
GATNNCDA: A Method Based on Graph Attention Network and Multi-Layer Neural Network for Predicting circRNA-Disease Associations


Abstract
:1. Introduction
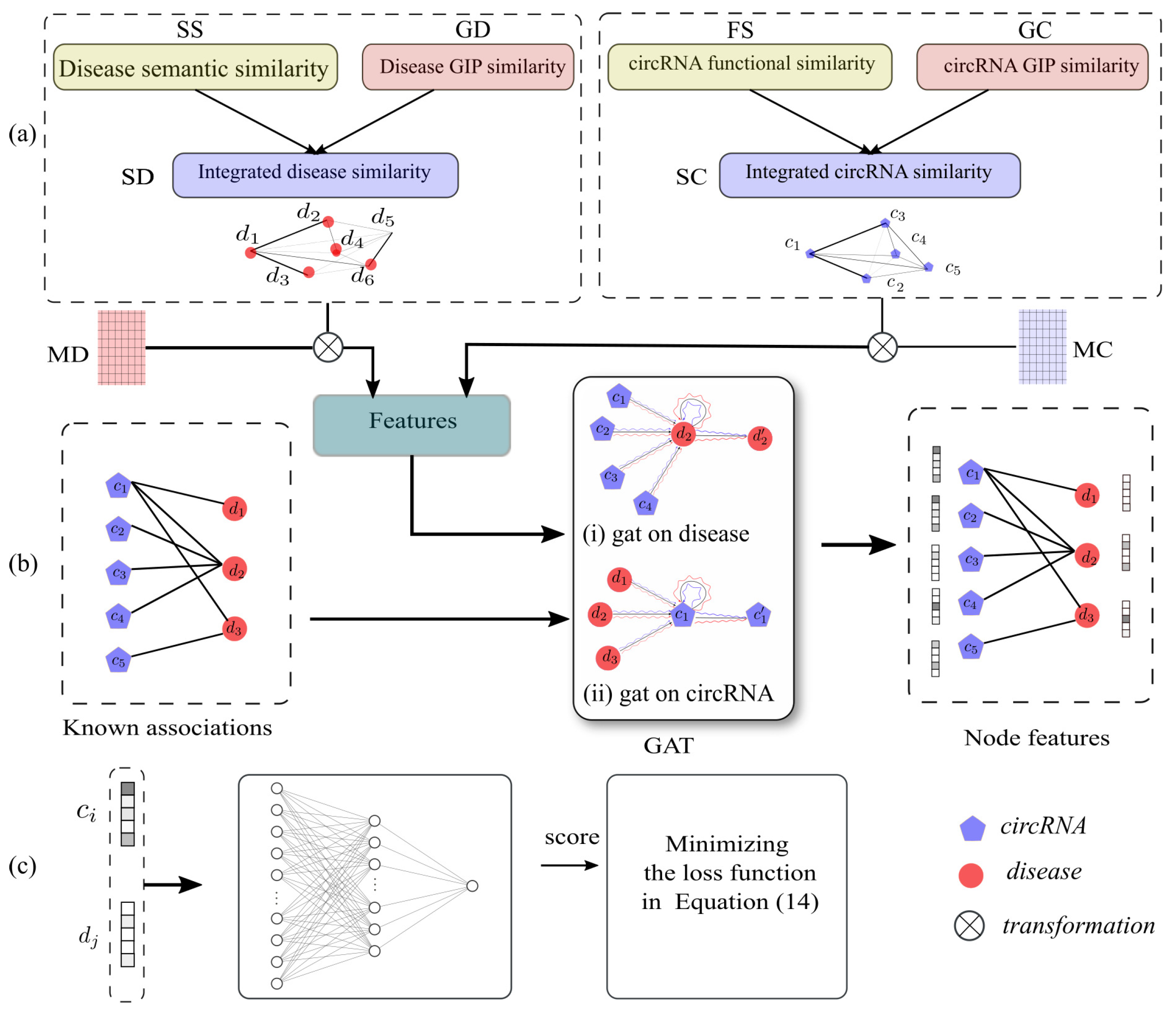
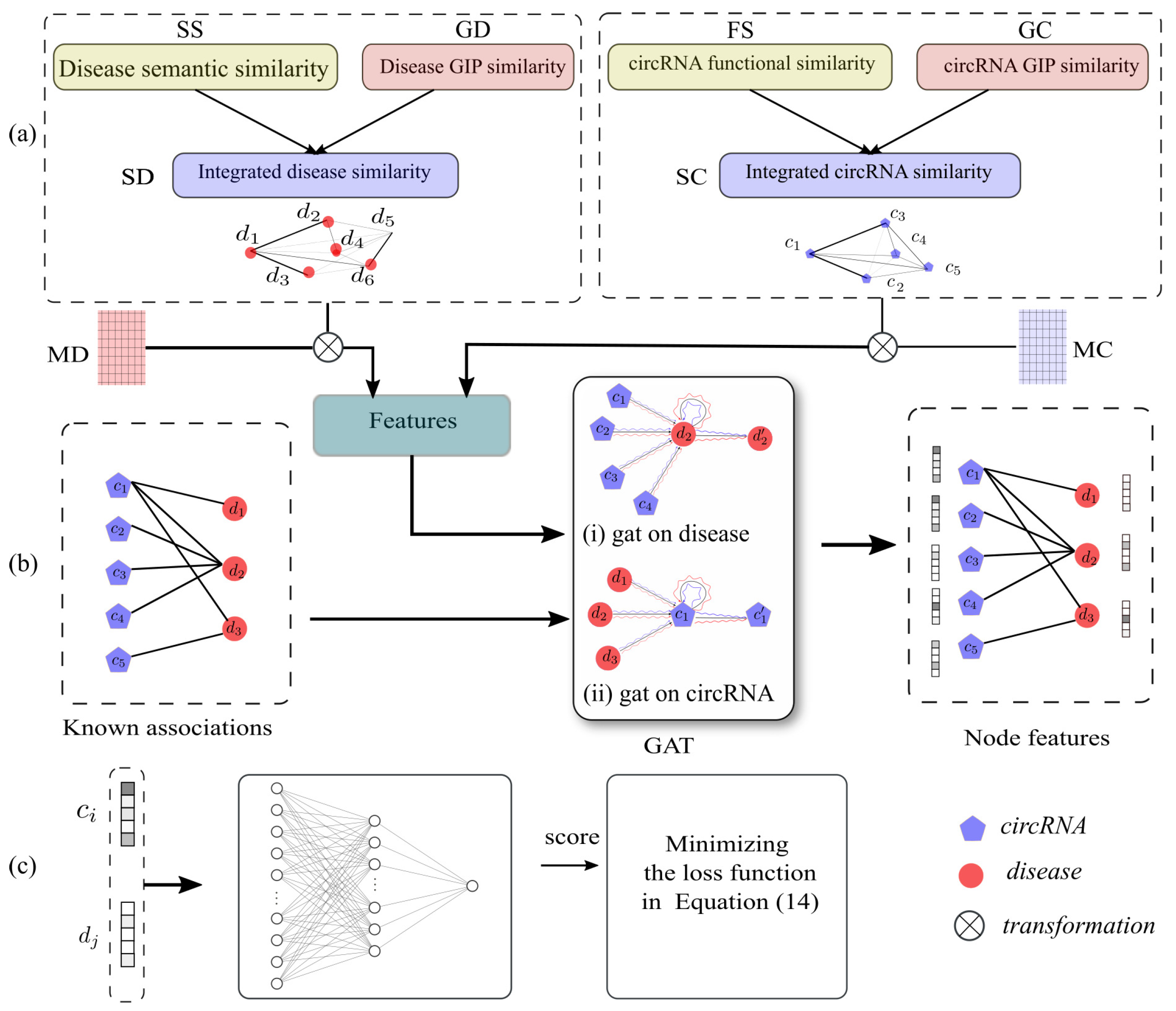
- We proposed an end-to-end framework for inferring disease-related circRNAs, which can effectively and accurately infer the potential associations between circRNAs and diseases.
- We made use of GAT to extract low-dimensional dense representations of circRNAs and diseases, and these presentations had rich structural and semantic information of the heterogeneous circRNA–disease graph.
- We proposed a NN-based classifier, and applied a sampling strategy to construct balanced samples. In addition, we designed cross-entropy loss with L2 regularization to make the training process fast and robust.
- We demonstrated the predictive performance of our method by extensive experiments via fivefold cross validation and case studies, and achieved competitive results on CircR2Disease and circRNADisease datasets.
2. Results and Discussion
2.1. Experiments Settings
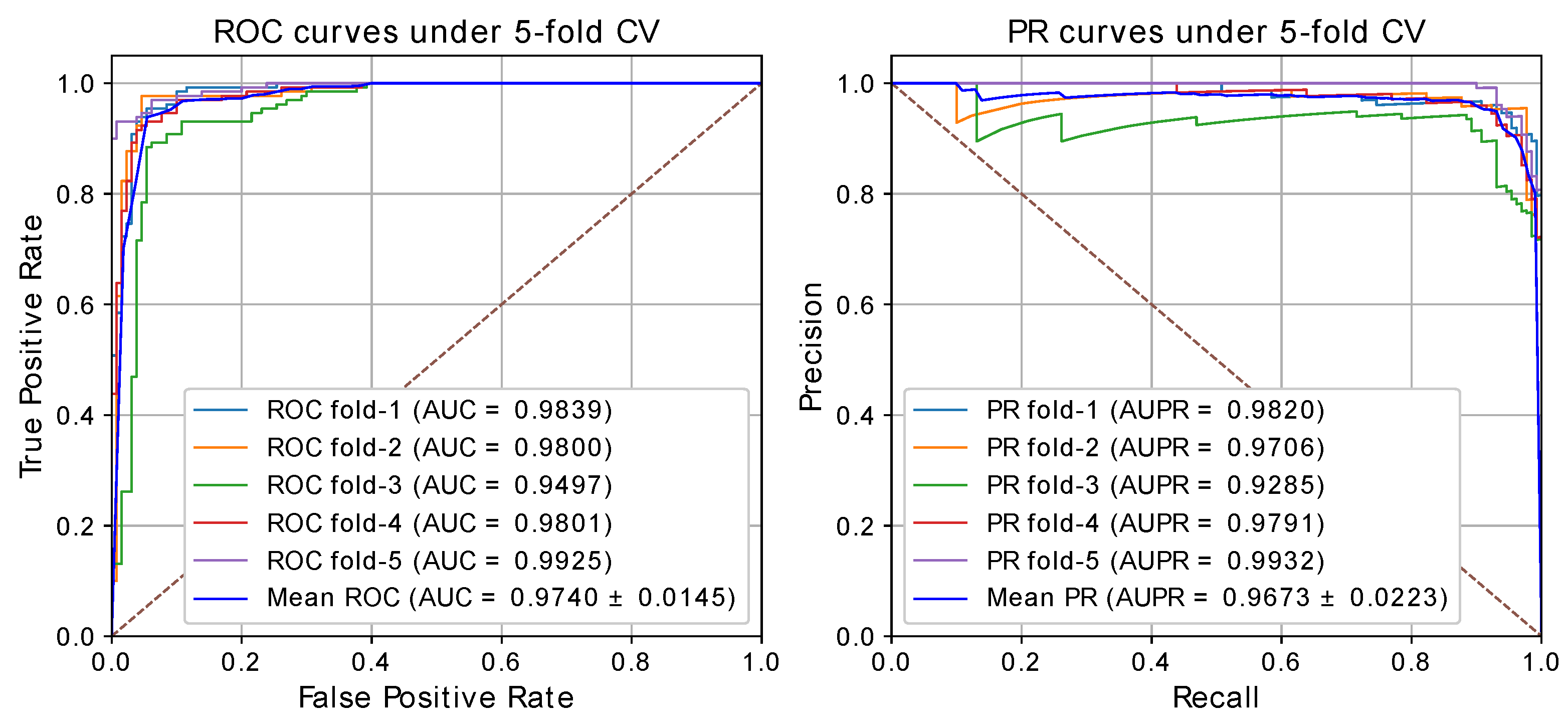
2.2. Performance Analysis
2.3. Comparison with Other Methods
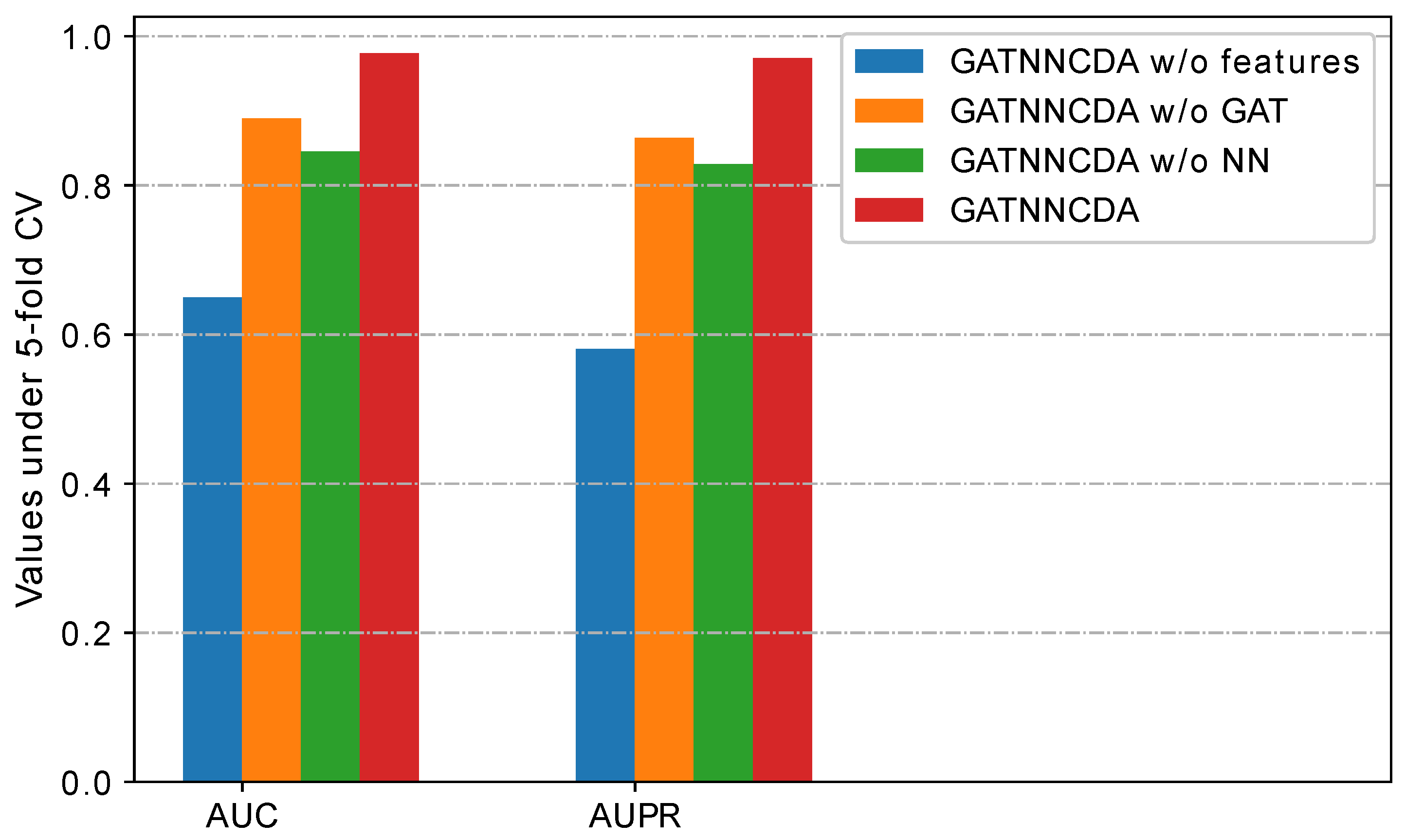
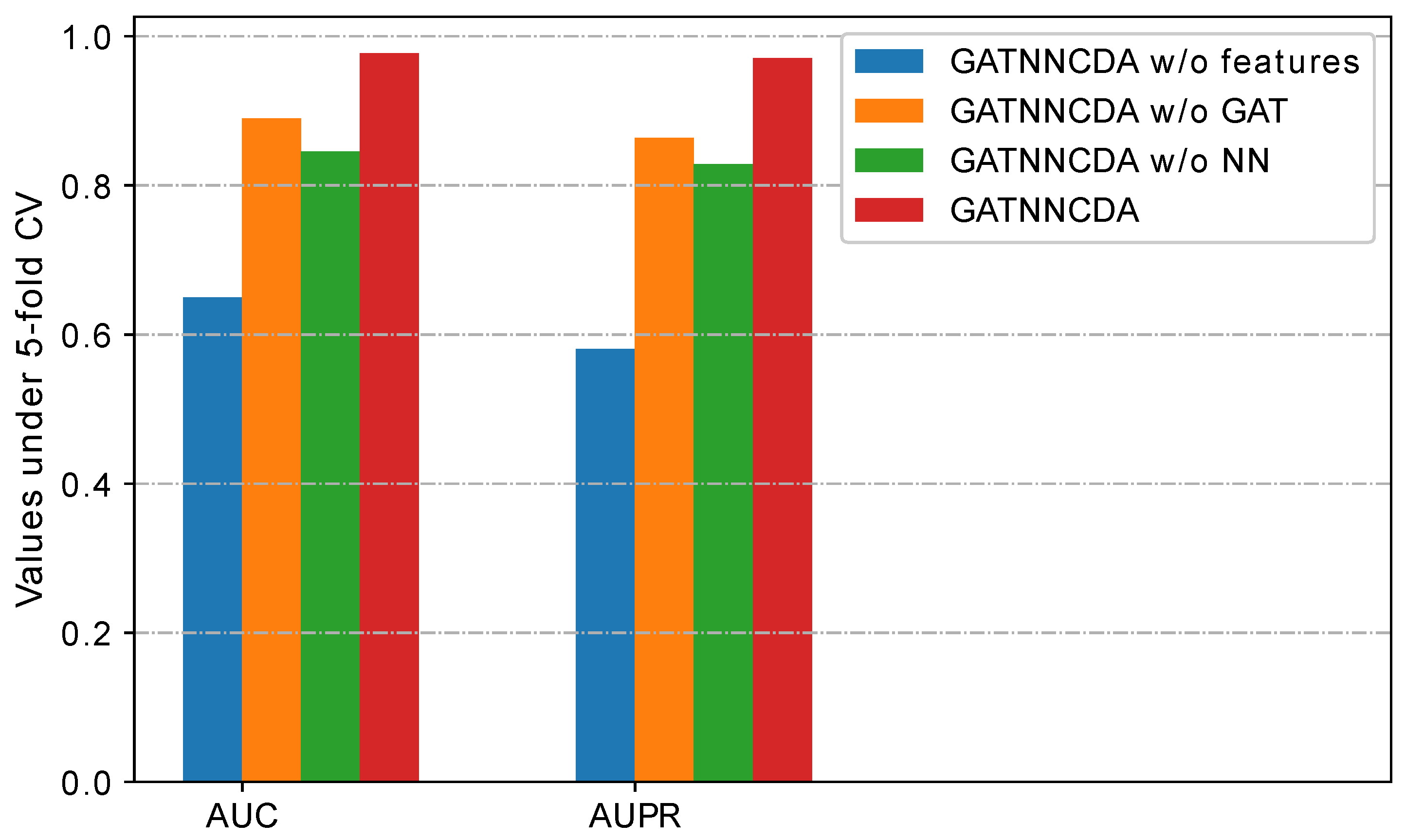
2.4. Ablation Study
- GATNNCDA w/o features: It uses randomly initialized and as initial node features, instead of integrated similarities.
- GATNNCDA w/o GAT: It removes the GAT from GATNNCDA, and uses the integrated similarities as features and a two-layer NN as a predictor.
- GATNNCDA w/o NN: It uses dot production to calculate the prediction score, instead of a two-layer NN as a predictor.
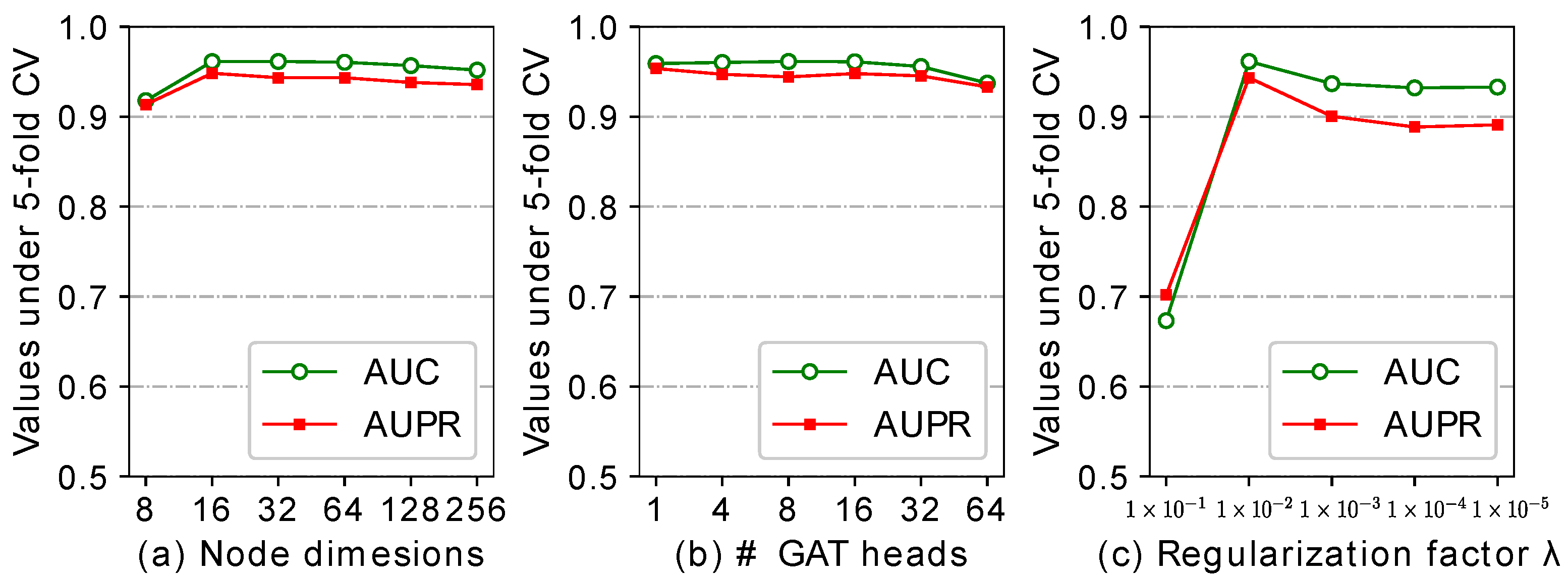
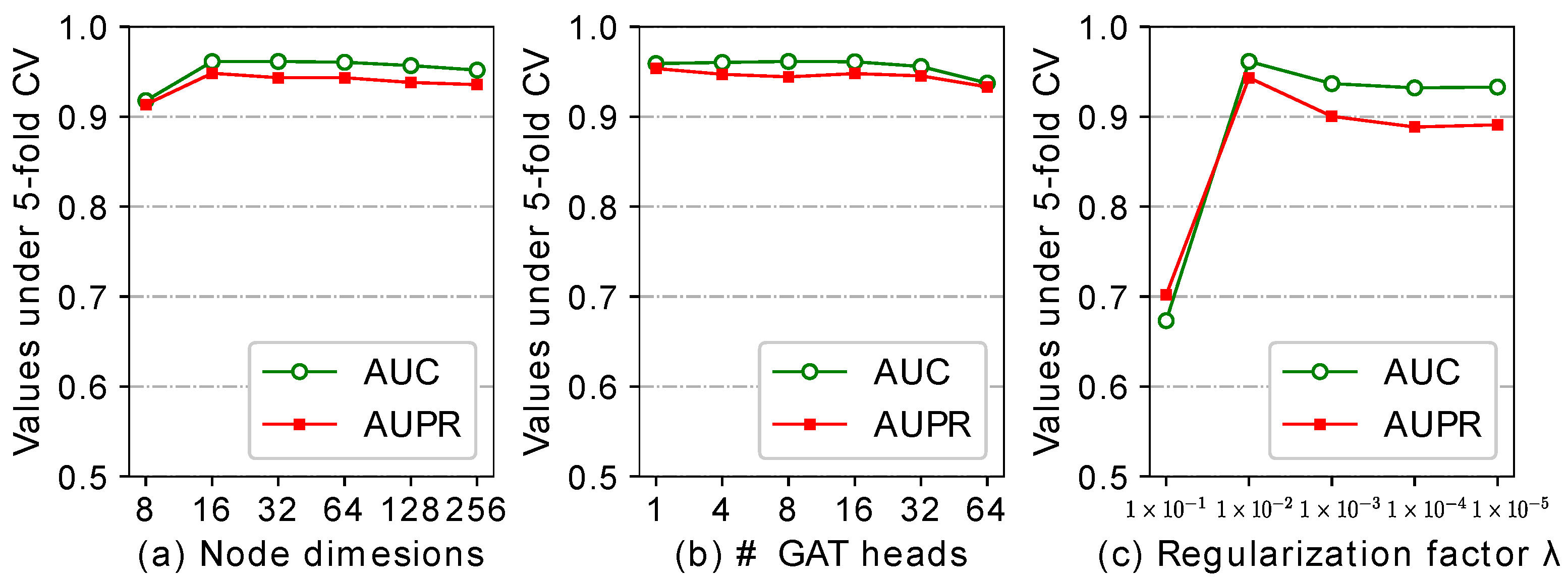
2.5. Effect of the Parameters
2.6. Case Studies
3. Materials and Methods
3.1. Known circRNA-Disease Associations
3.2. Disease Semantic Similarity
3.3. circRNA Functional Similarity
3.4. Gaussian Interaction Profile Kernel Similarity for Disease
3.5. Integrated Similarities for circRNA and Disease
3.6. Feature Extraction Based on Graph Attention Network
3.7. circRNA-Disease Association Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Meng, S.; Zhou, H.; Feng, Z.; Xu, Z.; Tang, Y.; Li, P.; Wu, M. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol. Cancer 2017, 16, 1–8. [Google Scholar] [CrossRef]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single stranded covalently closed circular RNA molecules existing as highly base paired rod like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [Green Version]
- Coca-Prados, M.; Hsu, M.T. Electron microscopic evidence for circular form of RNA in the cytoplasm of eukaryotic cells. Nature 1979, 280, 339–340. [Google Scholar]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L.; et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; Dan, R.; et al. The landscape of circular RNA in cancer. Cell 2020, 176, 869–881.e13. [Google Scholar] [CrossRef] [Green Version]
- Zeng, K.; Chen, X.; Xu, M.; Liu, X.; Hu, X.; Xu, T.; Sun, H.; Pan, Y.; He, B.; Wang, S. CircHIPK3 promotes colorectal cancer growth and metastasis by sponging miR-7 article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Chen, S.; Li, T.; Zhao, Q.; Xiao, B.; Guo, J. Using circular RNA hsa_circ_0000190 as a new biomarker in the diagnosis of gastric cancer. Clin. Chim. Acta 2017, 466, 167–171. [Google Scholar] [CrossRef]
- Shang, X.; Li, G.; Liu, H.; Li, T.; Liu, J.; Zhao, Q.; Wang, C. Comprehensive circular RNA profiling reveals that hsa-circ-0005075, a new circular RNA biomarker, is involved in hepatocellular crcinoma development. Medicine 2016, 95, e3811. [Google Scholar] [CrossRef]
- Qin, M.; Liu, G.; Huo, X.; Tao, X.; Sun, X.; Ge, Z.; Yang, J.; Fan, J.; Liu, L.; Qin, W. Hsa-circ-0001649: A circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark. 2016, 16, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Lukiw, W.J. Circular RNA (circRNA) in Alzheimer’s disease (AD). Front. Genet. 2013, 4, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fan, H.; Sun, J.; Ni, M.; Zhang, L.; Chen, C.; Hong, X.; Fang, F.; Zhang, W.; Ma, P. Circular RNA expression profile of Alzheimer’s disease and its clinical significance as biomarkers for the disease risk and progression. Int. J. Biochem. Cell Biol. 2020, 123, 105747. [Google Scholar] [CrossRef]
- Chen, L.L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. CircBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, S.; Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A database for functional circular RNAs. Database 2019, 2019, baz003. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Lei, X.; Fang, Z.; Jiang, Q.; Wu, F.X. CircR2Disease: A manually curated database for experimentally supported circular RNAs associated with various diseases. Database 2018, 2018, bay044. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Zhang, L.; Zheng, M.; Sun, X.; Lu, Y.; Liu, P. Circ2Disease: A manually curated database of experimentally validated circRNAs in human disease. Sci. Rep. 2018, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Lei, X.; Fang, Z.; Chen, L.; Wu, F.X. Pwcda: Path weighted method for predicting circrna-disease associations. Int. J. Mol. Sci. 2018, 19, 3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Wang, J.; Wu, F.X. DWNN-RLS: Regularized least squares method for predicting circRNA–disease associations. BMC Bioinform. 2018, 19, 520. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Lei, X.; Wu, F.X. Prediction of circRNA–disease associations using KATZ model based on heterogeneous networks. Int. J. Biol. Sci. 2018, 14, 1950–1959. [Google Scholar] [CrossRef]
- Xiao, Q.; Luo, J.; Dai, J. Computational Prediction of Human Disease- Associated circRNAs Based on Manifold Regularization Learning Framework. IEEE J. Biomed. Health Inform. 2019, 23, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Deepthi, K.; Jereesh, A.S. Inferring Potential circRNA–disease Associations via Deep Autoencoder-Based Classification. Mol. Diagn. Ther. 2021, 25, 87–97. [Google Scholar] [CrossRef]
- Li, G.; Luo, J.; Wang, D.; Liang, C.; Xiao, Q.; Ding, P.; Chen, H. Potential circRNA–disease association prediction using DeepWalk and network consistency projection. J. Biomed. Inform. 2020, 112, 103624. [Google Scholar] [CrossRef]
- Wang, L.; You, Z.H.; Li, Y.M.; Zheng, K.; Huang, Y.A. GCNCDA: A new method for predicting circRNA–disease associations based on Graph Convolutional Network Algorithm. PLoS Comput. Biol. 2020, 16, e7568. [Google Scholar] [CrossRef]
- Bian, C.; Lei, X.J.; Wu, F.X. GATCDA: Predicting circRNA–disease associations based on graph attention network. Cancers 2021, 13, 2595. [Google Scholar] [CrossRef]
- Lei, X.; Fang, Z.; Guo, L. Predicting circRNA–disease associations based on improved collaboration filtering recommendation system with multiple data. Front. Genet. 2019, 10, 897. [Google Scholar] [CrossRef]
- Le, N.Q.; Do, D.T.; Hung, T.N.; Lam, L.H.; Huynh, T.T.; Nguyen, N.T. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int. J. Mol. Sci. 2020, 21, 9070. [Google Scholar] [CrossRef]
- Ho Thanh Lam, L.; Le, N.H.; Van Tuan, L.; Tran Ban, H.; Nguyen Khanh Hung, T.; Nguyen, N.T.; Huu Dang, L.; Le, N.Q. Machine Learning Model for Identifying Antioxidant Proteins Using Features Calculated from Primary Sequences. Biology 2020, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An imperative style, high-performance deep learning library. arXiv 2019, arXiv:1912.01703. [Google Scholar]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with pytorch geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Kingma, D.P.; Ba, J.L. Adam: A method for stochastic optimization. In Proceedings of the 3rd International Conference on Learning Representations, ICLR 2015—Conference Track Proceedings, Diego, CA, USA, 7–9 May 2015. [Google Scholar]
- Li, G.; Yue, Y.; Liang, C.; Xiao, Q.; Ding, P.; Luo, J. NCPCDA: Network consistency projection for circRNA–disease association prediction. RSC Adv. 2019, 9, 33222–33228. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; You, Z.H.; Huang, Y.A.; Huang, D.S.; Chan, K.C. An efficient approach based on multi-sources information to predict circRNA–disease associations using deep convolutional neural network. Bioinformatics 2020, 36, 4038–4046. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, B. iCircDA-MF: Identification of circRNA–disease associations based on matrix factorization. Briefings Bioinform. 2019, 21, 1356–1367. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.H.; Liu, H. BNPMDA: Bipartite network projection for MiRNA–Disease association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. In Proceedings of the 5th International Conference on Learning Representations, ICLR 2017—Conference Track Proceedings, Toulon, France, 24–26 April 2017; pp. 1–14. [Google Scholar]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [Green Version]
- Velicković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Fold | Accuracy | Precision | Recall | F1-Score | AUC | AUPR |
|---|---|---|---|---|---|---|
| 1 | 0.9346 | 0.9821 | 0.9692 | 0.9368 | 0.9839 | 0.9820 |
| 2 | 0.9346 | 0.9720 | 0.9769 | 0.9373 | 0.9800 | 0.9706 |
| 3 | 0.9077 | 0.9305 | 0.9308 | 0.9098 | 0.9497 | 0.9285 |
| 4 | 0.9308 | 0.9793 | 0.9692 | 0.9333 | 0.9801 | 0.9791 |
| 5 | 0.9500 | 0.9933 | 0.9615 | 0.9506 | 0.9925 | 0.9932 |
| Average | 0.9315 | 0.9714 | 0.9615 | 0.9336 | 0.9742 | 0.9707 |
| Test Fold | Accuracy | Precision | Recall | F1-Score | AUC | AUPR |
|---|---|---|---|---|---|---|
| 1 | 0.9478 | 0.9799 | 1.0000 | 0.9504 | 0.9826 | 0.9794 |
| 2 | 0.9627 | 0.9944 | 0.9701 | 0.9630 | 0.9938 | 0.9943 |
| 3 | 0.9776 | 0.9719 | 1.0000 | 0.9781 | 0.9831 | 0.9703 |
| 4 | 0.9776 | 0.9879 | 0.9851 | 0.9778 | 0.9895 | 0.9877 |
| 5 | 0.9531 | 0.9921 | 1.0000 | 0.9552 | 0.9922 | 0.9920 |
| Average | 0.9638 | 0.9852 | 0.9910 | 0.9649 | 0.9882 | 0.9848 |
| Models | AUC |
|---|---|
| DWNN-RLS [26] | 0.8854 |
| PWCDA [25] | 0.8900 |
| KATZHCDA [27] | 0.7936 |
| NCPCDA [39] | 0.9201 |
| AE-RF [29] | 0.9486 |
| Wang’s method [40] | 0.8667 |
| iCircDA-MF [41] | 0.9178 |
| GCNCDA [31] | 0.9090 |
| GATNNCDA [42] | 0.9011 |
| GATNNCDA-best | 0.9742 |
| GATNNCDA-average | 0.9613 |
| Rank | circRNA | Evidence | Rank | circRNA | Evidence |
|---|---|---|---|---|---|
| 1 | hsa_circ_0007534 | II | 11 | hsa_circ_0068033 | I; II |
| 2 | hsa_circ_0011946 | II | 12 | circamotl1hsa_circ_0004214 | I; II |
| 3 | hsa_circ_0093859 | II | 13 | hsa_circ_0006528 | I; II |
| 4 | circrna-000911 | II | 14 | hsa_circ_0002874 | I; II> |
| 5 | circrna-001283 | PMID:29431182 | 15 | hsa_circ_0001667 | I; II |
| 6 | circrna-001175 | II | 16 | hsa_circ_0085495 | I; II |
| 7 | circrna-100438 | PMID:29431182 | 17 | hsa_circ_0086241 | I; II |
| 8 | hsa_circ_0001982 | I; II | 18 | hsa_circ_0092276 | I; II |
| 9 | hsa_circ_0001785 | I | 19 | hsa_circ_0003838 | I; II |
| 10 | hsa_circ_0108942 | I; II | 20 | circvrk1 | I; II |
| Rank | circRNA | Evidence |
|---|---|---|
| 1 | circc3p1 | II |
| 2 | hsa_circ_0067531 | II |
| 3 | circarsp91hsa_circ_0085154 | II |
| 4 | circmto1hsa_circrna_0007874hsa_circrna_104135 | II |
| 5 | hsa_circ_0005986 | I; II |
| 6 | hsa_circrna_100338circsnx27 | PMID:28710406 |
| 7 | hsa_circrna_104075 | I; II |
| 8 | hsa_circrna_102049 | PMID:28710406 |
| 9 | circrna_000839 | II |
| 10 | circzkscan1hsa_circ_0001727 | I; II |
| 11 | hsa_circ_0004018 | I; II |
| 12 | hsa_circ_0005075 | II |
| 13 | hsa_circrna_100571 | PMID: 29609527 |
| 14 | hsa_circrna_400031 | PMID:29609527 |
| 15 | hsa_circrna_102032 | PMID: 29609527 |
| 16 | hsa_circrna_103096 | PMID:29609527 |
| 17 | hsa_circrna_102347 | PMID:29609527 |
| 18 | hsa_circrna_000167hsa_circ_0000518 | unknown |
| 19 | hsa_circ_0000520 | PMID:27258521 |
| 20 | hsa_circ_0000172 | unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, C.; Liu, Z.; Wang, Y.; Ni, J.; Zheng, C. GATNNCDA: A Method Based on Graph Attention Network and Multi-Layer Neural Network for Predicting circRNA-Disease Associations. Int. J. Mol. Sci. 2021, 22, 8505. https://doi.org/10.3390/ijms22168505
Ji C, Liu Z, Wang Y, Ni J, Zheng C. GATNNCDA: A Method Based on Graph Attention Network and Multi-Layer Neural Network for Predicting circRNA-Disease Associations. International Journal of Molecular Sciences. 2021; 22(16):8505. https://doi.org/10.3390/ijms22168505
Chicago/Turabian StyleJi, Cunmei, Zhihao Liu, Yutian Wang, Jiancheng Ni, and Chunhou Zheng. 2021. "GATNNCDA: A Method Based on Graph Attention Network and Multi-Layer Neural Network for Predicting circRNA-Disease Associations" International Journal of Molecular Sciences 22, no. 16: 8505. https://doi.org/10.3390/ijms22168505
APA StyleJi, C., Liu, Z., Wang, Y., Ni, J., & Zheng, C. (2021). GATNNCDA: A Method Based on Graph Attention Network and Multi-Layer Neural Network for Predicting circRNA-Disease Associations. International Journal of Molecular Sciences, 22(16), 8505. https://doi.org/10.3390/ijms22168505