
Different Patterns of mRNA Nuclear Retention during Meiotic Prophase in Larch Microsporocytes

 , , ,
, , ,  and
and
Abstract
1. Introduction
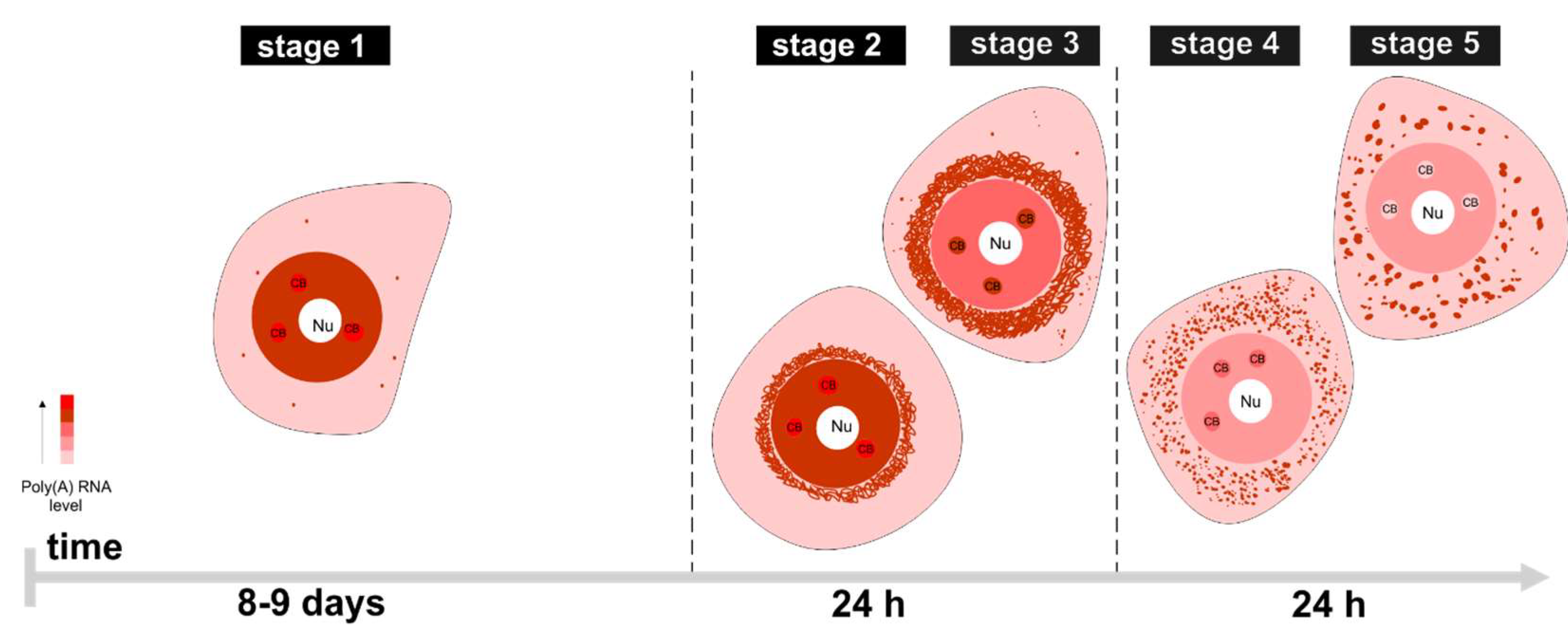
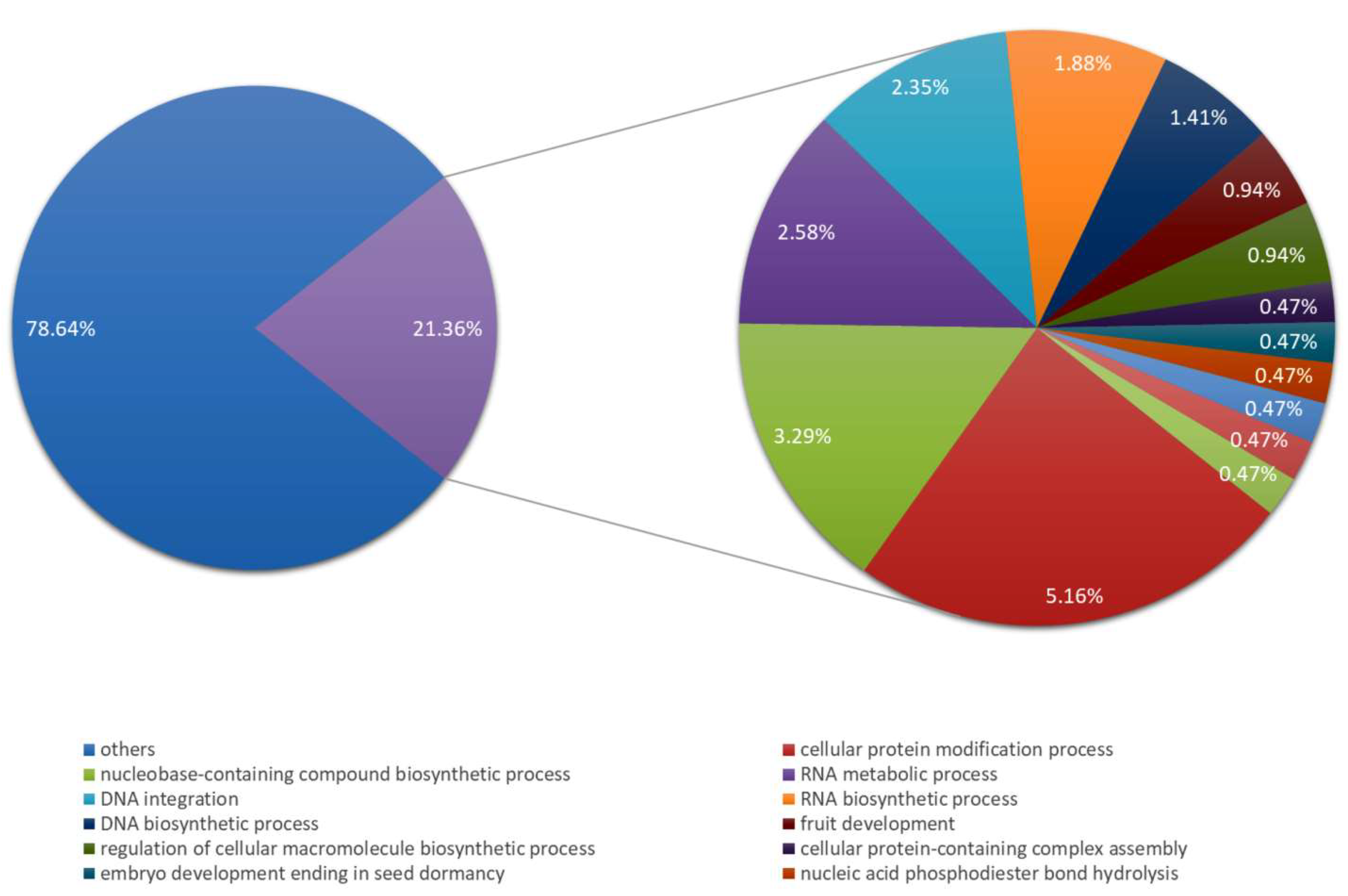
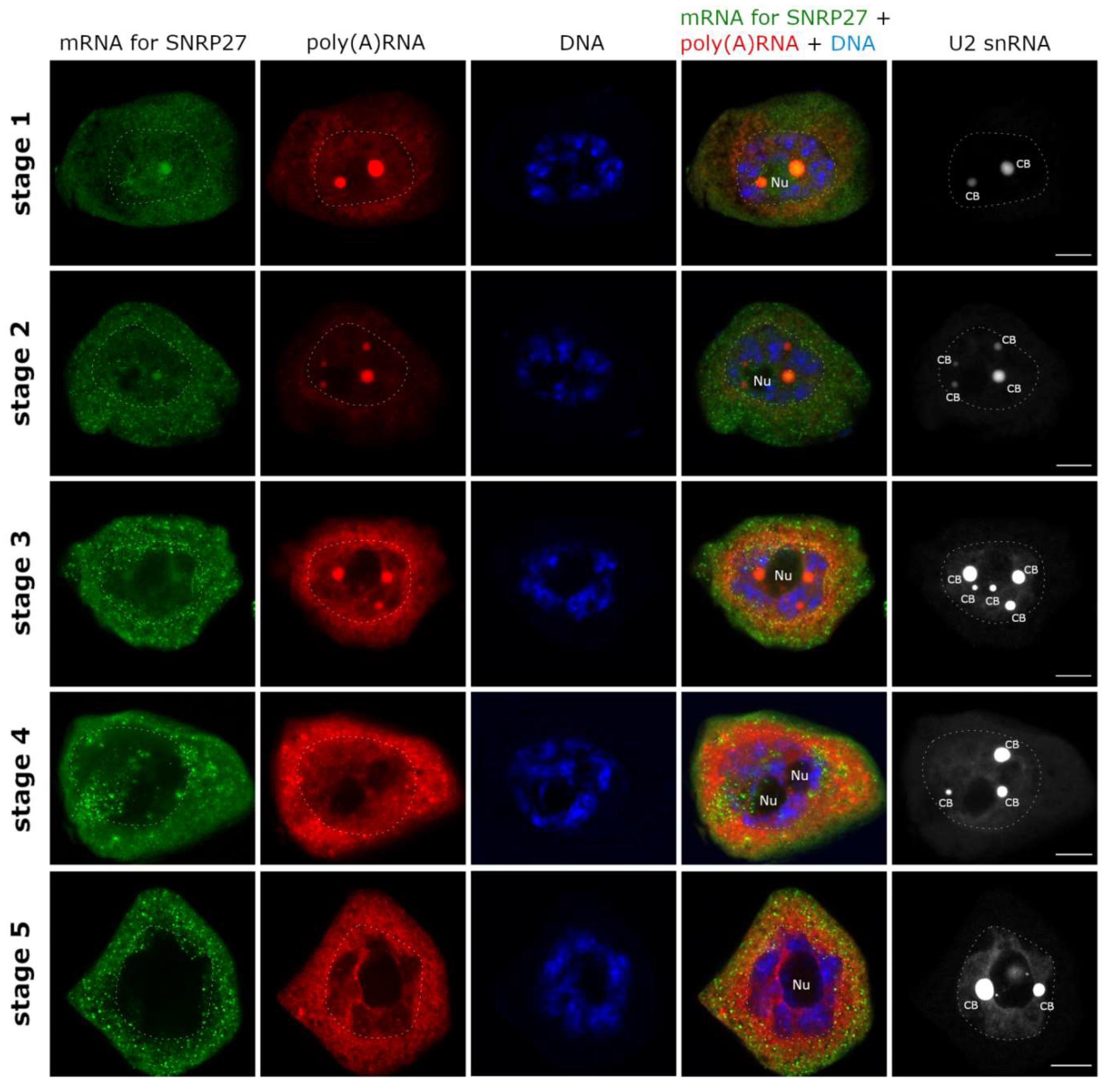
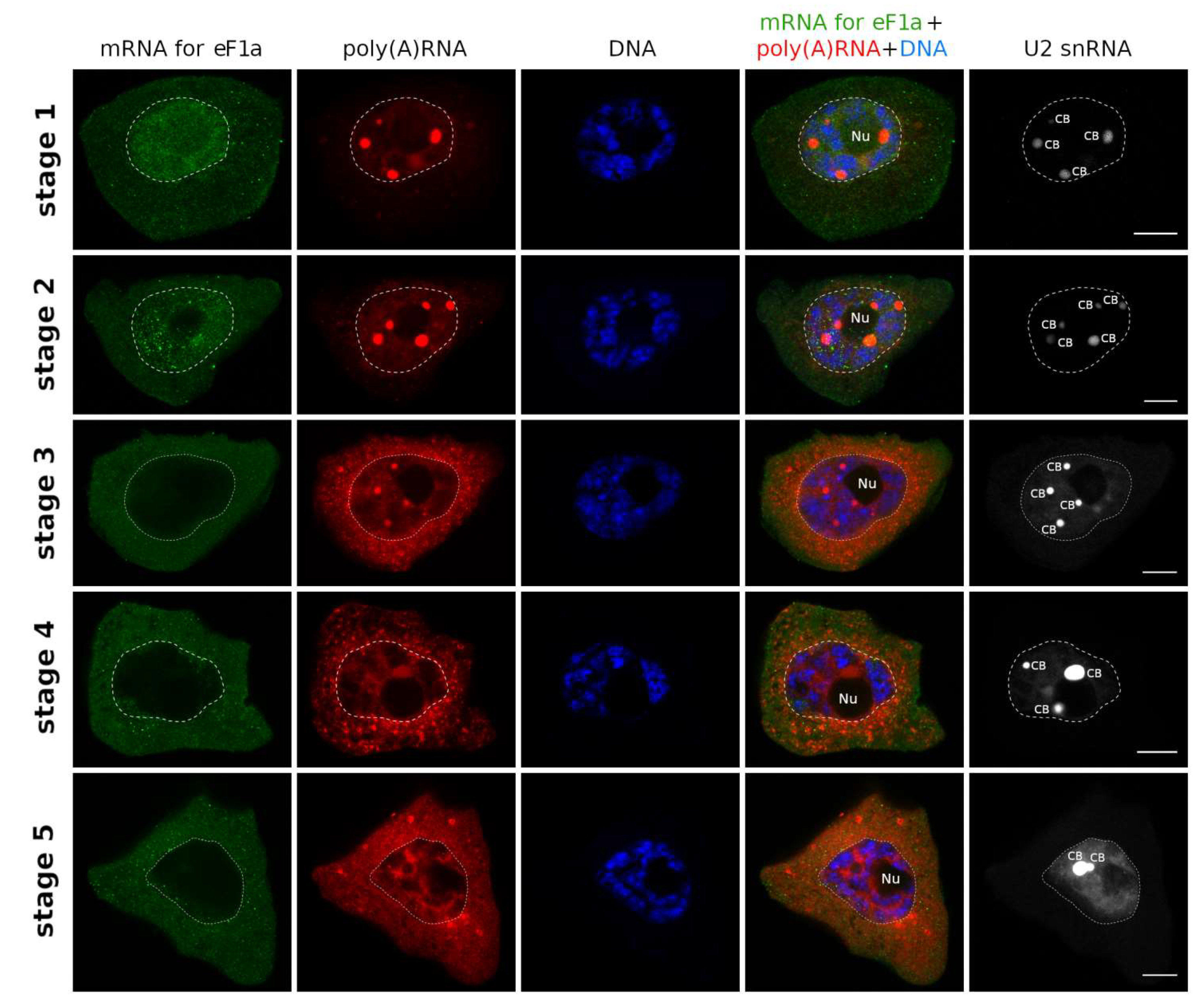
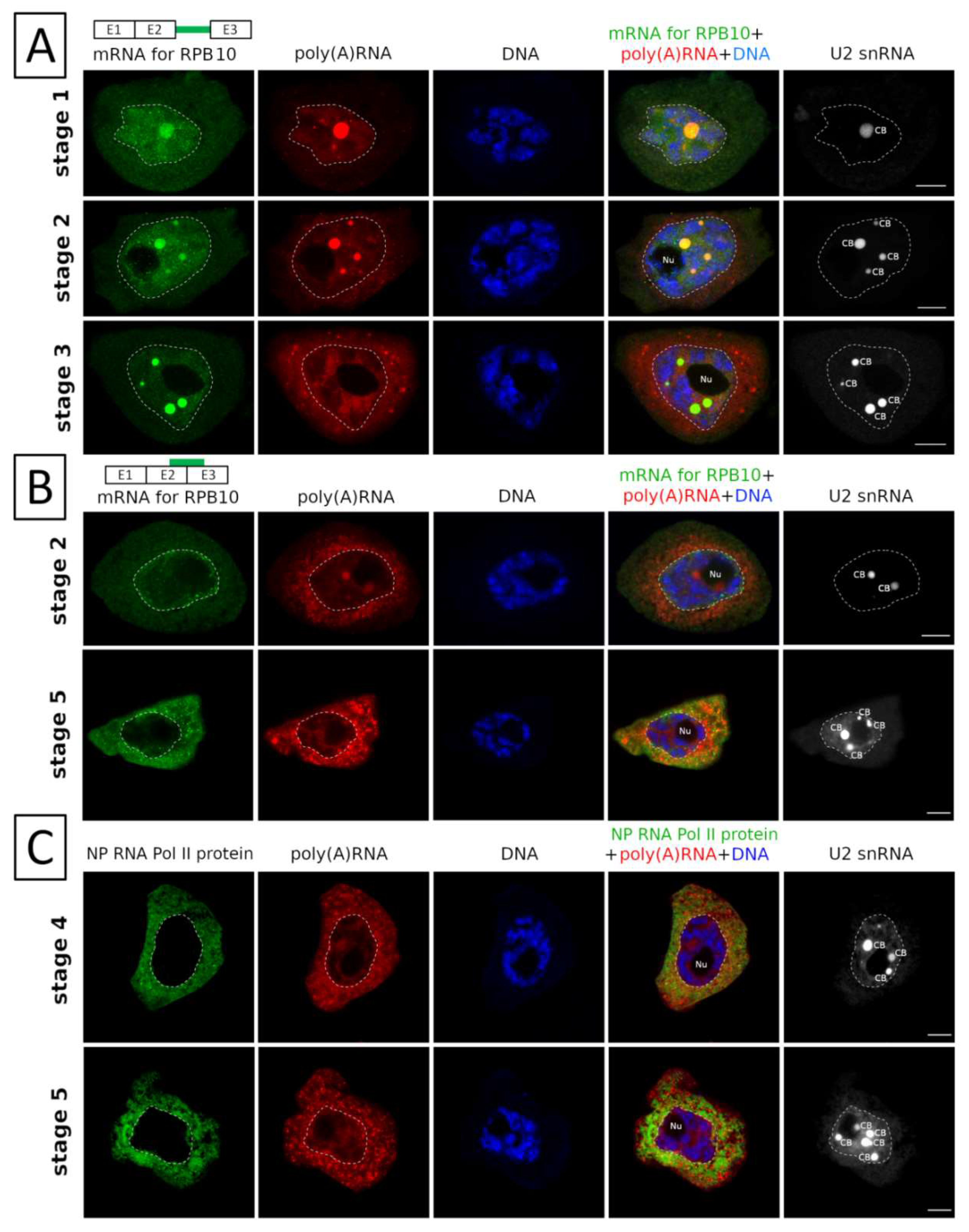
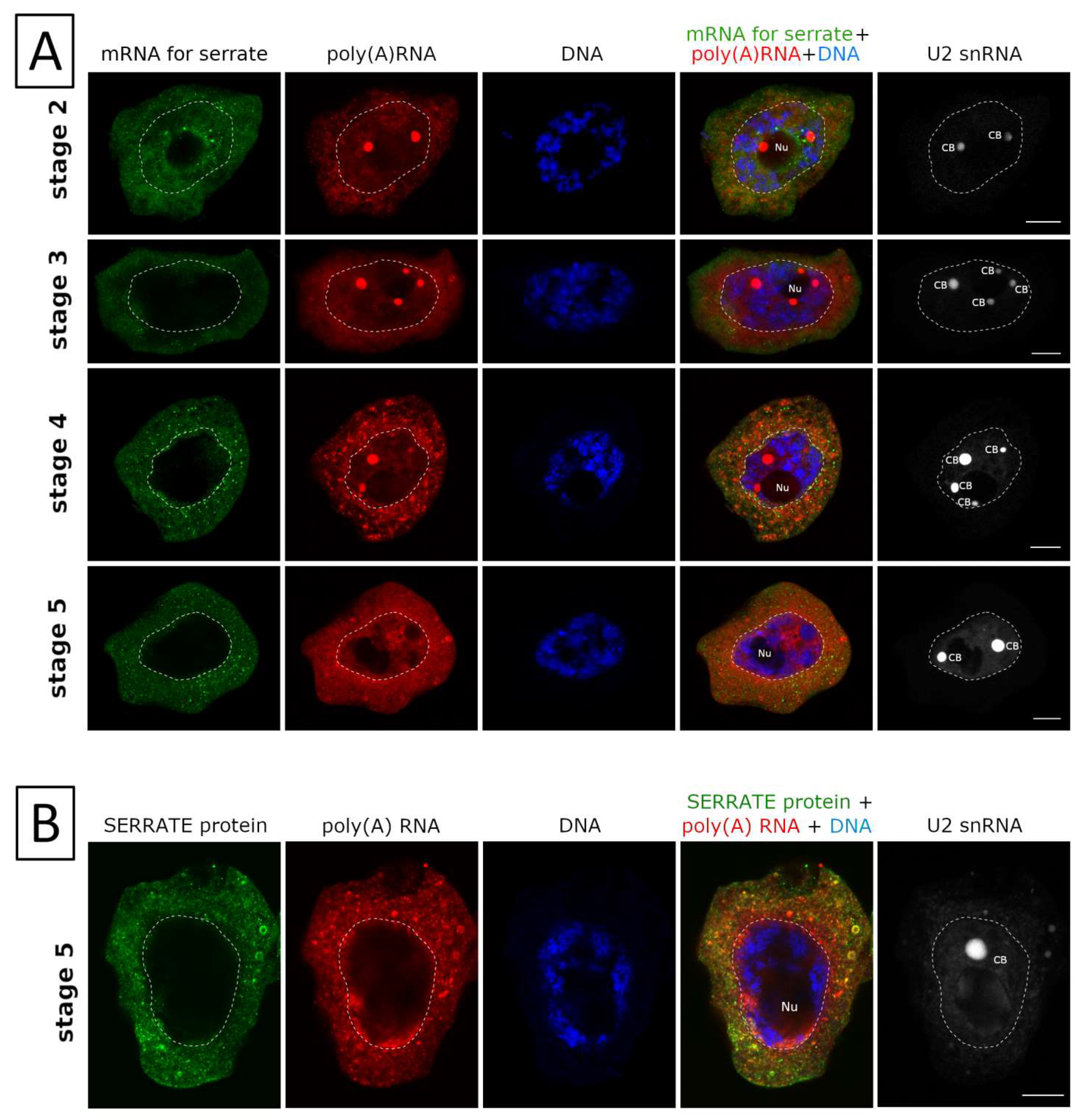
2. Results
3. Discussion
3.1. mRNAs Differ in Nuclear Retention Times and Rates of Export to Cytoplasm
3.2. Two Patterns of Nuclear mRNA Retention in Larch and the Importance of This Process at the Time of the Prophase of the 1st Meiotic Division
4. Materials and Methods
4.1. Plant Materials and Isolation of Meiotic Protoplasts
4.2. FISH in Multiplex Reactions
4.3. Immunodetection of Non-Phosphorylated RNA Pol II and SERRATE Proteins
4.4. Design of Multi-Labelling Immunofluorescence-FISH Reactions
4.5. Confocal Microscopy
4.6. Protoplasts of a Single Bud and Single Branch, Analysis of Synchronicity and Duration of Stages
4.7. Libraries Preparation and Sequencing
4.8. Bioinformatic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability Statement
Acknowledgments
Conflicts of Interest
References
- McAdams, H.H.; Arkin, A. Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 814–819. [Google Scholar] [CrossRef]
- Hasty, J.; Pradines, J.; Dolnik, M.; Collins, J.J. Noise-based switches and amplifiers for gene expression. Proc. Natl. Acad. Sci. USA 2000, 97, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Thattai, M.; van Oudenaarden, A. Intrinsic noise in gene regulatory networks. Proc. Natl. Acad. Sci. USA 2001, 98, 8614–8619. [Google Scholar] [CrossRef] [PubMed]
- Ross, I.L.; Browne, C.M.; Hume, D.A. Transcription of individual genes in eukaryotic cells occurs randomly and infrequently. Immunol. Cell Biol. 1994, 72, 177–185. [Google Scholar] [CrossRef]
- Walters, M.C.; Fiering, S.; Eidemiller, J.; Magis, W.; Groudine, M.; Martin, D.I.K. Enhancers increase the probability but not the level of gene expression. Proc. Natl. Acad. Sci. USA 1995, 92, 7125–7129. [Google Scholar] [CrossRef]
- Blake, W.J.; Balazsi, G.; Kohanski, M.A.; Isaacs, F.J.; Murphy, K.F.; Kuang, Y.; Cantor, C.R.; Walt, D.R.; Collins, J.J. Phenotypic Consequences ofPromoter-Mediated Transcriptional Noise. Mol. Cell 2006, 24, 853–865. [Google Scholar] [CrossRef]
- Urban, E.A.; Johnston, R.J. Buffering and Amplifying Transcriptional Noise During Cell Fate Specification. Front. Genet. 2018, 9, 591. [Google Scholar] [CrossRef] [PubMed]
- Barroso, G.V.; Puzovic, N.; Dutheil, J.Y. The Evolution of Gene-Specific Transcriptional Noise Is Driven by Selection at the Pathway Level. Genetics 2018, 1, 173–189. [Google Scholar] [CrossRef]
- Mugler, A.; Kittisopikul, M.; Hayden, L.; Liu, J.; Wiggins, C.H.; Süel, G.M.; Walczak, A.M. Noise Expands the Response Range of the Bacillus subtilis Competence Circuit. PLoS Comput. Biol. 2016, 12, e1004793. [Google Scholar] [CrossRef]
- Hansen, M.K.; Weinberger, L.S. Post-Transcriptional Noise Control. Bioessays 2019, 41, e1900044. [Google Scholar] [CrossRef]
- Averbeck, N.; Sunder, S.; Sample, N.; Wise, J.A.; Leatherwood, J. Negative control contributes to an extensive program of meiotic splicing in fission yeast. Mol. Cell 2005, 18, 491–498. [Google Scholar] [CrossRef]
- Boothby, T.C.; Zipper, R.S.; van der Weele, C.M.; Wolniak, S.M. Removal of Retained Introns Regulates Translation in the Rapidly Developing Gametophyte of Marsileavestita. Dev. Cell 2013, 24, 517–529. [Google Scholar] [CrossRef]
- Rudzka, M.; Hyjek-Składanowska, M.; Wróblewska-Ankiewicz, P.; Majewska, K.; Gołębiewski, M.; Sikora, M.; Smoliński, D.J.; Kołowerzo-Lubnau, A. Nuclear retention of pre-mRNA involving Cajal bodies during meiotic prophase in plants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Halpern, K.B.; Caspi, I.; Lemze, D.; Levy, M.; Landen, S.; Elinav, E.; Ulitsky, I.; Itzkovit, S. Nuclear Retention of mRNA in Mammalian Tissues. Cell Rep. 2015, 13, 2653–2662. [Google Scholar]
- Prasanth, K.V.; Prasanth, S.G.; Xuan, Z.; Hearn, S.; Freier, S.M.; Bennett, C.F.; Zhang, M.Q.; Spector, D.L. Regulating gene expression through RNA nuclear retention. Cell 2005, 123, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, K.; Kataoka, N.; Hagiwara, M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J. Cell Biol. 2011, 195, 27–40. [Google Scholar] [CrossRef]
- Wegener, M.; Müller-McNicoll, M. Nuclear retention of mRNAs—Quality control, gene regulation and human disease. Semin. Cell Dev. Biol. 2017, 79, 131–142. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Lee, E.S. Sequence Determinants for Nuclear Retention and Cytoplasmic Export of mRNAs and lncRNAs. Front. Genet. 2018, 9, 440. [Google Scholar] [CrossRef]
- Kallehauge, T.B.; Robert, M.C.; Bertrand, E.; Jensen, T.H. Nuclear retention prevents premature cytoplasmic appearance of mRNA. Mol. Cell 2012, 48, 145–152. [Google Scholar] [CrossRef]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Takemura, R.; Takeiwa, T.; Taniguchi, I.; McCloskey, A.; Ohno, M. Multiple factors in the early splicing complex are involved in the nuclear retention of pre-mRNAs in mammalian cells. Genes Cells 2011, 16, 1035–1049. [Google Scholar] [CrossRef]
- Das, S.; Biswas, S.; Chaudhuri, S.; Bhattacharyya, A.; Das, B. A Nuclear Zip Code in SKS1 mRNA Promotes Its Slow Export, Nuclear Retention, and Degradation by the Nuclear Exosome/DRN in Saccharomyces cerevisiae. J. Mol. Biol. 2019, 431, 3626–3646. [Google Scholar] [CrossRef] [PubMed]
- Edens, B.M.; Vissers, C.; Su, J.; Arumugam, S.; Xu, Z.; Shi, H.; Miller, N.; Ringeling, F.R.; Ming, G.L.; He, C.; et al. FMRP Modulates Neural Differentiation through m6 A-Dependent mRNA Nuclear Export. Cell Rep. 2019, 28, 845–854. [Google Scholar] [CrossRef]
- Niedojadło, J.; Dełeńko, K.; Niedojadło, K. Regulation of poly(A) RNA retention in the nucleus as a survival strategy of plants during hypoxia. RNA Biol. 2016, 13, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Kołowerzo-Lubnau, A.; Niedojadło, J.; Świdziński, M.; Bednarska-Kozakiewicz, E.; Smoliński, D.J. Transcriptional activity in diplotene larch microsporocytes, with emphasis on the diffuse stage. PLoS ONE 2015, 10, e0117337. [Google Scholar] [CrossRef]
- Sturrock, M.; Li, S.; Shahrezaei, V. The influence of nuclear compartmentalisation on stochastic dynamics of self-repressing gene expression 2017. J. Theor. Biol. 2017, 424, 55–72. [Google Scholar] [CrossRef][Green Version]
- Battich, N.; Stoeger, T.; Pelkmans, L. Control of Transcript Variability in Single Mammalian Cells. Cell 2015, 163, 1596–1610. [Google Scholar] [CrossRef] [PubMed]
- Monteuuis, G.; Wong, J.J.L.; Bailey, C.G.; Schmitz, U.; Rasko, J.E.J. The changing paradigm of intron retention: Regulation, ramifications and recepis. Nucleic Acids Res. 2019, 47, 11497–11513. [Google Scholar] [CrossRef]
- Naro, C.; Jolly, A.; Di Persio, S.; Bielli, P.; Setterblad, N.; Alberdi, A.J.; Vicini, E.; Geremia, R.; De la Grange, P.; Sette, C. An Orchestrated Intron Retention Program in Meiosis Controls Timely Usage of Transcripts during Germ Cell Differentiation. Dev. Cell 2017, 41, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.-L.; Ritchie, W.; Ebner, O.A.; Selbach, M.; Wong, J.W.H.; Huang, Y.; Gao, D.; Pinello, N.; Gonzalez, M.; Baidya, K.; et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Shalgi, R.; Hurt, J.A.; Lindquist, S.; Burge, C.B. Report widespread inhibi-tion of posttranscriptional splicing shapes the cellular transcriptomefollowing heat shock. Cell Rep. 2014, 7, 1362–1370. [Google Scholar] [CrossRef]
- Kołowerzo, A.; Smoliński, D.J.; Bednarska, E. Poly(A) RNA a new component of Cajal bodies. Protoplasma 2009, 236, 13–19. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full- length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Smolinski, D. Complete diplotene transcriptome of larch microsporocytes. Mendeley Data, V1 2021. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | Sequence |
|---|---|
| total poly(A)—RNA | 5′ Cy3 T(T)29 |
| U2 snRNA | 5′ Cy5 ATATTAAACTGATAAGAACAGATACTACACTTG 3′ |
| mRNA for eF1a | 5′ DIG TGGTGACCTTTGCACCAGTGGGATCCTTCT 3′ |
| mRNA for ACT | 5′DIG TCTTCTGGAGCAACTCGAAGCTCATTGTAGA 3′ |
| mRNA for eIF5b | 5′DIG TCTTCACACCAAGTTCATCTGCAAGCTCACGAG 3′ |
| mRNA for SDH | 5′ DIG ACTTGTTCGTCTCTTCATAGACCTCCCCACCAAT 3′ |
| mRNA for CLATH | 5′DIG TGCAAGATTCACAGCAAGCTCTAGATTATTCAGC 3′ |
| mRNA for SNRP27 | 5′DIG TCGGAATCCCCAATTTCTTCATCATCTC3′ |
| mRNA for RPB10 intron 2 | 5′Dig AAAAGCAAAGATAAGACGATTGATG3′ |
| mRNA for RPB10 on the border of exon2/exon3 | 5′Dig CACTCCTCTCTAAAGTGTTGTAATTCAAAA 3′ |
| mRNA for PEP | 5′Dig TGTAACCCGTGCATAAGGTCCACTCGAACGTGC 3′ |
| mRNA for DNJ | 5′Dig TCTTTAAGGGCATCCTCTCCATACTGGTCA 3′ |
| mRNA for PABP4 | 5′Dig ACTCCATAAAGCCATAGCCCTCTGACTGTCCA 3′ |
| mRNA for RS6 | 5′Dig TTCCTTTGCAGAGTCAATGGTGTCACAAGCC 3′ |
| mRNA for PER40 | 5′Dig TGAATGTTGCACAACGTGCCTTTCCTATTGTA 3′ |
| mRNA for SERRATE | 5′Dig GTTCCATAGTAATCCAATCCATGAACTCGCCA 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majewska, K.; Wróblewska-Ankiewicz, P.; Rudzka, M.; Hyjek-Składanowska, M.; Gołębiewski, M.; Smoliński, D.J.; Kołowerzo-Lubnau, A. Different Patterns of mRNA Nuclear Retention during Meiotic Prophase in Larch Microsporocytes. Int. J. Mol. Sci. 2021, 22, 8501. https://doi.org/10.3390/ijms22168501
Majewska K, Wróblewska-Ankiewicz P, Rudzka M, Hyjek-Składanowska M, Gołębiewski M, Smoliński DJ, Kołowerzo-Lubnau A. Different Patterns of mRNA Nuclear Retention during Meiotic Prophase in Larch Microsporocytes. International Journal of Molecular Sciences. 2021; 22(16):8501. https://doi.org/10.3390/ijms22168501
Chicago/Turabian StyleMajewska, Karolina, Patrycja Wróblewska-Ankiewicz, Magda Rudzka, Malwina Hyjek-Składanowska, Marcin Gołębiewski, Dariusz Jan Smoliński, and Agnieszka Kołowerzo-Lubnau. 2021. "Different Patterns of mRNA Nuclear Retention during Meiotic Prophase in Larch Microsporocytes" International Journal of Molecular Sciences 22, no. 16: 8501. https://doi.org/10.3390/ijms22168501
APA StyleMajewska, K., Wróblewska-Ankiewicz, P., Rudzka, M., Hyjek-Składanowska, M., Gołębiewski, M., Smoliński, D. J., & Kołowerzo-Lubnau, A. (2021). Different Patterns of mRNA Nuclear Retention during Meiotic Prophase in Larch Microsporocytes. International Journal of Molecular Sciences, 22(16), 8501. https://doi.org/10.3390/ijms22168501