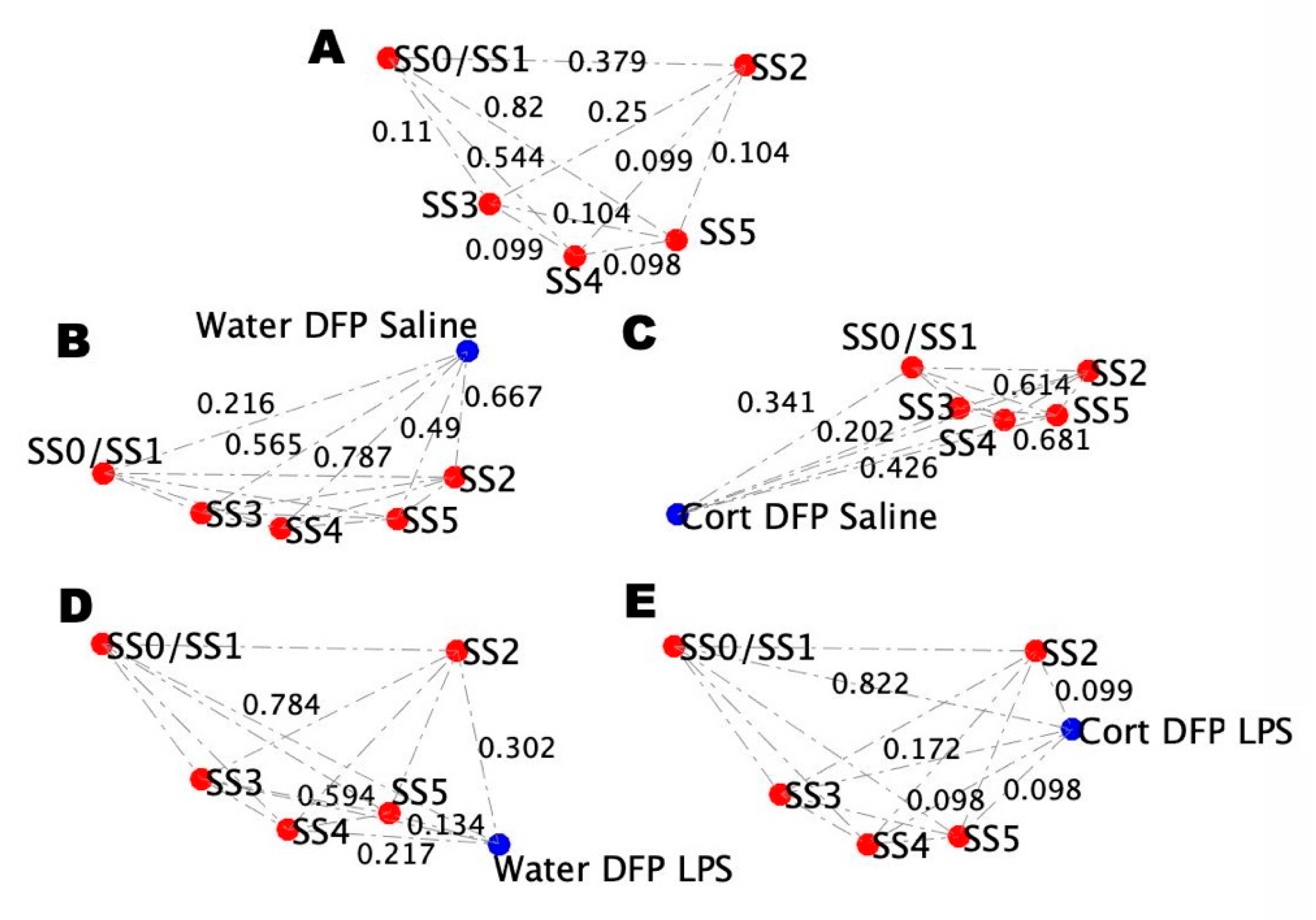
2.4. Alignment of the Blood Profiles with the Model-Predicted States
2.5. Simulated Treatment Courses
3. Discussion
4. Materials and Methods
4.1. Discrete Ternary Logical Analysis
4.2. Monte Carlo Simulation of State Evolution
4.3. Participants/Procedure
4.4. Human Blood Analysis
4.5. Animals and Dosing
4.6. Mouse Serum Cytokine Analysis
4.7. qRT-PCR Analysis of Mouse Brain Cytokines
4.8. Comparison of the Gene Expression Data to the Model-Predicted States
4.9. Simulating Intervention Courses
4.10. Genetic Algorithm for Optimizing Treatment Course
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Binns, J.; Bloom, F.; Bunker, J.; Crawford, F.; Golomb, B.; Graves, J.; Hardie, A.; Knox, M.L.; Meggs, W.J.; Nettleman, M.D. Research Advisory Committee on Gulf War Veterans’ Illnesses. Gulf War Illness and the Health of Gulf War Veterans: Research Update and Recommendations, 2009–2013; US Government Printing Office: Washington, DC, USA, 2014.
- Golier, J.A.; Schmeidler, J.; Legge, J.; Yehuda, R. Twenty-four Hour Plasma Cortisol and Adrenocorticotropic Hormone in Gulf War Veterans: Relationships to Posttraumatic Stress Disorder and Health Symptoms. Biol. Psychiatry 2007, 62, 1175–1178. [Google Scholar] [CrossRef]
- Golier, J.A.; Caramanica, K.; Michaelides, A.C.; Makotkine, I.; Schmeidler, J.; Harvey, P.D.; Yehuda, R. A randomized, double-blind, placebo-controlled, crossover trial of mifepristone in Gulf War veterans with chronic multisymptom illness. Psychoneuroendocrinology 2016, 64, 22–30. [Google Scholar] [CrossRef]
- Golier, J.A.; Schmeidler, J.; Yehuda, R. Pituitary response to metyrapone in Gulf War veterans: Relationship to deployment, PTSD and unexplained health symptoms. Psychoneuroendocrinology 2009, 34, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Golier, J.A.; Schmeidler, J.; Legge, J.; Yehuda, R. Enhanced cortisol suppression to dexamethasone associated with Gulf War deployment. Psychoneuroendocrinology 2006, 31, 1181–1189. [Google Scholar] [CrossRef]
- Broderick, G.; Ben-Hamo, R.; Vashishtha, S.; Efroni, S.; Nathanson, L.; Barnes, Z.; Fletcher, M.A.; Klimas, N. Altered immune pathway activity under exercise challenge in Gulf War Illness: An exploratory analysis. Brain Behav. Immun. 2013, 28, 159–169. [Google Scholar] [CrossRef]
- Janulewicz, P.; Krengel, M.; Quinn, E.; Heeren, T.; Toomey, R.; Killiany, R.; Zundel, C.; Ajama, J.; O’Callaghan, J.; Steele, L.; et al. The Multiple Hit Hypothesis for Gulf War Illness: Self-Reported Chemical/Biological Weapons Exposure and Mild Traumatic Brain Injury. Brain Sci. 2018, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Amourette, C.; Lamproglou, I.; Barbier, L.; Fauquette, W.; Zoppe, A.; Viret, R.; Diserbo, M. Gulf War illness: Effects of repeated stress and pyridostigmine treatment on blood–brain barrier permeability and cholinesterase activity in rat brain. Behav. Brain Res. 2009, 203, 207–214. [Google Scholar] [CrossRef]
- Lamproglou, I.; Barbier, L.; Diserbo, M.; Fauvelle, F.; Fauquette, W.; Amourette, C. Repeated stress in combination with pyridostigmine: Part I: Long-term behavioural consequences. Behav. Brain Res. 2009, 197, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Diserbo, M.; Lamproglou, I.; Amourette, C.; Peinnequin, A.; Fauquette, W. Repeated stress in combination with pyridostigmine: Part II: Changes in cerebral gene expression. Behav. Brain Res. 2009, 197, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, A.P.; James, L.M.; Mahan, M.; Joseph, J.; Georgopoulos, A.; Engdahl, B. Reduced Human Leukocyte Antigen (HLA) Protection in Gulf War Illness (GWI). EBioMedicine 2016, 3, 79–85. [Google Scholar] [CrossRef]
- Radhakrishnan, K.; Hauser, E.; Polimanti, R.; Helmer, D.; Provenzale, D.; McNeil, R.; Maffucci, A.; Quaden, R.; Zhao, H.; Whitbourne, S.; et al. Genomics of Gulf War Illness in U.S. Veterans Who Served during the 1990–1991 Persian Gulf War: Methods and Rationale for Veterans Affairs Cooperative Study. Brain Sci. 2021, 11, 845. [Google Scholar] [CrossRef] [PubMed]
- Alshelh, Z.; Albrecht, D.S.; Bergan, C.; Akeju, O.; Clauw, D.J.; Conboy, L.; Edwards, R.R.; Kim, M.; Lee, Y.C.; Protsenko, E.; et al. In-vivo imaging of neuroinflammation in veterans with Gulf War illness. Brain Behav. Immun. 2020, 87, 498–507. [Google Scholar] [CrossRef]
- Engdahl, B.E.; James, L.M.; Miller, R.D.; Leuthold, A.C.; Lewis, S.M.; Carpenter, A.F.; Georgopoulos, A.P. A Magnetoencephalographic (MEG) Study of Gulf War Illness (GWI). EBioMedicine 2016, 12, 127–132. [Google Scholar] [CrossRef]
- Rayhan, R.U.; Stevens, B.W.; Raksit, M.P.; Ripple, J.A.; Timbol, C.R.; Adewuyi, O.; VanMeter, J.; Baraniuk, J.N. Exercise Challenge in Gulf War Illness Reveals Two Subgroups with Altered Brain Structure and Function. PLoS ONE 2013, 8, e63903. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. Gulf War Illnesses: Causes and Treatments. Armed Forces Med Dev. 2001, 2, 41–44. [Google Scholar]
- Nicolson, G.L.; Nasralla, M.Y.; Franco, R.; Nicolson, N.L.; Erwin, R.; Ngwenya, R.; Berns, P.A. Diagnosis and Integrative Treatment of Intracellular Bacterial Infections in Chronic Fatigue and Fibromyalgia Syndromes, Gulf War Illness, Rheumatoid Arthritis, and Other Chronic Illnesses. Clin. Pract. Altern. Med. 2000, 1, 92–106. [Google Scholar]
- Weiner, M.W.; Meyerhoff, D.J.; Neylan, T.C.; Hlavin, J.; Ramage, E.R.; McCoy, D.; Studholme, C.; Cardens, V.; Marmar, C.; Truran, D.; et al. The relationship between Gulf War illness, brain N-acetylaspartate, and post-traumatic stress disorder. Mil. Med. 2011, 176, 896–902. [Google Scholar] [CrossRef]
- Jeffrey, M.; Collado, F.; Kibler, J.; DeLucia, C.; Messer, S.; Klimas, N.; Craddock, T.J.A. Post-traumatic stress impact on health outcomes in Gulf War Illness. BMC Psychol. 2021, 9, 1–9. [Google Scholar] [CrossRef]
- Jeffrey, M.G.; Krengel, M.; Kibler, J.L.; Zundel, C.; Klimas, N.G.; Sullivan, K.; Craddock, T.J.A. Neuropsychological Findings in Gulf War Illness: A Review. Front. Psychol. 2019, 10, 2088. [Google Scholar] [CrossRef]
- Plantinga, L.; Bremner, J.D.; Miller, A.H.; Jones, D.P.; Veledar, E.; Goldberg, J.; Vaccarino, V. Association between posttraumatic stress disorder and inflammation: A twin study. Brain Behav. Immun. 2013, 30, 125–132. [Google Scholar] [CrossRef]
- Hoge, E.; Brandstetter, K.; Moshier, S.; Pollack, M.; Wong, K.; Simon, N. Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depression Anxiety 2009, 26, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Liu, T.; Guo, J.-C.; Jiang, X.-L.; Chen, F.; Gao, Y.-S. Study on serum cytokine levels in posttraumatic stress disorder patients. Asian Pac. J. Trop. Med. 2012, 5, 323–325. [Google Scholar] [CrossRef]
- Baker, D.G.; Nievergelt, C.; O’Connor, D.T. Biomarkers of PTSD: Neuropeptides and immune signaling. Neuropharmacology 2012, 62, 663–673. [Google Scholar] [CrossRef]
- Khaiboullina, S.; DeMeirleir, K.L.; Rawat, S.; Berk, G.S.; Gaynor-Berk, R.S.; Mijatovic, T.; Blatt, N.; Rizvanov, A.; Young, S.G.; Lombardi, V.C. Cytokine expression provides clues to the pathophysiology of Gulf War illness and myalgic encephalomyelitis. Cytokine 2015, 72, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Broderick, G.; Kreitz, A.; Fuite, J.; Fletcher, M.A.; Vernon, S.D.; Klimas, N. A pilot study of immune network remodeling under challenge in Gulf War Illness. Brain Behav. Immun. 2011, 25, 302–313. [Google Scholar] [CrossRef]
- Friedman, A.; Kaufer, D.; Shemer, J.; Hendler, I.; Soreq, H.; Tur-Kaspa, I. Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat. Med. 1996, 2, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Esposito, P.; Chandler, N.; Kandere, K.; Basu, S.; Jacobson, S.; Connolly, R.; Tutor, D.; Theoharides, T.C. Corticotropin-Releasing Hormone and Brain Mast Cells Regulate Blood-Brain-Barrier Permeability Induced by Acute Stress. J. Pharmacol. Exp. Ther. 2002, 303, 1061–1066. [Google Scholar] [CrossRef]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joëls, M. Rapid non-genomic effects of corticosteroids through the membrane-associated MR and GR and their role in the central stress. J. Endocrinol. 2011, 2, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef]
- Mikics, E.; Kruk, M.R.; Haller, J. Genomic and non-genomic effects of glucocorticoids on aggressive behavior in male rats. Psychoneuroendocrinology 2004, 29, 618–635. [Google Scholar] [CrossRef]
- Rankin, J.; Walker, J.J.; Windle, R.; Lightman, S.L.; Terry, J.R. Characterizing Dynamic Interactions between Ultradian Glucocorticoid Rhythmicity and Acute Stress Using the Phase Response Curve. PLoS ONE 2012, 7, e30978. [Google Scholar] [CrossRef]
- Walker, J.J.; Spiga, F.; Waite, E.; Zhao, Z.; Kershaw, Y.; Terry, J.R.; Lightman, S.L. The origin of glucocorticoid hormone oscillations. PLoS Biol. 2012, 10, e1001341. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.J.; Terry, J.R.; Lightman, S.L. Origin of ultradian pulsatility in the hypothalamic–pituitary–adrenal axis. Proc. R. Soc. B Biol. Sci. 2010, 277, 1627–1633. [Google Scholar] [CrossRef]
- Vinther, F.; Andersen, M.; Ottesen, J.T. The minimal model of the hypothalamic–pituitary–adrenal axis. J. Math. Biol. 2011, 63, 663–690. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Vernon, S.D.; Broderick, G. Model-Based Therapeutic Correction of Hypothalamic-Pituitary-Adrenal Axis Dysfunction. PLoS Comput. Biol. 2009, 5, e1000273. [Google Scholar] [CrossRef]
- Gupta, S.; Aslakson, E.; Gurbaxani, B.M.; Vernon, S.D. Inclusion of the glucocorticoid receptor in a hypothalamic pituitary adrenal axis model reveals bistability. Theor. Biol. Med. Model. 2007, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Craddock, T.J.; Fritsch, P.; Rice, M.A., Jr.; Del Rosario, R.M.; Miller, D.B.; Fletcher, M.A.; Klimas, N.G.; Broderick, G. A role for homeostatic drive in the perpetuation of complex chronic illness: Gulf War Illness and chronic fatigue syndrome. PLoS ONE 2014, 9, e84839. [Google Scholar] [CrossRef]
- Fritsch, P.; Craddock, T.J.A.; del Rosario, R.M.; Rice, M.A.; Smylie, A.; Folcik, V.A.; de Vries, G.; Fletcher, M.A.; Klimas, N.G.; Broderick, G. Succumbing to the laws of attraction: Exploring the sometimes pathogenic versatility of discrete immune logic. Syst. Biomed. 2014, 1, 179–194. [Google Scholar] [CrossRef]
- Mendoza, L.; Xenarios, I. A method for the generation of standardized qualitative dynamical systems of regulatory networks. Theor. Biol. Med. Model. 2006, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Thieffry, D.; Kaufman, M. Dynamical behaviour of biological regulatory networks—I. Biological role of feedback loops and practical use of the concept of the loop-characteristic state. Bull. Math. Biol. 1995, 57, 247–276. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Kaufman, M. Multistationarity, the basis of cell differentiation and memory. II. Logical analysis of regulatory networks in terms of feedback circuits. Chaos Interdiscip. J. Nonlinear Sci. 2001, 11, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R. Regulatory networks seen as asynchronous automata: A logical description. J. Theor. Biol. 1991, 153, 1–23. [Google Scholar] [CrossRef]
- Whitley, D.; Garrett, D.; Watson, J.-P. Quad search and hybrid genetic algorithms. In Proceedings of the Genetic and Evolutionary Computation Conference, Chicago, IL, USA, 12–16 July 2003; Cantu-Paz, E., Foster, J.A., Deb, K., Davis, L.D., Roy, R., O’Reilly, U.-M., Beyer, H.-G., Standish, R., Kendall, G., Wilson, S., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Day, J.D.; Rubin, J.; Vodovotz, Y.; Chow, C.C.; Reynolds, A.; Clermont, G. A reduced mathematical model of the acute inflammatory response II. Capturing scenarios of repeated endotoxin administration. J. Theor. Biol. 2006, 242, 237–256. [Google Scholar] [CrossRef]
- Reynolds, A.; Rubin, J.; Clermont, G.; Day, J.D.; Vodovotz, Y.; Ermentrout, G.B. A reduced mathematical model of the acute inflammatory response: I. Derivation of model and analysis of anti-inflammation. J. Theor. Biol. 2006, 242, 220–236. [Google Scholar] [CrossRef]
- McDonald, S.D.; Thompson, N.L.; Stratton, K.J.; Calhoun, P.S. Diagnostic accuracy of three scoring methods for the Davidson Trauma Scale among U.S. military Veterans. J. Anxiety Disord. 2014, 28, 160–168. [Google Scholar] [CrossRef]
- Storey, J.D. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2002, 64, 479–498. [Google Scholar] [CrossRef]
- White, R.F.; Steele, L.; O’Callaghan, J.P.; Sullivan, K.; Binns, J.H.; Golomb, B.A.; Bloom, F.E.; Bunker, J.A.; Crawford, F.; Graves, J.C.; et al. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: Effects of toxicant exposures during deployment. Cortex 2016, 74, 449–475. [Google Scholar] [CrossRef] [PubMed]
- Locker, A.R.; Michalovicz, L.T.; Kelly, K.A.; Miller, J.V.; Miller, D.B.; O’Callaghan, J.P. Corticosterone primes the neuroinflammatory response to Gulf War Illness-relevant organophosphates independently of acetylcholinesterase inhibition. J. Neurochem. 2017, 142, 444–455. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Kelly, K.; Locker, A.R.; Miller, D.B.; Lasley, S.M. Corticosterone primes the neuroinflammatory response to DFP in mice: Potential animal model of Gulf War Illness. J. Neurochem. 2015, 133, 708–721. [Google Scholar] [CrossRef]
- Koo, B.-B.; Michalovicz, L.T.; Calderazzo, S.; Kelly, K.; Sullivan, K.; Killiany, R.J.; O’Callaghan, J.P. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of Gulf War Illness. Brain Behav. Immun. 2018, 67, 42–46. [Google Scholar] [CrossRef]
- Craddock, T.J.A.; Michalovicz, L.; Kelly, K.A.; Rice, M., Jr.; Miller, D.B.; Klimas, N.; Morris, M.; O’Callaghan, J.P.; Broderick, G. A Logic Model of Neuronal-Glial Interaction Suggests Altered Homeostatic Regulation in the Perpetuation of Neuroinflammation. Front. Cell. Neurosci. 2018, 12, 336. [Google Scholar] [CrossRef]
- Miller, J.V.; LeBouf, R.; A Kelly, K.; Michalovicz, L.T.; Ranpara, A.; Locker, A.R.; Miller, D.B.; O’Callaghan, J.P. The Neuroinflammatory Phenotype in a Mouse Model of Gulf War Illness is Unrelated to Brain Regional Levels of Acetylcholine as Measured by Quantitative HILIC-UPLC-MS/MS. Toxicol. Sci. 2018, 165, 302–313. [Google Scholar] [CrossRef]
- Jones, B.C.; Miller, D.B.; Lu, L.; Zhao, W.; Ashbrook, D.G.; Xu, F.; Mulligan, M.K.; Williams, R.W.; Zhuang, D.; Torres-Rojas, C.; et al. Modeling the Genetic Basis of Individual Differences in Susceptibility to Gulf War Illness. Brain Sci. 2020, 10, 143. [Google Scholar] [CrossRef]
- Xu, F.; Ashbrook, D.; Gao, J.; Starlard-Davenport, A.; Zhao, W.; Miller, D.B.; O’Callaghan, J.P.; Williams, R.W.; Jones, B.C.; Lu, L. Genome-wide transcriptome architecture in a mouse model of Gulf War Illness. Brain Behav. Immun. 2020, 89, 209–223. [Google Scholar] [CrossRef]
- Ashbrook, D.G.; Hing, B.; Michalovicz, L.T.; Kelly, K.A.; Miller, J.V.; De Vega, W.C.; Miller, D.B.; Broderick, G.; O’Callaghan, J.P.; McGowan, P.O. Epigenetic impacts of stress priming of the neuroinflammatory response to sarin surrogate in mice: A model of Gulf War illness. J. Neuroinflammation 2018, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, F.; Starlard-Davenport, A.; Miller, D.B.; O’Callaghan, J.P.; Jones, B.C.; Lu, L. Exploring the Role of Chemokine Receptor 6 (Ccr6) in the BXD Mouse Model of Gulf War Illness. Front. Neurosci. 2020, 14, 818. [Google Scholar] [CrossRef] [PubMed]
- Belgrad, J.; Dutta, D.; Bromley-Coolidge, S.; Kelly, K.A.; Michalovicz, L.T.; Sullivan, K.A.; O’Callaghan, J.P.; Fields, R.D. Oligodendrocyte involvement in Gulf War Illness. Glia 2019, 67, 2107–2124. [Google Scholar] [CrossRef] [PubMed]
- Lacagnina, M.J.; Li, J.; Lorca, S.; Rice, K.C.; Sullivan, K.; O’Callaghan, J.P.; Grace, P.M. A role for neuroimmune signaling in a rat model of Gulf War Illness-related pain. Brain Behav. Immun. 2021, 91, 418–428. [Google Scholar] [CrossRef]
- Michalovicz, L.T.; Locker, A.R.; Kelly, K.A.; Miller, J.V.; Barnes, Z.; Fletcher, M.A.; Miller, D.B.; Klimas, N.G.; Morris, M.; Lasley, S.M.; et al. Corticosterone and pyridostigmine/DEET exposure attenuate peripheral cytokine expression: Supporting a dominant role for neuroinflammation in a mouse model of Gulf War Illness. NeuroToxicology 2019, 70, 26–32. [Google Scholar] [CrossRef]
- Cheng, C.-H.; Koo, B.-B.; Calderazzo, S.; Quinn, E.; Aenlle, K.; Steele, L.; Klimas, N.; Krengel, M.; Janulewicz, P.; Toomey, R.; et al. Alterations in high-order diffusion imaging in veterans with Gulf War Illness is associated with chemical weapons exposure and mild traumatic brain injury. Brain Behav. Immun. 2020, 89, 281–290. [Google Scholar] [CrossRef]
- Kansagara, D.; Emily, G.G.; Ayers, C.K.; Winchell, K.A.; Freeman, M.; Press, A.M.; Paynte, R.; Kansagara, D.; Nugent, S.M. Biological Measures and Diagnostic Tools for Gulf War Illness: A Systematic Review. Evid. Synth. Program 2020. [Google Scholar]
- Gean, E.G.; Ayers, C.K.; Winchell, K.A.; Freeman, M.; Press, A.M.; Paynter, R.; Kansagara, D.; Nugent, S.M. Biological measures and diagnostic tools for Gulf War Illness—A systematic review. Life Sci. 2021, 275, 119360. [Google Scholar] [CrossRef]
- National Library of Medicine US. TNF and Glucocorticoid Antagonist for GWI Associated Multi-symptom Disease Homeostasis Reset (E/M) Identifier: NCT04254627. Available online: https://clinicaltrials.gov/ct2/show/NCT04254627 (accessed on 5 February 2020).
- Craddock, T.J.A.; Del Rosario, R.R.; Rice, M.; Zysman, J.P.; Fletcher, M.A.; Klimas, N.G.; Broderick, G. Achieving Remission in Gulf War Illness: A Simulation-Based Approach to Treatment Design. PLoS ONE 2015, 10, e0132774. [Google Scholar] [CrossRef] [PubMed]
- Ishøy, T.; Andersson, A.-M.; Suadicani, P.; Guldager, B.; Appleyard, M.; Gyntelberg, F.; Skakkebaek, N.E. Major reproductive health characteristics in male Gulf War Veterans. The Danish Gulf War Study. Dan. Med Bull. 2001, 48, 29–32. [Google Scholar]
- Ishoy, T.; Suadicani, P.; Andersson, A.-M.; Guldager, B.; Appleyard, M.; Skakkebæk, N.; Gyntelberg, F. Prevalence of male sexual problems in the Danish Gulf War Study. Scand. J. Sexol. 2001, 4, 43–45. [Google Scholar]
- Ishøy, T.; Suadicani, P.; Guldager, B.; Appleyard, M.; O Hein, H.; Gyntelberg, F. State of health after deployment in the Persian Gulf. The Danish Gulf War Study. Dan. Med Bull. 1999, 46, 416–419. [Google Scholar]
- McDiarmid, M.A.; Squibb, K.; Engelhardt, S.; Oliver, M.; Gucer, P.; Wilson, P.D.; Kane, R.; Kabat, M.; Kaup, B.; Anderson, L.; et al. Surveillance of Depleted Uranium Exposed Gulf War Veterans: Health Effects Observed in an Enlarged "Friendly Fire" Cohort. J. Occup. Environ. Med. 2001, 43, 991–1000. [Google Scholar] [CrossRef]
- McDiarmid, M.A.; Engelhardt, S.M.; Dorsey, C.D.; Oliver, M.; Gucer, P.; Gaitens, J.M.; Kane, R.; Cernich, A.; Kaup, B.; Hoover, D.; et al. Longitudinal Health Surveillance in a Cohort of Gulf War Veterans 18 Years After First Exposure to Depleted Uranium. J. Toxicol. Environ. Heal. Part A 2011, 74, 678–691. [Google Scholar] [CrossRef] [PubMed]
- McDiarmid, M.A.; Engelhardt, S.M.; Oliver, M.; Gucer, P.; Wilson, P.D.; Kane, R.; Cernich, A.; Kaup, B.; Anderson, L.; Hoover, D.; et al. Health Surveillance Of Gulf War I Veterans Exposed to Depleted Uranium: Updating The Cohort. Heal. Phys. 2007, 93, 60–73. [Google Scholar] [CrossRef]
- McDiarmid, M.A.; Engelhardt, S.; Oliver, M.; Gucer, P.; Wilson, P.D.; Kane, R.; Kabat, M.; Kaup, B.; Anderson, L.; Hoover, D.; et al. Health Effects of Depleted Uranium on Exposed Gulf War Veterans: A 10-Year Follow-Up. J. Toxicol. Environ. Heal. Part A 2004, 67, 277–296. [Google Scholar] [CrossRef] [PubMed]
- McDiarmid, M.A.; Keogh, J.P.; Hooper, F.J.; McPhaul, K.; Squibb, K.; Kane, R.; DiPino, R.; Kabat, M.; Kaup, B.; Anderson, L.; et al. Health Effects of Depleted Uranium on Exposed Gulf War Veterans. Environ. Res. 2000, 82, 168–180. [Google Scholar] [CrossRef] [PubMed]
- McDiarmid, M.A.; Gaitens, J.M.; Hines, S.; Breyer, R.; Wong-You-Cheong, J.J.; Engelhardt, S.M.; Oliver, M.; Gucer, P.; Kane, R.; Cernich, A.; et al. The Gulf War depleted uranium cohort at 20 years: Bioassay results and novel approaches to fragment surveillance. Health Phys. 2013, 104, 347–361. [Google Scholar] [CrossRef] [PubMed]
- McDiarmid, M.A.; Engelhardt, S.M.; Oliver, M.; Gucer, P.; Wilson, P.D.; Kane, R.; Kabat, M.; Kaup, B.; Anderson, L.; Hoover, D.; et al. Biological monitoring and surveillance results of Gulf War I veterans exposed to depleted uranium. Int. Arch. Occup. Environ. Heal. 2005, 79, 11–21. [Google Scholar] [CrossRef]
- Joshi, S.C.; Mathur, R.; Gulati, N. Testicular toxicity of chlorpyrifos (an organophosphate pesticide) in albino rat. Toxicol. Ind. Heal. 2007, 23, 439–444. [Google Scholar] [CrossRef]
- Peiris, D.C.; Dhanushka, T. Low doses of chlorpyrifos interfere with spermatogenesis of rats through reduction of sex hormones. Environ. Sci. Pollut. Res. 2017, 24, 20859–20867. [Google Scholar] [CrossRef] [PubMed]
- Alaa-Eldin, E.A.; El-Shafei, D.A.; Abouhashem, N.S. Individual and combined effect of chlorpyrifos and cypermethrin on reproductive system of adult male albino rats. Environ. Sci. Pollut. Res. 2016, 24, 1532–1543. [Google Scholar] [CrossRef]
- Kudavidanage, E.P.; Dissanayake, D.M.I.; Keerthirathna, W.L.R.; Nishshanke, N.L.W.; Peiris, L.D.C. Commercial Formulation of Chlorpyrifos Alters Neurological Behaviors and Fertility. Biology 2020, 9, 49. [Google Scholar] [CrossRef]
- Li, J.; Ren, F.; Li, Y.; Luo, J.; Pang, G. Chlorpyrifos Induces Metabolic Disruption by Altering Levels of Reproductive Hormones. J. Agric. Food Chem. 2019, 67, 10553–10562. [Google Scholar] [CrossRef]
- White, H.D.; Robinson, T.D. A novel use for testosterone to treat central sensitization of chronic pain in fibromyalgia patients. Int. Immunopharmacol. 2015, 27, 244–248. [Google Scholar] [CrossRef]
- White, H.D.; Brown, L.A.; Gyurik, R.J.; Manganiello, P.D.; Robinson, T.D.; Hallock, L.S.; Lewis, L.D.; Yeo, K.-T.J. Treatment of pain in fibromyalgia patients with testosterone gel: Pharmacokinetics and clinical response. Int. Immunopharmacol. 2015, 27, 249–256. [Google Scholar] [CrossRef]
- Rice, M.A., Jr.; Craddock, T.J.A.; Folcik, V.A.; del Rosario, R.M.; Barnes, Z.M.; Klimas, N.G.; Fletcher, M.A.; Zysman, J.; Broderick, G. Gulf War Illness: Is there lasting damage to the endocrine-immune circuitry? Syst. Biomed. 2014, 2, 80–89. [Google Scholar] [CrossRef][Green Version]
- Maes, M.; Lin, A.-H.; Delmeire, L.; Van Gastel, A.; Kenis, G.; De Jongh, R.; Bosmans, E. Elevated serum interleukin-6 (IL-6) and IL-6 receptor concentrations in posttraumatic stress disorder following accidental man-made traumatic events. Biol. Psychiatry 1999, 45, 833–839. [Google Scholar] [CrossRef]
- Lima, B.B.; Hammadah, M.; Wilmot, K.; Pearce, B.D.; Shah, A.; Levantsevych, O.; Kaseer, B.; Obideen, M.; Gafeer, M.M.; Kim, J.H.; et al. Posttraumatic stress disorder is associated with enhanced interleukin-6 response to mental stress in subjects with a recent myocardial infarction. Brain Behav. Immun. 2019, 75, 26–33. [Google Scholar] [CrossRef]
- Passos, I.C.; Vasconcelos-Moreno, M.P.; Costa, L.G.; Kunz, M.; Brietzke, E.; Quevedo, J.; Salum, G.; Magalhães, P.V.; Kapczinski, F.; Kauer-Sant’Anna, M. Inflammatory markers in post-traumatic stress disorder: A systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2015, 2, 1002–1012. [Google Scholar] [CrossRef]
- Cheng, Y.; Jope, R.S.; Beurel, E. A pre-conditioning stress accelerates increases in mouse plasma inflammatory cytokines induced by stress. BMC Neurosci. 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Tao, W.; Dong, Y.; Su, Q.; Wang, H.; Chen, Y.; Xue, W.; Chen, C.; Xia, B.; Duan, J.; Chen, G. Liquiritigenin reverses depression-like behavior in unpredictable chronic mild stress-induced mice by regulating PI3K/Akt/mTOR mediated BDNF/TrkB pathway. Behav. Brain Res. 2016, 308, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Tagliari, B.; Tagliari, A.P.; Schmitz, F.; Da Cunha, A.A.; Dalmaz, C.; Wyse, A.T.S. Chronic Variable Stress Alters Inflammatory and Cholinergic Parameters in Hippocampus of Rats. Neurochem. Res. 2010, 36, 487–493. [Google Scholar] [CrossRef]
- Reader, B.; Jarrett, B.; McKim, D.; Wohleb, E.; Godbout, J.; Sheridan, J. Peripheral and central effects of repeated social defeat stress: Monocyte trafficking, microglial activation, and anxiety. Neuroscience 2015, 289, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Gamble-George, J.C.; Baldi, R.; Halladay, L.; Kocharian, A.; Hartley, N.; Silva, C.G.; Roberts, H.; Haymer, A.; Marnett, L.J.; Holmes, A.; et al. Author response: Cyclooxygenase-2 inhibition reduces stress-induced affective pathology. Elife 2016, 5, e14137. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.-Y.; He, Z.; Zannas, A.S.; Hahn, O.; Kühne, C.; Reichel, J.M.; Binder, E.B.; Wotjak, C.T.; Khaitovich, P.; Turck, C.W. Fluoxetine treatment prevents the inflammatory response in a mouse model of posttraumatic stress disorder. J. Psychiatr. Res. 2016, 76, 74–83. [Google Scholar] [CrossRef]
- Ebenezer, P.J.; Wilson, C.B.; Wilson, L.D.; Nair, A.R.; J, F. The Anti-Inflammatory Effects of Blueberries in an Animal Model of Post-Traumatic Stress Disorder (PTSD). PLoS ONE 2016, 11, e0160923. [Google Scholar] [CrossRef]
- Young, M.B.; Howell, L.L.; Hopkins, L.; Moshfegh, C.; Yu, Z.; Clubb, L.; Seidenberg, J.; Park, J.; Swiercz, A.P.; Marvar, P.J. A peripheral immune response to remembering trauma contributes to the maintenance of fear memory in mice. Psychoneuroendocrinology 2018, 94, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Nisenbaum, R.; Stewart, G.; Thompson, W.W.; Robin, L.; Washko, R.M.; Noah, D.L.; Barrett, D.H.; Randall, B.; Herwaldt, B.L. Chronic multisymptom illness affecting Air Force veterans of the Gulf War. JAMA 1998, 280, 981–988. [Google Scholar] [CrossRef]
- Reeves, W.C.; Lloyd, A.; Vernon, S.D.; Klimas, N.; Jason, L.A.; Bleijenberg, G.; Evengard, B.; White, P.D.; Nisenbaum, R.; Unger, E.R.; et al. Identification of ambiguities in the 1994 chronic fatigue syndrome research case definition and recommendations for resolution. BMC Health Serv. Res. 2003, 3, 1–9. [Google Scholar] [CrossRef]
- Collins, J.F.; Donta, S.T.; Engel, C.C., Jr.; Baseman, J.B.; Dever, L.L.; Taylor, T.; Boardman, K.D.; Martin, S.E.; Wiseman, A.L.; Feussner, J.R. The antibiotic treatment trial of Gulf War Veterans’ Illnesses: Issues, design, screening, and baseline characteristics. Control. Clin. Trials 2002, 23, 333–353. [Google Scholar] [CrossRef]
- Kelly, K.A.; Michalovicz, L.T.; Miller, J.V.; Castranova, V.; Miller, D.B.; O’Callaghan, J.P. Prior exposure to corticosterone markedly enhances and prolongs the neuroinflammatory response to systemic challenge with LPS. PLoS ONE 2018, 13, e0190546. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.B. 400: A method for combining non-independent, one-sided tests of significance. Biometrics 1975, 31, 987–992. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Total | GWI | GWIH | GWIL | HC | p3 | p2 |
|---|---|---|---|---|---|---|---|
| n | 89 | 51 | 32 | 19 | 38 | ||
| Mean Age (y) | 43.7 ± 7.4 | 43.8 ± 5.7 | 44.4 ± 5.0 | 42.8 ± 6.7 | 43.5 ± 9.3 | 0.991 | 0.979 |
| Mean BMI | 29.5 ± 5.4 | 30.3 ± 4.6 | 30.3 ± 4.1 | 30.2 ± 5.4 | 28.5 ± 5.4 | 0.958 | 0.803 |
| Race | 0.318 | 0.245 | |||||
| Asian | 2.2% | 3.9% | 3.1% | 5.3% | 0.0% | ||
| Black | 22.5% | 27.5% | 34.4% | 15.8% | 15.8% | ||
| Hispanic | 46.1% | 39.2% | 40.6% | 36.8% | 55.3% | ||
| White | 28.1% | 29.4% | 21.9% | 42.1% | 26.3% | ||
| Other | 1.1% | 0.0% | 0.0% | 0.0% | 2.6% | ||
| Marital Status | 0.755 | 0.838 | |||||
| Married | 62.9% | 64.7% | 71.9% | 52.6% | 60.5% | ||
| Widowed | 1.1% | 0.0% | 0.0% | 0.0% | 2.6% | ||
| Divorced | 12.4% | 13.7% | 12.5% | 15.8% | 10.5% | ||
| Separated | 3.4% | 3.9% | 3.1% | 5.3% | 2.6% | ||
| Never Married | 9.0% | 7.8% | 9.4% | 5.3% | 10.5% | ||
| Not Answered | 11.2% | 9.8% | 3.1% | 21.1% | 13.2% | ||
| Employed | 75.3% | 70.6% | 68.8% | 73.7% | 81.6% | 0.456 | 0.234 |
| Mean Schooling (y) | 15.5 ± 2.5 | 15.3 ± 2.6 | 15.7 ± 2.8 | 15.4 ± 2.0 | 15.8 ± 2.8 | 0.994 | 0.907 |
| Treatment | tstart | tstop |
|---|---|---|
| Increase Testosterone | 1 | 43 |
| Inhibit CK1 | 21 | 83 |
| Inhibit GRD | 75 | 99 |
| Treatment | tstart | tstop |
|---|---|---|
| Inhibit MK6 | 1 | 100 |
| Inhibit CK1 | 8 | 40 |
| Inhibit GRD | 77 | 99 |
| Treatment | tstart | tstop |
|---|---|---|
| Inhibit MK6 | 1 | 96 |
| Inhibit GRD | 7 | 93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrera Arias, F.J.; Aenlle, K.; Abreu, M.; Holschbach, M.A.; Michalovicz, L.T.; Kelly, K.A.; Klimas, N.; O’Callaghan, J.P.; Craddock, T.J.A. Modeling Neuroimmune Interactions in Human Subjects and Animal Models to Predict Subtype-Specific Multidrug Treatments for Gulf War Illness. Int. J. Mol. Sci. 2021, 22, 8546. https://doi.org/10.3390/ijms22168546
Carrera Arias FJ, Aenlle K, Abreu M, Holschbach MA, Michalovicz LT, Kelly KA, Klimas N, O’Callaghan JP, Craddock TJA. Modeling Neuroimmune Interactions in Human Subjects and Animal Models to Predict Subtype-Specific Multidrug Treatments for Gulf War Illness. International Journal of Molecular Sciences. 2021; 22(16):8546. https://doi.org/10.3390/ijms22168546
Chicago/Turabian StyleCarrera Arias, Francisco J., Kristina Aenlle, Maria Abreu, Mary A. Holschbach, Lindsay T. Michalovicz, Kimberly A. Kelly, Nancy Klimas, James P. O’Callaghan, and Travis J. A. Craddock. 2021. "Modeling Neuroimmune Interactions in Human Subjects and Animal Models to Predict Subtype-Specific Multidrug Treatments for Gulf War Illness" International Journal of Molecular Sciences 22, no. 16: 8546. https://doi.org/10.3390/ijms22168546
APA StyleCarrera Arias, F. J., Aenlle, K., Abreu, M., Holschbach, M. A., Michalovicz, L. T., Kelly, K. A., Klimas, N., O’Callaghan, J. P., & Craddock, T. J. A. (2021). Modeling Neuroimmune Interactions in Human Subjects and Animal Models to Predict Subtype-Specific Multidrug Treatments for Gulf War Illness. International Journal of Molecular Sciences, 22(16), 8546. https://doi.org/10.3390/ijms22168546