
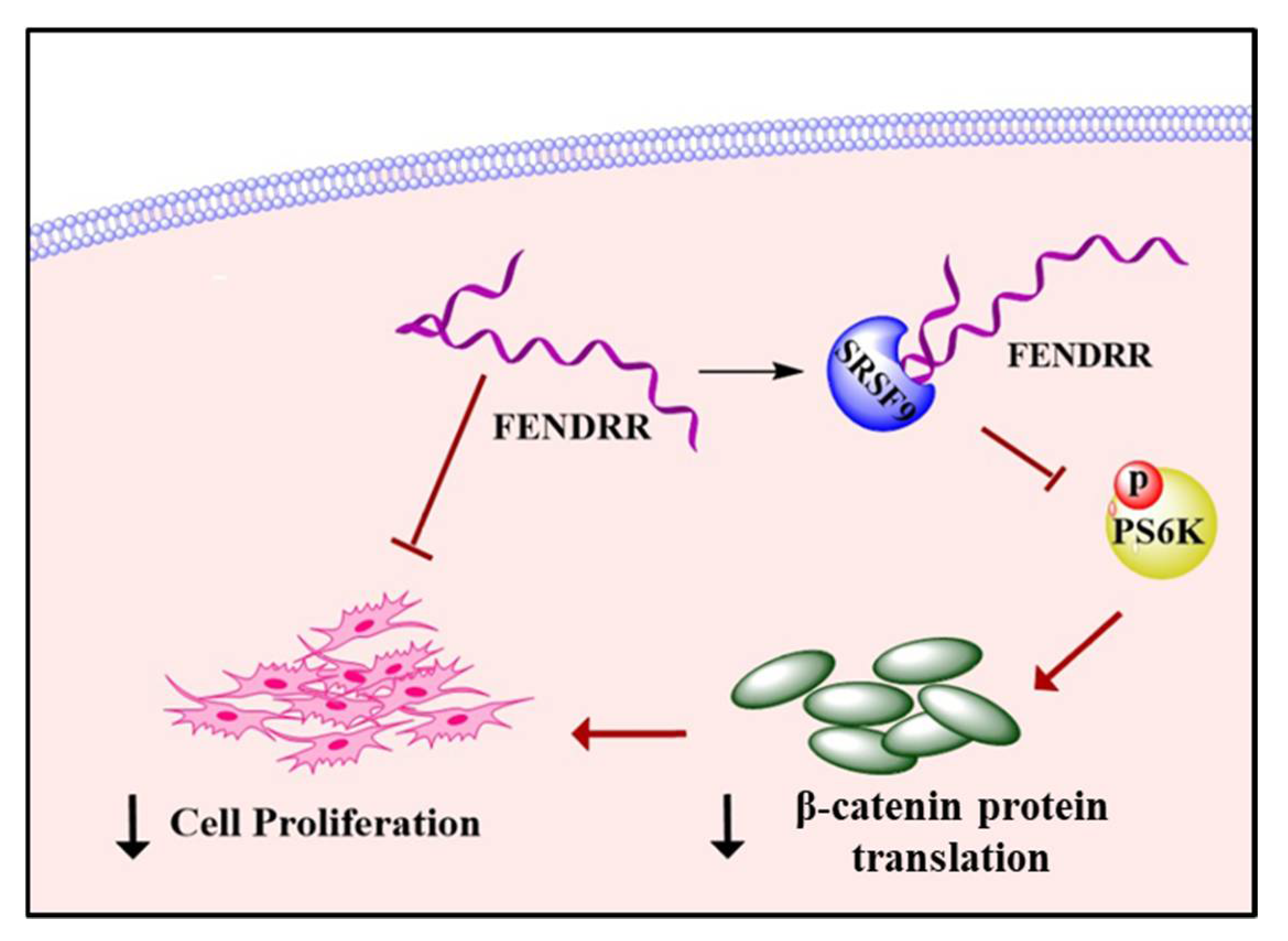
Long Noncoding RNA FENDRR Inhibits Lung Fibroblast Proliferation via a Reduction of β-Catenin

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
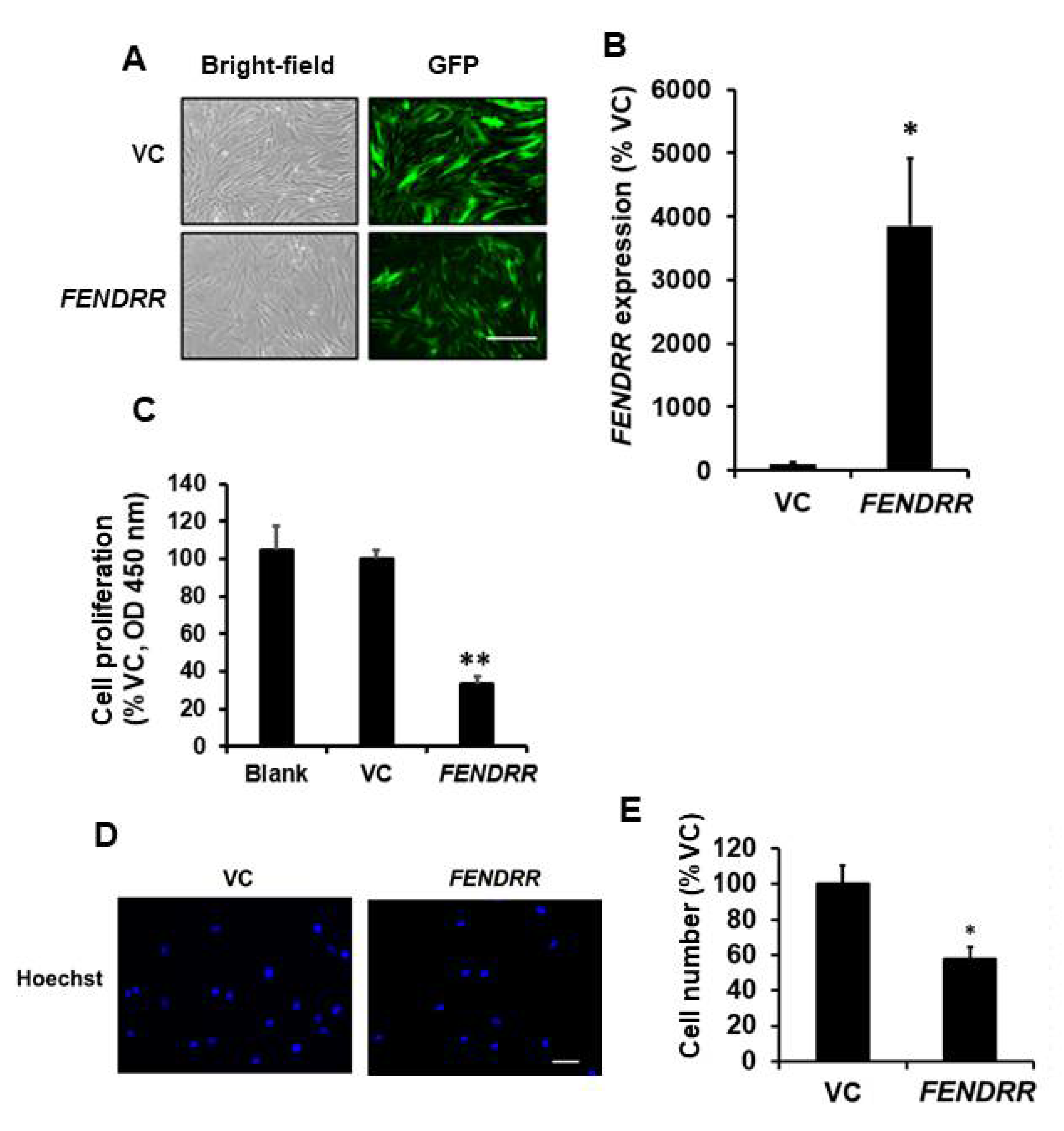
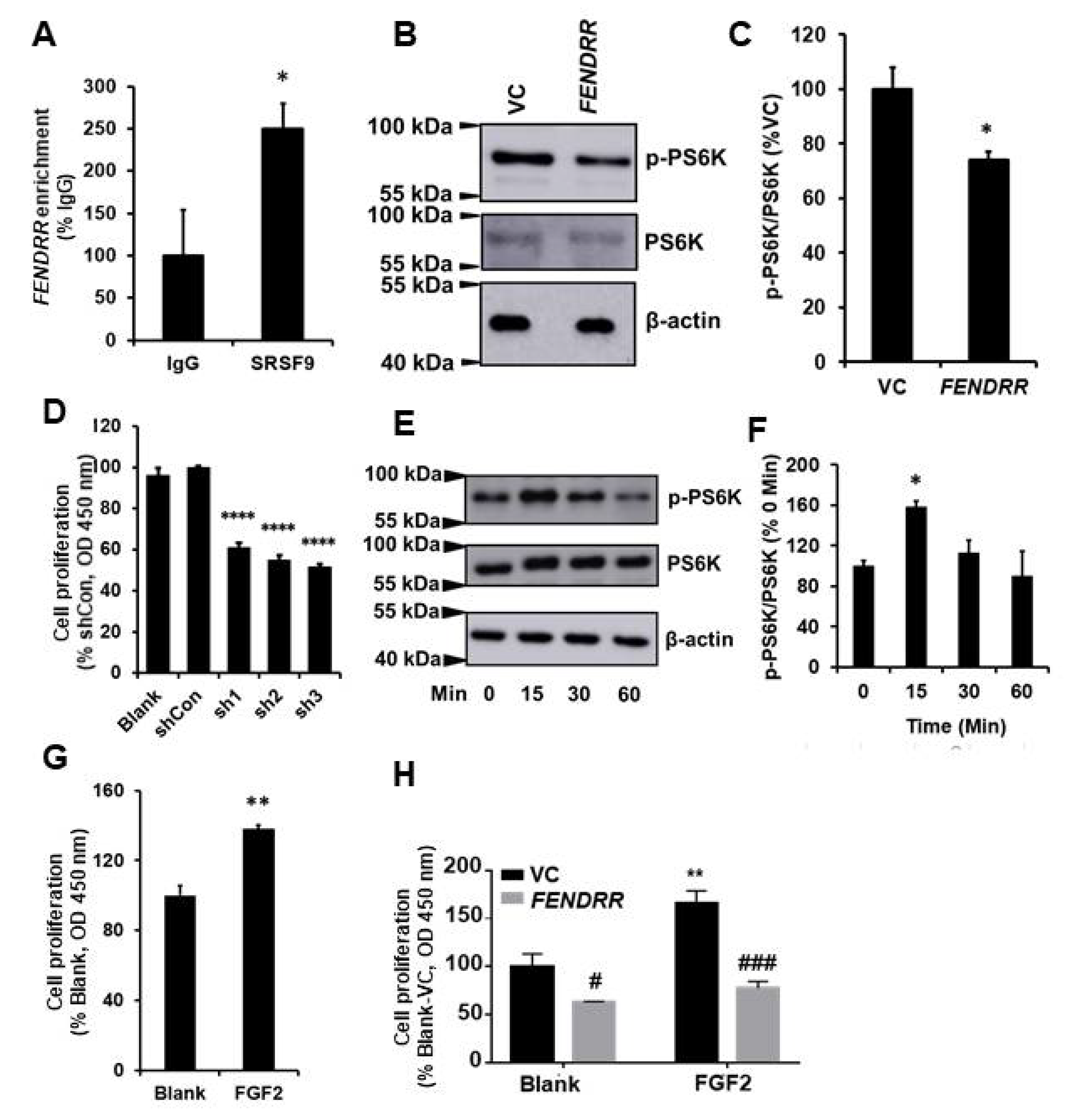
2.1. FENDRR Inhibits Fibroblast Proliferation
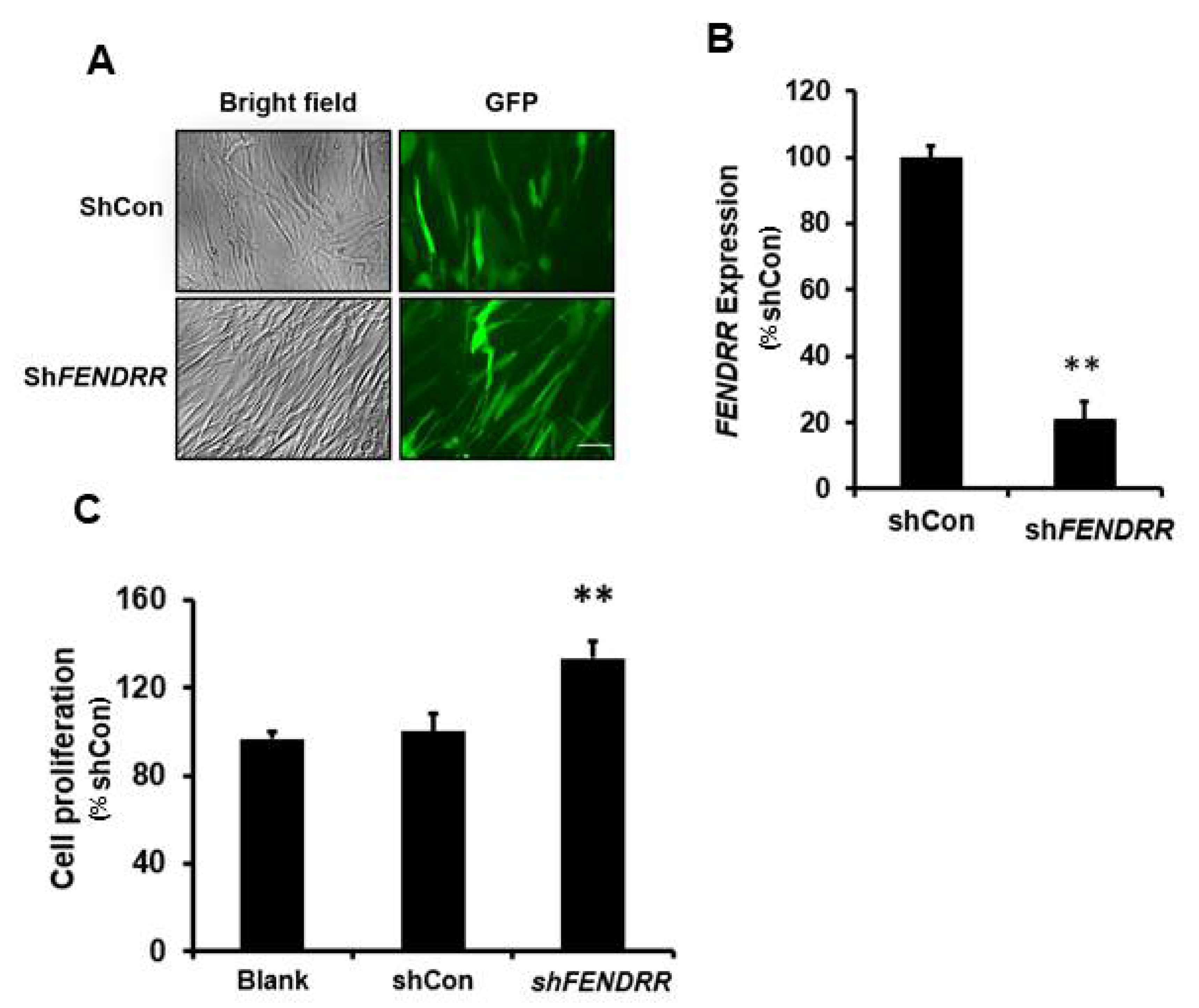
2.2. Silencing of FENDRR Increases Fibroblast Proliferation
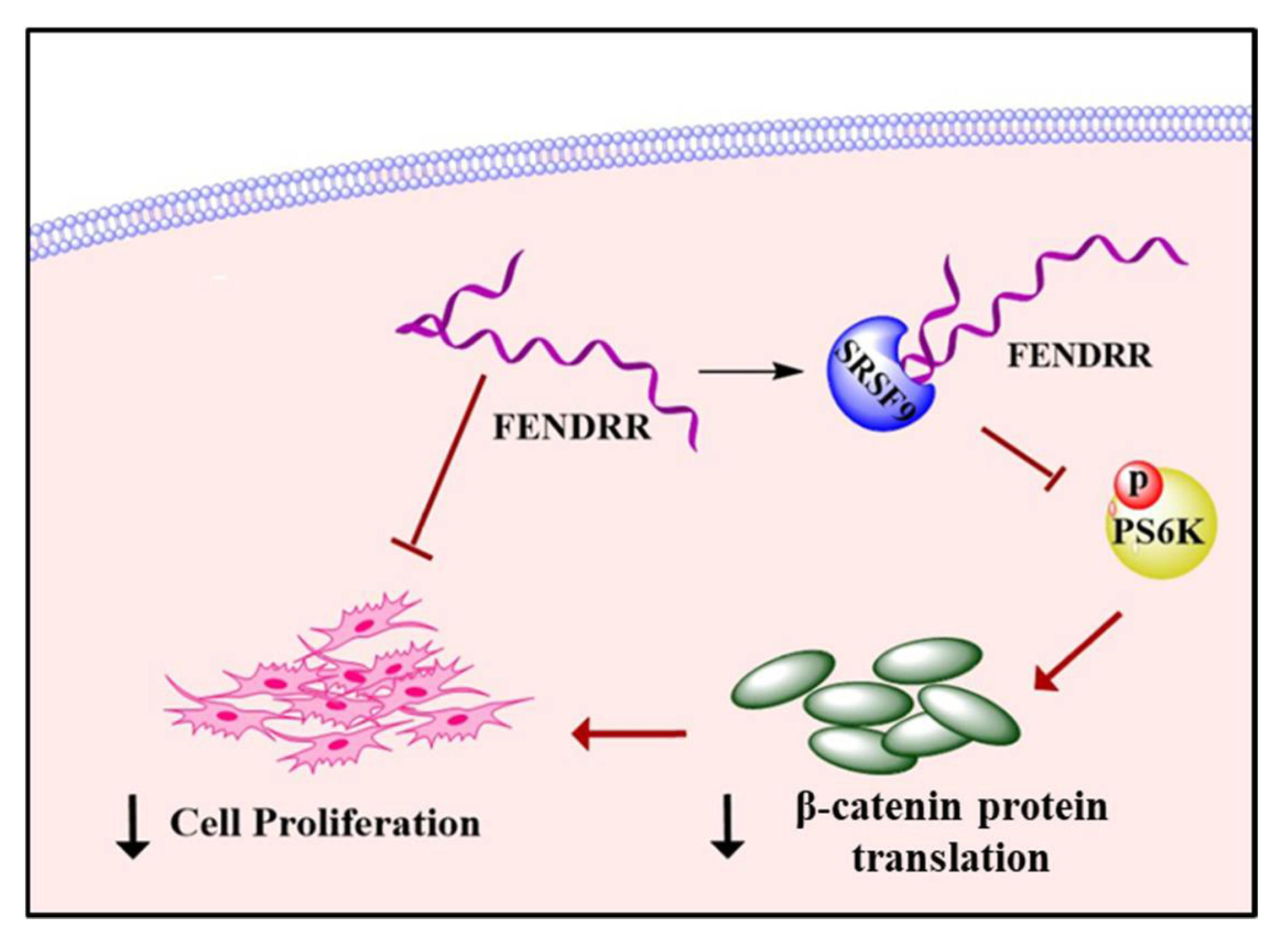
2.3. FENDRR Binds SRSF9 and Inhibits the Phosphorylation of PS6K
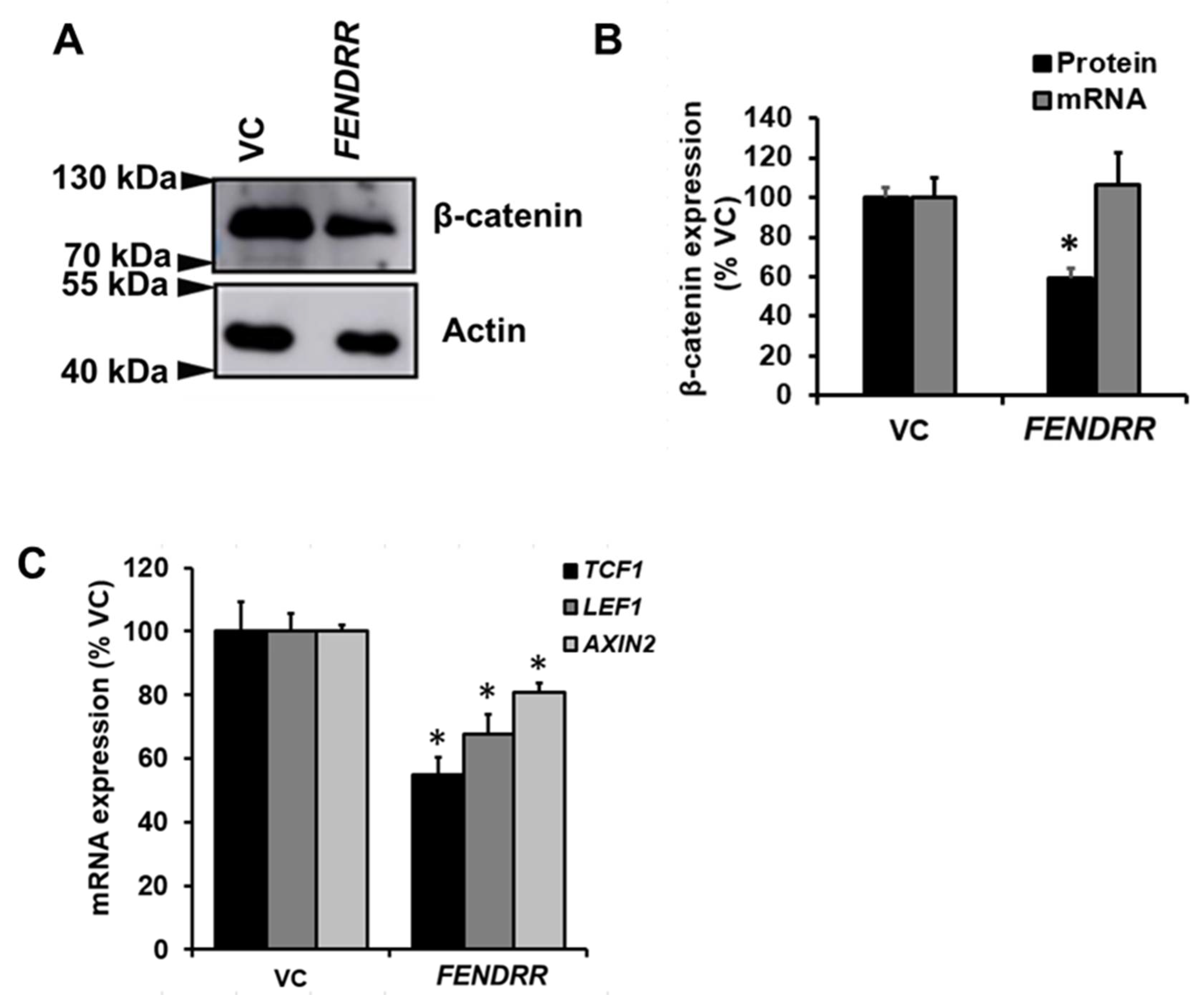
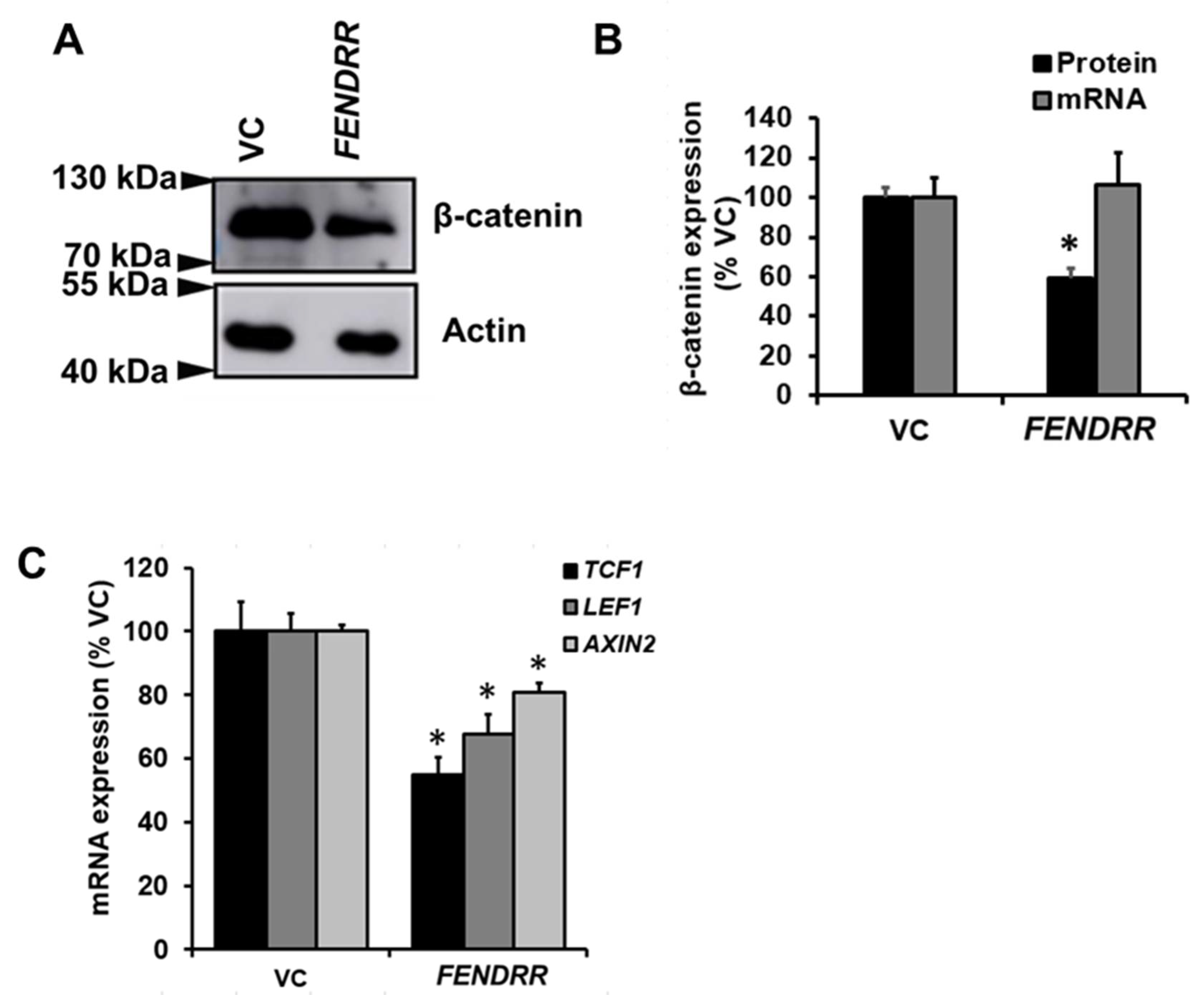
2.4. FENDRR Reduces β-Catenin Protein Level
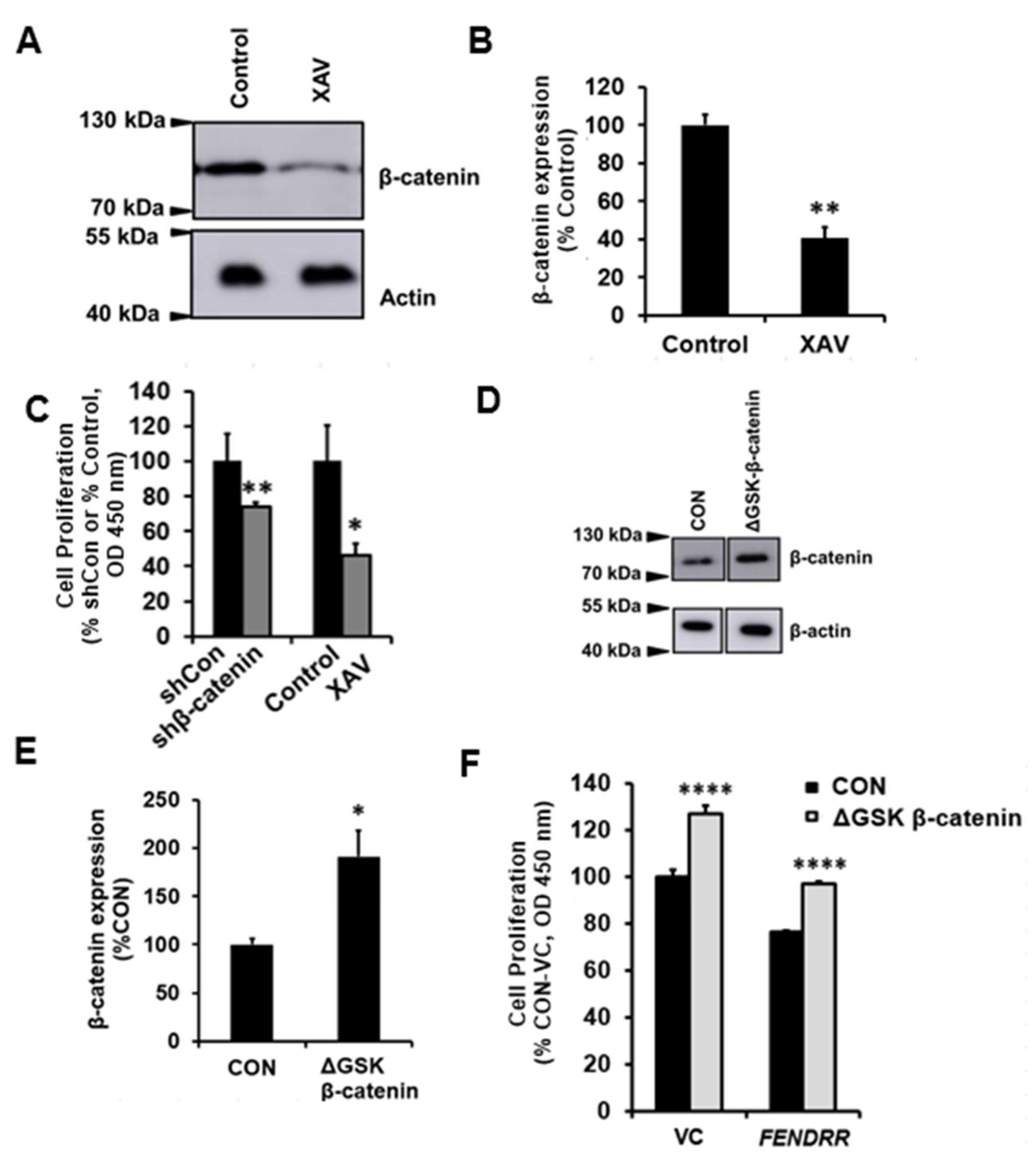
2.5. Silencing and Overexpression of β-Catenin Reduces or Increases Fibroblast Proliferation
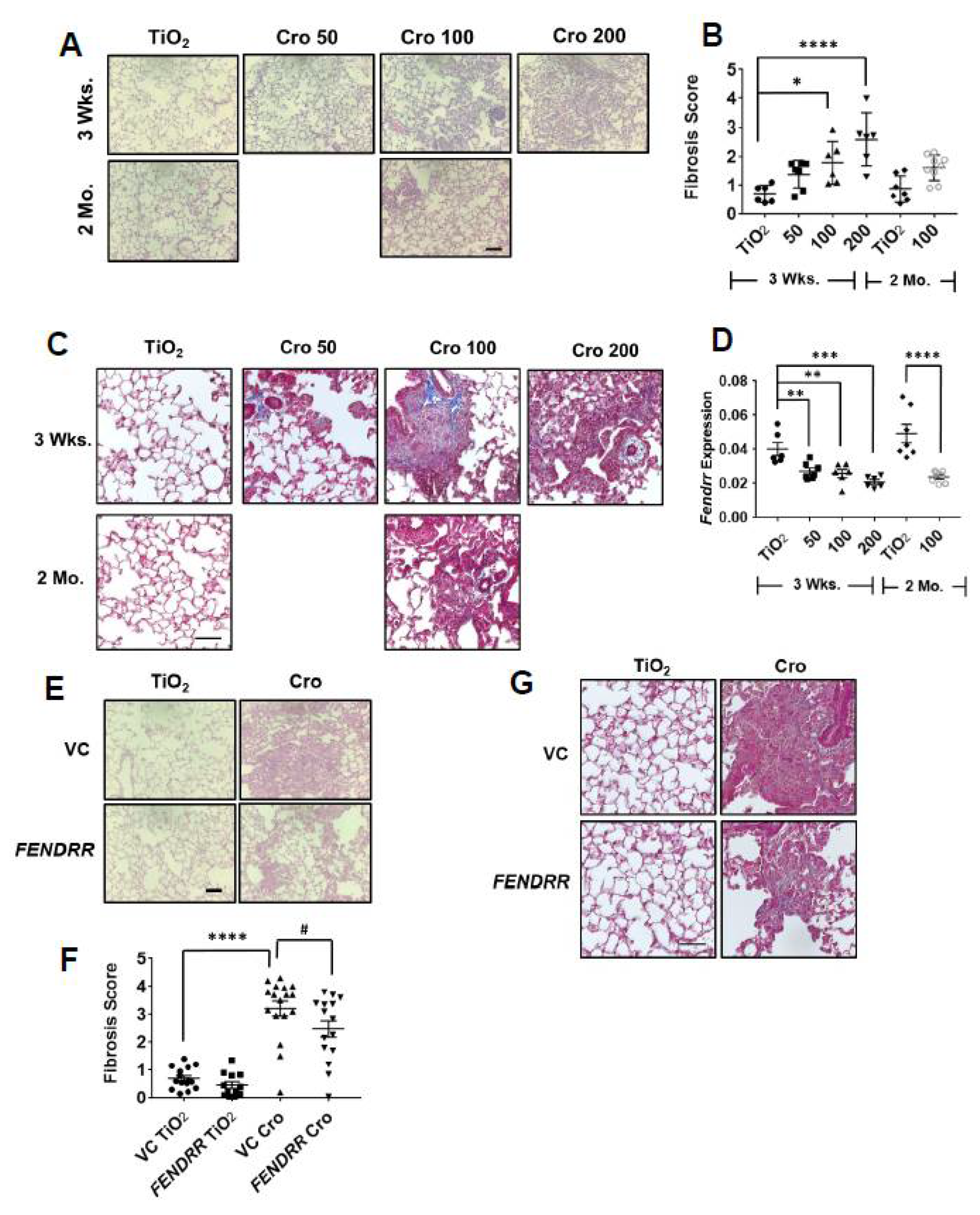
2.6. FENDRR Attenuates Asbestos-Induced Pulmonary Fibrosis in Mice
2.7. RNA Sequencing Analysis Identifies Seven Cell Proliferation-Related Genes That Are Up-Regulated by Asbestos, but Attenuated by FENDRR
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Vector Construction and Virus Preparation
4.3. Generation of Stable Cells Expressing FENDRR
4.4. RNA Isolation and Real-Time PCR
4.5. RNA Immunoprecipitation (RIP)
4.6. Western Blot
4.7. Nucleofection
4.8. Cell Proliferation Assay
4.9. A Mouse Model of Asbestos-Induced Pulmonary Fibrosis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Selman, M.; Pardov, A. Idiopathic pulmonary fibrosis: An epithelial/fibroblastic cross-talk disorder. Respir. Res. 2002, 3, 1–8. [Google Scholar] [CrossRef]
- Katzenstein, A.-L.A.; Myers, J.L. Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 1998, 157, 1301–1315. [Google Scholar] [CrossRef]
- Huang, C.; Yang, Y.; Liu, L. Interaction of long noncoding RNAs and microRNAs in the pathogenesis of idiopathic pulmonary fibrosis. Physiol. Genom. 2015, 47, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, P.; Herrmann, B.G. The long non-coding RNA Fendrr links epigenetic control mechanisms to gene regulatory networks in mammalian embryogenesis. RNA Biol. 2013, 10, 1579–1585. [Google Scholar] [CrossRef] [Green Version]
- Imam, H.; Bano, A.S.; Patel, P.; Holla, P.; Jameel, S. The lncRNA NRON modulates HIV-1 replication in a NFAT-dependent manner and is differentially regulated by early and late viral proteins. Sci. Rep. 2015, 5, 8639. [Google Scholar] [CrossRef]
- More, S.; Zhu, Z.; Lin, K.; Huang, C.; Pushparaj, S.; Liang, Y.; Sathiaseelan, R.; Yang, X.; Liu, L. Long non-coding RNA PSMB8-AS1 regulates influenza virus replication. RNA Biol. 2019, 16, 340–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzillotti, C.; De Mattei, M.; Mazziotta, C.; Taraballi, F.; Rotondo, J.C.; Tognon, M.; Martini, F. Long Non-coding RNAs and MicroRNAs Interplay in Osteogenic Differentiation of Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2021, 9, 646032. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cheng, Z.; Dai, L.; Jiang, T.; Jia, L.; Jing, X.; An, L.; Wang, H.; Liu, M. Knockdown of Long Noncoding RNA H19 Represses the Progress of Pulmonary Fibrosis through the Transforming Growth Factor β/Smad3 Pathway by Regulating MicroRNA 140. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Guo, Z.; Xie, W.; Jin, W.; Zhu, D.; Chen, S.; Ren, T. The lncRNA H19 Mediates Pulmonary Fibrosis by Regulating the miR-196a/COL1A1 Axis. Inflammation 2018, 41, 896–903. [Google Scholar] [CrossRef]
- Wan, X.; Tian, X.; Du, J.; Lu, Y.; Xiao, Y. Long non-coding RNA H19 deficiency ameliorates bleomycin-induced pulmonary inflammation and fibrosis. Respir. Res. 2020, 21, 290. [Google Scholar] [CrossRef]
- Wu, Q.; Han, L.; Yan, W.; Ji, X.; Han, R.; Yang, J.; Yuan, J.; Ni, C. miR-489 inhibits silica-induced pulmonary fibrosis by targeting MyD88 and Smad3 and is negatively regulated by lncRNA CHRF. Sci. Rep. 2016, 6, 30921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Xu, P.; Meng, C.; Song, C.; Blackwell, T.S.; Li, R.; Li, H.; Zhang, J.; Lv, C. lncITPF Promotes Pulmonary Fibrosis by Targeting hnRNP-L Depending on Its Host Gene ITGBL1. Mol. Ther. 2019, 27, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Liang, Y.; Zeng, X.; Yang, X.; Xu, D.; Gou, X.; Sathiaseelan, R.; Senavirathna, L.K.; Wang, P.; Liu, L. Long Noncoding RNA FENDRR Exhibits Antifibrotic Activity in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 440–453. [Google Scholar] [CrossRef]
- Munteanu, M.C.; Huang, C.; Liang, Y.; Sathiaseelan, R.; Zeng, X.; Liu, L. Long non-coding RNA FENDRR regulates IFNγ-induced M1 phenotype in macrophages. Sci. Rep. 2020, 10, 13672. [Google Scholar] [CrossRef]
- Zheng, H.; Krishnan, A.R.; Zou, A.E.; Ongkeko, W.M. The role of long noncoding RNA FOXF1-AS1 as a tumor suppressor in non-small cell lung cancer. Transl. Cancer Res. 2016, 5, S1440–S1442. [Google Scholar] [CrossRef]
- Herrera, A.; Cuadros, M.; Rodriguez, M.I.; Rodriguez, S.; Torres, R.; Estecio, M.; Coira, I.F.; Loidi, C.; Saiz, M.; Carmona-Saez, P. The value of lncRNA FENDRR and FOXF1 as a prognostic factor for survival of lung adenocarcinoma. Oncotarget 2017, 5, 1172–1185. [Google Scholar]
- He, W.; Zhong, G.; Wang, P.; Jiang, C.; Jiang, N.; Huang, J. Downregulation of long noncoding RNA FENDRR predicts poor prognosis in renal cell carcinoma. Oncol. Lett. 2019, 17, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Du, W. LncRNA FENDRR attenuates colon cancer progression by repression of SOX4 protein. Onco Targets Ther. 2019, 12, 4287–4295. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.P.; Huang, M.D.; Xia, R.; Liu, X.X.; Sun, M.; Yin, L.; Chen, W.M.; Han, L.; Zhang, E.B.; Kong, R.; et al. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J. Hematol. Oncol. 2014, 7, 63. [Google Scholar] [CrossRef]
- Munteanu, M.C.; Sethuraman, S.N.; Singh, M.P.; Malayer, J.; Ranjan, A. LncRNA FENDRR Expression Correlates with Tumor Immunogenicity. Genes 2021, 12, 897. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Huang, B.; Shi, Z.; Han, J.; Wang, Y.; Huangfu, J.; Wu, W. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing β-catenin biosynthesis. EMBO Mol. Med. 2013, 5, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur. Respir. Rev. 2013, 22, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.P.; Flozak, A.S.; Russell, S.; Wei, J.; Jain, M.; Mutlu, G.M.; Budinger, G.R.S.; Feghali-Bostwick, C.A.; Varga, J.; Gottardi, C.J. Nuclear β-catenin is increased in systemic sclerosis pulmonary fibrosis and promotes lung fibroblast migration and proliferation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.-S.; Liu, C.-C.; Lin, J.-H.; Hsu, T.-W.; Hsu, J.-W.; Su, K.; Hung, S.-C. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef]
- Masckauchan, T.N.; Agalliu, D.; Vorontchikhina, M.; Ahn, A.; Parmalee, N.L.; Li, C.M.; Khoo, A.; Tycko, B.; Brown, A.M.; Kitajewski, J. Wnt5a signaling induces proliferation and survival of endothelial cells in vitro and expression of MMP-1 and Tie-2. Mol. Biol. Cell 2006, 17, 5163–5172. [Google Scholar] [CrossRef]
- Yang, C.-M.; Ji, S.; Li, Y.; Fu, L.-Y.; Jiang, T.; Meng, F.-D. β-Catenin promotes cell proliferation, migration, and invasion but induces apoptosis in renal cell carcinoma. Onco Targets Ther. 2017, 10, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Sinnberg, T.; Menzel, M.; Ewerth, D.; Sauer, B.; Schwarz, M.; Schaller, M.; Garbe, C.; Schittek, B. β-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS ONE 2011, 6, e23429. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Krainer, A.R. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. 2014, 12, 1195–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michlewski, G.; Sanford, J.R.; Cáceres, J.F. The Splicing Factor SF2/ASF Regulates Translation Initiation by Enhancing Phosphorylation of 4E-BP1. Mol. Cell 2008, 30, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Twyffels, L.; Gueydan, C.; Kruys, V. Shuttling SR proteins: More than splicing factors. FEBS J. 2011, 278, 3246–3255. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Enokida, H.; Chiyomaru, T.; Tatarano, S.; Hidaka, H.; Yamasaki, T.; Gotannda, T.; Tachiwada, T.; Nohata, N.; Yamane, T.; et al. Tumor suppressive microRNA-1 mediated novel apoptosis pathways through direct inhibition of splicing factor serine/arginine-rich 9 (SRSF9/SRp30c) in bladder cancer. Biochem. Biophys. Res. Commun. 2012, 417, 588–593. [Google Scholar] [CrossRef]
- Shanmugam, R.; Zhang, F.; Srinivasan, H.; Richard, J.L.C.; Liu, K.I.; Zhang, X.; Woo, C.W.A.; Chua, Z.H.M.; Buschdorf, J.P.; Meaney, M.J.; et al. SRSF9 selectively represses ADAR2-mediated editing of brain-specific sites in primates. Nucleic Acids Res. 2018, 46, 7379–7395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Huang, J.; Higgs, B.W.; Hu, Z.; Xiao, Z.; Yao, X.; Conley, S.; Zhong, H.; Liu, Z.; Brohawn, P.; et al. Genomic Landscape Survey Identifies SRSF1 as a Key Oncodriver in Small Cell Lung Cancer. PLoS Genet. 2016, 12, e1005895. [Google Scholar] [CrossRef]
- Barth, A.I.; Stewart, D.B.; Nelson, W.J. T cell factor-activated transcription is not sufficient to induce anchorage-independent growth of epithelial cells expressing mutant beta-catenin. Proc. Natl. Acad. Sci. USA 1999, 96, 4947–4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [Green Version]
- Rangarajan, S.; Kurundkar, A.; Kurundkar, D.; Bernard, K.; Sanders, Y.Y.; Ding, Q.; Antony, V.B.; Zhang, J.; Zmijewski, J.; Thannickal, V.J. Novel Mechanisms for the Antifibrotic Action of Nintedanib. Am. J. Respir. Cell Mol. Biol. 2016, 54, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.P.; Gottardi, C.J. β-catenin signaling: A novel mediator of fibrosis and potential therapeutic target. Curr. Opin. Rheumatol. 2011, 23, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; King, T.E.; Pardo, A. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Navarro, C.; Cano, C.; Cuadros, M.; Herrera-Merchan, A.; Molina, M.; Blanco, A. A Mechanistic Study of lncRNA Fendrr Regulation of FoxF1 Lung Cancer Tumor Supressor; Springer International Publishing: Cham, Switzerland, 2016; pp. 781–789. [Google Scholar]
- Miao, L.; Huang, Z.; Zengli, Z.; Li, H.; Chen, Q.; Yao, C.; Cai, H.; Xiao, Y.; Xia, H.; Wang, Y. Loss of long noncoding RNA FOXF1-AS1 regulates epithelial-mesenchymal transition, stemness and metastasis of non-small cell lung cancer cells. Oncotarget 2016, 7, 68339–68349. [Google Scholar] [CrossRef]
- Zhu, K.-P.; Zhang, C.-L.; Ma, X.-L. Antisense lncRNA FOXF1-AS1 Promotes Migration and Invasion of Osteosarcoma Cells Through the FOXF1/MMP-2/-9 Pathway. Int. J. Biol. Sci. 2017, 13, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.; Allemand, E.; Zhang, Z.; Karni, R.; Myers, M.P.; Krainer, A.R. Arginine methylation controls the subcellular localization and functions of the oncoprotein splicing factor SF2/ASF. Mol. Cell. Biol. 2010, 30, 2762–2774. [Google Scholar] [CrossRef] [Green Version]
- Cazalla, D.; Zhu, J.; Manche, L.; Huber, E.; Krainer, A.R.; Cáceres, J.F. Nuclear export and retention signals in the RS domain of SR proteins. Mol. Cell. Biol. 2002, 22, 6871–6882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, J.R.; Gray, N.K.; Beckmann, K.; Cáceres, J.F. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004, 18, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, J.R.; Ellis, J.D.; Cazalla, D.; Cáceres, J.F. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc. Natl. Acad. Sci. USA 2005, 102, 15042–15047. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Proud, C.G. mTORC1 signaling: What we still don’t know. J. Mol. Cell Biol. 2010, 3, 206–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karni, R.; Hippo, Y.; Lowe, S.W.; Krainer, A.R. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc. Natl. Acad. Sci. USA 2008, 105, 15323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubilewicz, A.; Hecquet, C.; Jeanny, J.-C.; Soubrane, G.; Courtois, Y.; Mascarelli, F. Two distinct signalling pathways are involved in FGF2-stimulated proliferation of choriocapillary endothelial cells: A comparative study with VEGF. Oncogene 2001, 20, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Lau, M.-T.; So, W.-K.; Leung, P.C.K. Fibroblast growth factor 2 induces E-cadherin down-regulation via PI3K/Akt/mTOR and MAPK/ERK signaling in ovarian cancer cells. PLoS ONE 2013, 8, e59083. [Google Scholar] [CrossRef]
- Masckauchán, T.N.; Shawber, C.J.; Funahashi, Y.; Li, C.M.; Kitajewski, J. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis 2005, 8, 43–51. [Google Scholar] [CrossRef]
- Karimkhanloo, H.; Mohammadi-Yeganeh, S.; Hadavi, R.; Koochaki, A.; Paryan, M. Potential role of miR-214 in β-catenin gene expression within hepatocellular carcinoma. Mol. Biol. Rep. 2020, 47, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.; Li, F.; Lin, Y.; Zhang, X.; Lv, Z.; Jiang, J. MiR-214 inhibits cell growth in hepatocellular carcinoma through suppression of β-catenin. Biochem. Biophys. Res. Commun. 2012, 428, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ooi, L.L.P.J.; Hui, K.M. MiR-214 Targets β-Catenin Pathway to Suppress Invasion, Stem-Like Traits and Recurrence of Human Hepatocellular Carcinoma. PLoS ONE 2012, 7, e44206. [Google Scholar] [CrossRef]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal models of fibrotic lung disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, D.W. Asbestos-induced lung diseases: An update. Transl. Res. J. Lab. Clin. Med. 2009, 153, 143–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, D.W.; Weitzman, S.A. The molecular basis of asbestos induced lung injury. Thorax 1999, 54, 638. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.; Brody, A.R. Increased TGF-beta1 in the lungs of asbestos-exposed rats and mice: Reduced expression in TNF-alpha receptor knockout mice. J. Environ. Pathol. Toxicol. Oncol. 2001, 20, 97–108. [Google Scholar] [CrossRef]
- Senavirathna, L.K.; Huang, C.; Pushparaj, S.; Xu, D.; Liu, L. Hypoxia and transforming growth factor β1 regulation of long non-coding RNA transcriptomes in human pulmonary fibroblasts. Physiol. Rep. 2020, 8, e14343. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Meininger, C.J.; Kelly, K.A.; Hawker, J.R., Jr.; Morris, S.M., Jr.; Wu, G. Activities of arginase I and II are limiting for endothelial cell proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R64–R69. [Google Scholar] [CrossRef]
- Heby, O. Role of polyamines in the control of cell proliferation and differentiation. Differentiation 1981, 19, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Torres-González, E.; Rojas, M.; Corredor, C.; Ritzenthaler, J.; Xu, J.; Roman, J.; Brigham, K.; Stecenko, A. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 35, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Haston, C.K.; Tomko, T.G.; Godin, N.; Kerckhoff, L.; Hallett, M.T. Murine candidate bleomycin induced pulmonary fibrosis susceptibility genes identified by gene expression and sequence analysis of linkage regions. J. Med. Genet. 2005, 42, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Jin, Y.Y.; Tang, Y.L.; Yang, H.J.; Zhou, X.Q.; Lei, Z. GPNMB silencing suppresses the proliferation and metastasis of osteosarcoma cells by blocking the PI3K/Akt/mTOR signaling pathway. Oncol. Rep. 2018, 39, 3034–3040. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Wang, L.F.; Chien, Y.W. Nitric oxide in the pathogenesis of diffuse pulmonary fibrosis. Free Radic. Biol. Med. 2007, 42, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Liu, X.; Liang, J.; Habiel, D.M.; Kulur, V.; Coelho, A.L.; Deng, N.; Xie, T.; Wang, Y.; Liu, N.; et al. PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 2018, 10, eaar8356. [Google Scholar] [CrossRef] [Green Version]
- Jian, W.; Zhang, X.; Wang, J.; Liu, Y.; Hu, C.; Wang, X.; Liu, R. Scinderin-knockdown inhibits proliferation and promotes apoptosis in human breast carcinoma cells. Oncol. Lett. 2018, 16, 3207–3214. [Google Scholar] [CrossRef]
- Wang, D.; Sun, S.Q.; Yu, Y.H.; Wu, W.Z.; Yang, S.L.; Tan, J.M. Suppression of SCIN inhibits human prostate cancer cell proliferation and induces G0/G1 phase arrest. Int. J. Oncol. 2014, 44, 161–166. [Google Scholar] [CrossRef]
- Liu, H.; Shi, D.; Liu, T.; Yu, Z.; Zhou, C. Lentivirus-mediated silencing of SCIN inhibits proliferation of human lung carcinoma cells. Gene 2015, 554, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Si, D.; Zhang, R.; Liu, J.; Gou, H.; Xia, Y.; Tian, D.; Dai, J.; Yang, K.; Liu, E.; et al. Deletion of SMARCA4 impairs alveolar epithelial type II cells proliferation and aggravates pulmonary fibrosis in mice. Genes Dis. 2017, 4, 204–214. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Königshoff, M.; Eickelberg, O. WNT signaling in lung disease: A failure or a regeneration signal? Am. J. Respir. Cell Mol. Biol. 2010, 42, 21–31. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; Kaminski, N. Idiopathic Pulmonary Fibrosis: Aberrant Recapitulation of Developmental Programs? PLoS Med. 2008, 5, e62. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Xiao, X.; Yang, Y.; Mishra, A.; Liang, Y.; Zeng, X.; Yang, X.; Xu, D.; Blackburn, M.R.; Henke, C.A.; et al. MicroRNA-101 attenuates pulmonary fibrosis by inhibiting fibroblast proliferation and activation. J. Biol. Chem. 2017, 292, 16420–16439, Erratum in 2019, 294, 6694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senavirathna, L.K.; Huang, C.; Yang, X.; Munteanu, M.C.; Sathiaseelan, R.; Xu, D.; Henke, C.A.; Liu, L. Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci. Rep. 2018, 8, 2709. [Google Scholar] [CrossRef] [PubMed]
- Gou, D.; Weng, T.; Wang, Y.; Wang, Z.; Zhang, H.; Gao, L.; Chen, Z.; Wang, P.; Liu, L. A novel approach for the construction of multiple shRNA expression vectors. J. Gene Med. 2007, 9, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, C.; Chintagari, N.R.; Bhaskaran, M.; Weng, T.; Guo, Y.; Xiao, X.; Liu, L. miR-375 regulates rat alveolar epithelial cell trans-differentiation by inhibiting Wnt/beta-catenin pathway. Nucleic Acids Res. 2013, 41, 3833–3844. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Senavirathna, L.K.; Gou, X.; Huang, C.; Liang, Y.; Liu, L. EZH2 enhances the differentiation of fibroblasts into myofibroblasts in idiopathic pulmonary fibrosis. Physiol. Rep. 2016, 4, e12915. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Cheresh, P.; Morales-Nebreda, L.; Kim, S.J.; Yeldandi, A.; Williams, D.B.; Cheng, Y.; Mutlu, G.M.; Budinger, G.R.; Ridge, K.; Schumacker, P.T.; et al. Asbestos-induced pulmonary fibrosis is augmented in 8-oxoguanine DNA glycosylase knockout mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 25–36. [Google Scholar] [CrossRef]
- Hübner, R.H.; Gitter, W.; El Mokhtari, N.E.; Mathiak, M.; Both, M.; Bolte, H.; Freitag-Wolf, S.; Bewig, B. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques 2008, 44, 507–517. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senavirathna, L.K.; Liang, Y.; Huang, C.; Yang, X.; Bamunuarachchi, G.; Xu, D.; Dang, Q.; Sivasami, P.; Vaddadi, K.; Munteanu, M.C.; et al. Long Noncoding RNA FENDRR Inhibits Lung Fibroblast Proliferation via a Reduction of β-Catenin. Int. J. Mol. Sci. 2021, 22, 8536. https://doi.org/10.3390/ijms22168536
Senavirathna LK, Liang Y, Huang C, Yang X, Bamunuarachchi G, Xu D, Dang Q, Sivasami P, Vaddadi K, Munteanu MC, et al. Long Noncoding RNA FENDRR Inhibits Lung Fibroblast Proliferation via a Reduction of β-Catenin. International Journal of Molecular Sciences. 2021; 22(16):8536. https://doi.org/10.3390/ijms22168536
Chicago/Turabian StyleSenavirathna, Lakmini Kumari, Yurong Liang, Chaoqun Huang, Xiaoyun Yang, Gayan Bamunuarachchi, Dao Xu, Quanjin Dang, Pulavendran Sivasami, Kishore Vaddadi, Maria Cristina Munteanu, and et al. 2021. "Long Noncoding RNA FENDRR Inhibits Lung Fibroblast Proliferation via a Reduction of β-Catenin" International Journal of Molecular Sciences 22, no. 16: 8536. https://doi.org/10.3390/ijms22168536
APA StyleSenavirathna, L. K., Liang, Y., Huang, C., Yang, X., Bamunuarachchi, G., Xu, D., Dang, Q., Sivasami, P., Vaddadi, K., Munteanu, M. C., Hewawasam, S., Cheresh, P., Kamp, D. W., & Liu, L. (2021). Long Noncoding RNA FENDRR Inhibits Lung Fibroblast Proliferation via a Reduction of β-Catenin. International Journal of Molecular Sciences, 22(16), 8536. https://doi.org/10.3390/ijms22168536